

Abstract
In addition to its role as a morphogen, Sonic hedgehog (Shh) has also been shown to function as a guidance factor that directly acts on the growth cones of various types of axons. However, the noncanonical signaling pathways that mediate the guidance effects of Shh protein remain poorly understood. We demonstrate that a novel signaling pathway consisting of protein kinase Cα (PKCα) and integrin-linked kinase (ILK) mediates the negative guidance effects of high concentration of Shh on retinal ganglion cell (RGC) axons. Shh rapidly increased Ca2+ level and activated PKCα and ILK in the growth cones of RGC axons. By in vitro kinase assay, PKCα was found to directly phosphorylate ILK on threonine-173 and -181. Inhibition of PKCα or expression of a mutant ILK with the PKCα phosphorylation sites mutated (ILK-DM), abolished the Shh-induced macropinocytosis, growth cone collapse and repulsive axon turning. In vivo, expression of a dominant negative PKCα or ILK-DM disrupted RGC axon pathfinding at the optic chiasm but not the projection toward the optic disc, supporting that this signaling pathway plays a specific role in Shh-mediated negative guidance effects.
Keywords: Shh, PKCα, ILK, axon guidance
INTRODUCTION
Sonic hedgehog (Shh), a well characterized morphogen, has recently been shown to act as a guidance factor to direct axonal projection. Shh signaling plays important roles in the pathfinding of commissural axons towards the floor plate, along the longitudinal axis of the spinal cord, and in the guidance of retinal ganglion cell (RGC) axons toward the optic disc and at the optic chiasm (Bourikas et al., 2005; Charron et al., 2003; Fabre et al., 2010; Kolpak et al., 2005; Sanchez-Camacho and Bovolenta, 2008; Trousse et al., 2001). We previously demonstrated that Shh plays a dual role in chick RGC axon growth and guidance, acting as a positive factor at lower concentrations and as a negative factor at higher concentrations in vitro (Kolpak et al., 2005; Kolpak et al., 2009). Shh can act directly on the growth cones in a transcription-independent manner, causing growth cone collapse and repulsive axon turning of chick RGCs through Rho GTPase (Kolpak et al., 2009), and inducing attractive axon turning of rat commissural neurons through the Src family kinase (Yam et al., 2009).
Protein kinase C (PKC) is a family of serine/threonine kinases that are classified into conventional PKCs (α, βI, βII, γ), novel PKCs (δ, ε, η, θ) and atypical PKCs (ζ, λ), based on their second messenger requirements (Steinberg, 2008). Conventional PKCs require Ca2+ and diacylglycerols (DAG) for their activation, while novel PKCs require DAG only and atypical PKCs require neither. PKCδ was shown to be involved in the Shh canonical signaling cascade, required for the transcriptional regulation of Gli and Patched-1 (Riobo et al., 2006). Activation of conventional and novel PKCs by phorbol myristate acetate (PMA) elicits both repulsive axon turning of Xenopus spinal neurons and chick RGCs (Kolpak et al., 2009; Xiang et al., 2002) and acute growth cone collapse of ganglia of Helisoma (Zhou et al., 2001). However, since PMA activates multiple PKC isoforms, the roles of specific PKC isoforms and their substrates in axon guidance are not completely understood.
Integrin-linked kinase (ILK), first identified in a yeast-two-hybrid screen as a direct binding protein to the cytoplasmic tail of β1 integrin, has been implicated in cancer cell growth and survival through modulation of downstream targets (Hannigan et al., 2005). By binding to PINCH, parvin and other proteins, ILK functions as an “adaptor” to provide a platform for coupling cell adhesion and growth factor signaling. In neurons, expression of dominant-negative constructs of ILK (E359K or S343A) inhibits neurite outgrowth (Ishii et al., 2001; Mills et al., 2003a) and neuronal polarity determination (Guo et al., 2007). However, the role of ILK in axon guidance has not been reported.
Here, we demonstrate that a novel signaling pathway composed of PKCα and ILK mediates the negative effects of a high concentration of Shh on chick RGC axons. Shh rapidly increased Ca2+ level, activated PKCα, leading to phosphorylation of ILK in the growth cones of RGC axons. Disruption of PKCα and ILK signaling pathway abolished the negative guidance effects of Shh on RGC axons and resulted in aberrant RGC axon pathfinding at the optic chiasm in vivo, demonstrating a critical role of this pathway in Shh-mediated axon guidance.
MATERIALS AND METHODS
Reagents and constructs
Gö6976, PKCβ inhibitor and Rottlerin were purchased from EMD chemicals. Anti-PKCα, βI, δ, ζ, μ and anti-Phospho-PKCα (Ser657) antibodies were obtained from Santa Cruz Biotechnology. Anti-Phospho-PKC (pan) and anti-Phospho-ILK (Thr173) were purchased from Cell Signaling and Abgent, respectively. Anti-phospho-integrin β1 (T788/789) and anti-phospho-PKCβI (Thr642) antibodies were from Invitrogen. Dominant-negative PKCα (Soh and Weinstein, 2003) and RCASBP-Y DV constructs were provided by Dr. B. Weinstein and Dr. W. Pavan through Addgene. Human Slit2 and pGEX-ILK-WT are gifts from Dr. Yi. Rao, Jane Wu (Northwestern Univ.) and Prof. Chuanyue Wu (Univ. of Pittsburgh), respectively. Mutations of ILK were generated by site-directed mutagenesis using QuikChange kit (Stratagene). To generate RCAS constructs, full length DN-PKCα and ILK-Double Mutants (ILK-DM) were first cloned in-frame into entry vector pENTR1A-GFP-N2 (a generous gift from Drs. E. Campeau and P. Kaufman, UMass. Med. Sch.)(Campeau et al., 2009), then a Gateway Cloning system (Invitrogen) was used to recombine target sequences into the retroviral vector RCASBP-Y DV. All constructs were verified by DNA sequencing. RCAS virus was prepared by transfection of a chicken fibroblast line, DF1, and concentrated by ultracentrifugation as described before (Chau et al., 2006).
RGC axon culture and time-lapse experiments
Fertilized White Leghorn eggs (Charles River Laboratories) were incubated in a moisturized 38°C incubator. Axon cultures were prepared as described previously (Kolpak et al., 2009). To prepare RCAS-virus infected RGC axon culture, RCAS viruses were microinjected into optic vesicles at E1.5 and then the embryos were returned to incubator until E6 or E7.
Time-lapse experiments were performed on a Carl Zeiss Axiovert 200 microscope equipped with a 37°C heated stage. Time-lapse images were recorded for 30 minutes at 1-minute intervals. To study the effect of PKC on Shh-induced growth cone collapse, cultures were pre-incubated with 100 nM Gö6976 (EMD Biosciences) or 50 nM PKCβ inhibitor for 30 minutes before adding vehicle, Shh (recombinant Shh-N, R&D system) or Slit2-conditioned supernatant. The Slit2-conditioned supernatant was prepared by transfection of human embryonic kidney HEK 293T cells with an expression construct encoding the human Slit2. Supernatant was used straight without dilution (Kolpak et al., 2009). Growth cone collapse was scored by a loss of lamellipodia and decrease of filopodia number to three or less per growth cone.
Cell fractionation and immunoblotting
Dissected E6 retinas were incubated in media for 15 minutes and then treated with either vehicle or 3.0 μg/ml Shh for the time indicated. After washing twice with ice-cold PBS, retinal lysates were prepared in Buffer A (20 mM Tris-HCl, pH 7.5, 0.25 M sucrose, 2 mM EGTA, 2 mM EDTA, protease and phosphotase inhibitor cocktails) by first passing through a needle and then sonicating. The lysates were centrifuged at 100,000 g for 1 hour and the supernatant was designated as cytosolic fraction. The pellet was re-suspended with buffer A containing 1% TritonX-100 on ice for 30 min. Following centrifugation as before, the supernatant was collected as detergent-soluble fraction. The pellet was dissolved with buffer A containing 1% SDS and designated as detergent-insoluble fraction. Protein concentration was determined by Bio-Rad detergent compatible protein assay. Equal amounts of protein were loaded onto the SDS-PAGE gel and standard western blot protocol was followed.
For analysis of PKC isoform phosphorylation, E6 chick retinas were harvested and treated with vehicle (0.1% BSA) or Shh (3.0μg/ml) for 5 min. Retinas were washed with ice-cold PBS and lysed with RIPA buffer followed by brief sonication. Then the lysates were subjected to western blot analysis with antibodies against phosphorylated PKCα, PKCβI or integrin β1. In some experiments, retinas were pre-incubated with Gö6976 (100 nM) or PKCβ inhibitor (50 nM) for 90 min before Shh treatment.
Protein purification and in vitro phosphorylation
E.coli strain BL21 (DE3) was used for expression of the glutathione S-transferase (GST) fusion proteins of ILK-WT and mutants. GST fusion proteins were purified by glutathione-agarose beads (Sigma) according to the manufacturer’s instruction. For in vitro phosphorylation assay, purified recombinant PKCα (0.1μg) (Millipore) and ILK (0.4μg) were mixed in 20 μl of the reaction mixture (20 mM HEPES, pH 7.4, 10 mM MgCl2, 1 mM CaCl2, 2.5 μM ATP, 0.125μg/ml phosphatidylserine, 200 nM PMA). After addition of 0.75μl of [γ-32P] ATP (10mCi/ml) (Perkin Elmer), the reaction mixture was incubated for 30 min at 30°C. Samples were analyzed on SDS-PAGE and then developed by Phosphoimager.
DiI labeling and immunofluorescent staining
RCAS viral stocks were microinjected into optic vesicles at E1.5 as described previously (Jin et al., 2003) and the embryos were returned to the incubator until E7. At E7, the lens and vitreous body of the right eyes were removed and a small amount of DiI (1mg/ml) was injected into the optic disc with a fine glass micropipette. The embryos were then fixed with 4% paraformaldehyde at 37°C for 2 to 3 weeks. After that, the embryos were imbedded in 3% agarose and sectioned at 150 μm on a vibratome. Sections were mounted on coverslips and examined on a Nikon Eclipse E600 microscope or Leica TBS SP2 confocal microscope.
Immunocytochemistry staining was carried out similarly as described previously (Kolpak et al., 2009). The background staining of the antibodies was determined by omitting the primary antibodies. Fluorescent images were acquired using a 63x objective on a Zeiss Axiovert 200 microscope. To quantify the specific fluorescent signal change in the growth cones and minimize the nonspecific effect due to growth cone size change, we analyzed total amount of fluorescence in the growth cone instead of fluorescence intensity. Concentration of fluorescent signal as a result of a decrease of growth cone size would affect fluorescence intensity but not the total fluorescence amount in the growth cone. Axons were randomly chosen throughout the coverslips and images were taken with a fixed exposure time (below saturation and with minimal background signal) for all samples. For each sample, axons (~40) were manually traced of their outlines within 20 μm from the tips of growth cones including the growth cones and distal segments of axons. Total amount of fluorescence within the outline was then quantified by measuring integrated density in each growth cone by ImageJ.
Axon turning assay, dextran internalization and calcium imaging
Dextran uptake, axon turning assay and data analyses were carried out similarly as in our previous study (Kolpak et al., 2009). For dextran internalization, RCAS-virus infected retinas were dissected at E6 or E7 and cultured overnight. The cultures were treated with 2.5mg/ml 10K tetramethylrhodamine dextran (Invitrogen) with 3.0 μg/ml Shh or vehicle for 2 mins at 37°C, washed, and then fixed in 4% paraformaldehyde. GFP-positive axons were photographed and analyzed.
To study the effect of PKC on Shh-induced axonal turning, RGC cultures were pre-incubated with 5nM Gö6976 (only inhibiting PKCα) (Martiny-Baron et al., 1993) for 1 hour or 50 nM PKCβ inhibitor for 30 minutes prior to the turning assays. For the experiments with the DN-PKCα and ILK-DM-expressing axons, RCAS-viruses infected RGC cultures were prepared as above and turning assay was performed on GFP-positive axons.
For calcium imaging, RGC cultures were loaded with 5 μM Fluo-3 AM (Invitrogen) for 30 minutes, rinsed with media prior to imaging. Imaging was initiated 20 seconds before the pulsing of picospritzer delivering vehicle or Shh protein, with a similar setup as used in the turning assay above. Images were acquired at 1 frame/s with excitation wavelength at 488 nm. In some experiments, 5 μM cyclopamine (Toronto Research Chemicals) was added to the media 30 minutes before application of Shh.
RESULTS
Shh rapidly activates PKCα in the RGC growth cones
As we and others previously showed (Kolpak et al., 2009; Trousse et al., 2001), minutes after the addition of Shh (2.5–3.0 μg/ml) into the culture media, chick RGC axons exhibited rapid growth cone collapse (67.0±2.3 % of total axons collapsed); a loss of lamellipodia and retrieval of filapodia occurred, followed by axon retraction (Fig. 1A). As several PKC isotypes (e.g. α, β, δ, ζ) have been shown to be expressed in RGC axons in mice and chick (Wong et al., 2004; Wu et al., 2003), specific inhibitors of PKC signaling were tested of their ability to inhibit the effect of Shh. Some of them, e.g. a PKCδ inhibitor Rottlerin, caused rapid growth cone collapse by themselves at the recommended concentrations, precluding their use in the study of Shh signaling (data not shown). Gö6976, a specific inhibitor of PKCα and β1 at 100 nM (Martiny-Baron et al., 1993), and a PKCβ-specific inhibitor (Lin et al., 2011; Tanaka et al., 2004), did not have any significant effect on RGC axons when added alone (Fig. 1B). However, pretreatment of the RGC culture with Gö6976 abolished the effect of Shh on RGC axons. Growth cones did not collapse after addition of Shh (10.8±2.7 % of growth cones collapsed) (Fig. 1B), rather remained dynamic with motile lamellipodia and filopodia, and no significant axon retraction was observed. In contrast, the PKCβ-specific inhibitor did not block the Shh-induced growth cone collapse or axon retraction (Fig. 1B). Another negative factor Slit2 also caused growth cone collapse in the chick RGC axons (Kolpak et al., 2009), but preincubation of Gö6976 was ineffective in inhibiting Slit2-induced growth cone collapse (Supplementary Fig.1). These data suggest that PKCα may play a specific role in RGC growth cone collapse in response to Shh.
Figure 1.
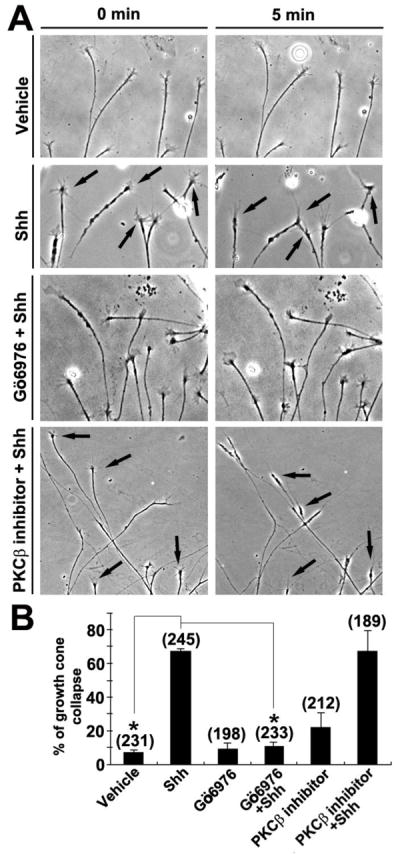
Shh-induced growth cone collapse is blocked by inhibition of PKCα. Time-lapse microscopy of RGC culture was initiated after addition of either vehicle or 3.0 μg/ml Shh. For some experiments, inhibitors to conventional PKC isoforms (Gö6976 or PKCβ inhibitor) were added to the cultures for 30 minutes prior to the time-lapse experiment. A, Bright field images of RGC axons at 0 and 5 min after the addition of vehicle control or Shh. Note that Gö6976 but not the PKCβ inhibitor abolished Shh-induced growth cone collapse. Arrows indicate collapsed growth cones. B, Percentages of RGC growth cones that have collapsed within 5 minutes of addition of vehicle or Shh. Growth cone collapse was defined as loss of lamellipodia and reduction of filopodia number to three or fewer per growth cone. Numbers in parentheses indicate the total number of axons scored from at least three independent experiments. Data are represented as mean±SEM. *p<0.0001, Student’s t test.
One of the hallmarks of activation of various PKC isoforms is the translocation of PKC protein from the cytosol to specialized cellular compartments (Dempsey et al., 2000; Rosse et al., 2010; Shirai and Saito, 2002). To examine the effect of Shh on PKC protein translocation, E6 chick retinas were dissected and incubated with Shh for 5 and 15 min. Retinal cells were then lysed and subjected to ultracentrifugation to separate the cytosolic proteins from the pellets. The pellets were subsequently extracted with TritonX-100 to further fractionate into TritonX-100 soluble membrane fraction and insoluble fraction which includes the cytoskeletons and likely TritonX-100-insoluble lipid rafts. Equal amount of total proteins in the fractions were run on SDS-PAGE and blotted with antibodies recognizing PKC isoforms including anti-PKCα, βI, δ, ζ or μ. Five minutes of Shh treatment lead to a significant increase in the amount of PKCα in the TritonX-100 soluble and TritonX-100 insoluble fractions (average 1.8 and 6.2 folds, respectively) (Fig. 2A,B). The translocation of other PKCs was not significant except for PKCμ which showed ~1.6 fold increase in the TritonX-100-insoluble fraction (Fig. 2B). The amount of PKCδ was detectable in cytosolic factions but too low to be detected in the non-cytosolic fractions. A corresponding decrease of each isotype in cytosolic fractions was not observed, possibly due to the presence of large amount of PKC protein isoforms in the cytosol. To examine whether Shh increased PKCα phosphorylation, another hallmark of PKC activation, E6 chick retinas were incubated with vehicle or Shh for 5 min, and the lysates were subjected to western blot analysis with antibodies against phospho-PKCα (Ser657) or phospho-PKCβI (Thr642). Shh treatment markedly increased the phosphoryation level of PKCα, but not that of PKCβI. It has been shown before that the phosphorylation of Ser657on PKCα can be inhibited by Go6976 (Ginnan et al., 2004). Pre-treatment of the retinas with Gö6976 decreased phospho-PKCα (Ser657) level in response to Shh, while the PKCβ inhibitor appeared to have no effect (Fig. 2C, C′).
Figure 2.

Shh preferentially activates the PKCα isoform. A, Chick retinal explants were treated with 3.0 μg/ml Shh for 5 and 15 min. Cell lysates were processed to result in cytosolic, Triton X-100 soluble and insoluble fractions. Western blots were carried out by using antibodies specific for various PKC isoforms and loading controls were shown blotted with GAPDH, Na+/K+-ATPase and α-tubulin antibodies. Representative gel image of three independent experiments is shown. B, Quantification of translocation of PKC isoforms to TritonX-100 soluble and TritonX-100 insoluble fractions. C, Chick retinal explants were treated with vehicle control, Shh alone or Shh in the presence of Gö6976 or PKCβ inhibitor for 5 mins. Western blots were carried out by using antibodies specific for phospho-PKCα (S657) or phospho-PKCβI (T642). Representative gel image of three independent experiments is shown. C′, Quantification of phospho-PKCα (S657) elevation in chick retinal explants treated with vehicle control, Shh alone or Shh in the presence of Gö6976 or PKCβ inhibitor for 5 mins. Data are represented as mean±SEM. *p<0.05, Student’s t test. D, E, Chick RGC axon cultures were treated with vehicle control, Shh alone or Shh in the presence of Gö6976 or PKCβ inhibitor for 2 mins. Representative immunofluorescent images of antibody staining specific for phospho-PKCα (S657) are shown. The amount of fluorescence in each distal axon was quantified by ImageJ (see Methods) and the mean values were calculated. Numbers in parentheses indicate the total number of axons measured from three independent experiments. F, Fluorescent images of Fluo-3 AM-loaded growth cone at 0″, 33″ and 104″ after the onset of Shh application from a micropipette positioned at ~150 μm from the growth cone. G, Average Fluo-3 fluorescence changes in the whole growth cones after application of vehicle (n=7), Shh (n=7) or Shh with cyclopamine (n=6) were depicted as percent changes normalized to the average fluorescent intensity before the onset of vehicle or Shh application (time 0) (ΔF/F). Data are represented as mean±SEM. *p<0.01, Student’s t test.
At E6, the newly differentiated RGCs and their elongating axons are exposed at the ganglion side of the retina whereas the undifferentiated cells are exposed at the ventricular side (Bao, 2008; Prada et al., 1991). We were not able to purify the RGCs, because the antibody recognizing the chicken Thy-1 required for the panning procedure for purification of RGCs is not available to us. To confirm that PKCα activation occurred in the RGC axon in response to Shh, we performed immunofluorescent staining with an antibody specific for phospho-PKCα(Ser657). While many axons in the control samples appeared negative for the staining, Shh treatment appeared to increase the number of axons containing the fluorescdent puncta positive for the anti-phospho-PKCα (Ser657) antibody staining. Although the fluorescent puncta were not restricted inside the growth cones, they appeared to be more concentrated in the growth cones, suggesting that the initial PKCα activation probably occurred inside the growth cones. For quantification of the results, total amount of fluorescence rather than fluorescence intensity within the growth cone and distal axonal shaft was analyzed, to minimize the effect of concentration of fluorescence due to growth cone size reduction associated with collapse (see Methods). Compared to the vehicle control, 2 min treatment of Shh significantly increased the level of phospho-PKCα (Ser657) in the RGC axons (Fig. 2D, E). Pretreatment of the RGC culture with Gö6976 at a low concentration of 5 nM, which reportedly inhibits PKCα only (Martiny-Baron et al., 1993), abolished the increase of phospho-PKCα (Ser657) in the RGC axons by Shh (Fig. 2D,E), while the PKCβ inhibitor appeared to have no effect. Similar results were obtained by using an anti-phospho-PKC (pan) antibody that recognizes PKCα, βI, βII, δ, ε, η and θ isoforms when phosphorylated on residues homologous to Ser657 of PKCα (Supplementary Fig. 2). The fact that we observed similar increase in the level of phospho-PKCα versus phsopho-PKC (pan) in response to Shh and the increase was abolished by pre-incubation with Gö6976 in both cases, suggests that PKCα is the main PKC isoform that is activated by Shh in the RGC axon.
Binding of Ca2+ to the N-terminal C2 domain is required for the activation of conventional PKCs (Steinberg, 2008). We therefore performed Ca2+ imaging using the cell-permeable Ca2+ indicator Fluo-3 AM. Shh or vehicle control was pulse-applied from a fine glass micropipette positioned at ~150 μm from the growth cone and at 45° angle to the direction of axon extension. Unlike the vehicle control (n=7), Shh rapidly increased Ca2+ level in the growth cones (Fig. 2F and G, p<0.05). The magnitude of Ca2+ increase [ΔF/F=(Ft-F0)/F0] by Shh ranged from ~25% to ~50% and the duration of the increase lasted from 50 seconds to a few minutes (n=7). Pretreatment with cyclopamine, a specific inhibitor of Shh signaling pathway, abolished the Ca2+ increase induced by Shh in the growth cones (n=6). These results demonstrate that Shh rapidly induces Ca2+ elevation in the RGC growth cones.
PKCα directly phosphorylates the integrin-linked kinase (ILK)
We next searched for proteins that act as downstream effectors of PKCα to mediate the effects of Shh in RGC axon guidance. The NetworKin algorithm predicts that PKCα may directly phosphorylate integrin-linked kinase (ILK) at threonine 173 (Linding et al., 2007). As ILK has been shown to be expressed in the neurites of hippocampal neurons, dorsal root ganglion and PC12 cells (Guo et al., 2007; Mills et al., 2003b), we tested the possibility that ILK was downstream of Shh-PKCα by staining the RGC axons with an antibody specifically recognizing ILK that is phosphorylated at threonine 173 (ILK-T173) (Fig 3A and Supplementary Fig. 3). Compared to vehicle control, 2 min treatment of Shh significantly increased the level of phosphorylated ILK-T173 in the RGC axons, which was inhibited by pretreatment of RGC culture with Gö6976 prior to Shh addition (Fig.3B). These data suggest that Shh increased phosphorylation of T173 on ILK through PKCα activation.
Figure 3.

Shh activates ILK through direct phosphorylation by PKCα. A, B, Chick RGC axon cultures were treated with vehicle control, Shh alone or Shh in the presence of Gö6976 for 2 mins. The immunofluorescent signals of phospho-ILK-T173 in each axon were quantified by ImageJ (see Methods) and the mean values were plotted. Numbers in parentheses indicate the total number of axons measured from three independent experiments. Data are represented as mean±SEM. *p<0.001, Student’s t test. C, In vitro kinase reactions were carried out by mixing the purified recombinant PKCα with wild type or ILK single mutant (ILK-T173A). Phosphorylation of ILK was analyzed on SDS-PAGE followed by autoradiography. The upper band shows the autophosphorylation of PKCα (81 kDa) and the lower band shows the phosphorylation of ILK-GST fusion protein (≈76kDa). Without PKCα, auto-phosphorylation of ILK-WT was undetectable. D, Double mutant (ILK-T173A/T181A) but not the other double mutations further reduced ILK phosphorylation to the background level. Equal loading of the gel was verified by western blot using an anti-GST antibody. E, Chick RGC axon cultures were treated with vehicle control or Shh for 2 mins. The immunofluorescent signal of phospho-β1-integrin (T788/T789) in each axon was quantified by ImageJ and the mean values were plotted. F, E6 chick retinas were treated with vehicle or Shh for 5 mins, cell lysates were run on SDS-PAGE gel and blotted with an antibody for phospho-β1-integrin (T788/T789).
Recombinant wild type ILK protein (ILK-WT), as well as a mutant ILK protein with threonine 173 replaced by alanine (ILK-T173A) were produced. In vitro kinase assay was carried out by incubation of purified recombinant PKCα with the ILK proteins without addition of any cellular extracts. As shown in Figure 3C, high level of phosphorylation was observed in ILK-WT and the phosphorylation level was decreased to ~50% in ILK-T173A mutant, suggesting that PKCα can directly phosphorylate ILK on T173 and on an additional unidentified site. A 3-D protein model of ILK was built by using Geno3D and AS2TS softwares and the potential ILK phosphorylation sites were mapped onto the 3-D model. Sites located on the surface of the protein and in close proximity to the T173 were selected for mutagenesis. Six additional sites (T172, T181, S186, S246, T310 and T332) were mutagenized to alanine in the ILK-T173A constructs to generate double mutants. As shown in Figure 3D, ILK double mutant T173A/T181A (ILK-DM) appeared not phosphorylated by PKCα in the in vitro phosphorylation assay, while other double mutants exhibited phosphorylation level similar to that of single mutant (ILK-T173A). Although mixed results were reported whether ILK can be auto-phosphorylated (Acconcia et al., 2007; Hannigan et al., 1996; Wickstrom et al., 2010), auto-phosphorylation of ILK was not detected in our assays (Fig. 3C).
ILK has been implicated in phosphorylation of β1 integrin cytoplasmic domain at threonine 788 and 789 (Hannigan et al., 2005; Hannigan et al., 1996), although direct evidence is still lacking. Since β1 integrin has been shown to be important for chemorepulsion of growth cones by MAG (Hines et al., 2010), we examined whether Shh increased the phosphorylation of T788/T789 on β1 integrin. Both immunofluorescent staining on RGC axons and western blot analysis using whole retina showed that phosphoryation level of β1 integrin at T788/T789 appeared un-affected by Shh treatment (Fig 3E, F). However, this does not rule out that other sites (e.g. S785) may be phosphorylated by ILK.
PKCα-ILK signaling is required for the effects of Shh on RGC growth cone collapse, repulsive axon turning and macropinocytosis
Though large portion of previous research focused on the roles of cytoskeleton in axon guidance, recent studies carried out in our lab and others suggested that asymmetric removal/addition of the growth cone surface area by clathrin and macropinosome-mediated vesicle uptake/release contribute to the growth cone collapse and turning (Hines et al., 2010; Tojima et al., 2006; Tojima et al., 2010). Shh induced macropinocytosis in the RGC growth cones and inhibition of macropinocytosis appeared to block the growth cone collapse elicited by Shh (Kolpak et al., 2009). As different PKC isoforms have been shown to regulate macropinocytosis in different cellular contexts (see Discussion), we assessed the role of PKCα and ILK in Shh-induced macropinocytosis. A dominant-negative PKCα (K368R) with a GFP fusion at C-terminus was constructed into a replication-competent retroviral RCAS vector (RCAS-DN-PKCα). The K368R mutation at the ATP-binding site of the PKCα abolishes its kinase activity (Ohno et al., 1990). In addition, ILK-DM (T173A/T181A) fused with GFP at the C-terminus was also cloned into the RCAS vector (RCAS-ILK-DM). Retroviral stocks were produced and injected into optic vesicles in the chick embryos at E1.5. Injected embryos were returned to incubator for further development until E6 or E7 when the retinas were dissected. Wide spread infection was observed on flat-mount retinas (Fig. 7 A, C, E).
Figure 7.

Intraretinal projection of the RGC axons was not affected by expression of DN-PKCα or ILK-DM. The optic vesicles of the chick embryos were injected with RCAS-GFP, RCAS-DN- PKCα or RCAS-ILK-DM at E1.5. Retinas were harvested at E7, flat-mounted with the ganglion side up, and wide-spread GFP expression was observed (A, C, E). Immunofluorescent staining with an anti-neurofilament antibody (B, D, F ) showed the trajectories of the RGC axons. In all cases, the projection patterns of the RGC axons toward the optic disc appeared normal (some minor aberrant appearance is due to the curvature of the retinal surface). Arrows indicate the direction to the optic disc.
The RGC axon cultures were prepared from the injected samples and dextran uptake assays were carried out by incubation with fluorescently-conjugated dextran together with vehicle control or Shh for 2 min. Axons expressing the DN-PKCα, ILK-DM or control GFP were identified based on GFP fusion protein expression and selected for scoring the dextran uptake. As shown in Figure 4A, dextran-labeled macropinosomes were predominantly present in the growth cone. The control RCAS-GFP-infected axons showed similar percentages of axons containing dex+ vesicles as those in the uninfected samples, with or without the addition of Shh (Kolpak et al., 2009) (data not shown). Samples infected with RCAS-DN-PKCα or RCAS-ILK-DM showed similar rates of dextran uptake in the basal condition without Shh, as those infected with the RCAS-GFP (Fig. 4A′). However, the RCAS-DN-PKCα and RCAS-ILK-DM infected axons showed a significant reduction in Shh-induced macropinocytosis compared to RCAS-GFP-infected samples (Fig. 4A′). Consistently, expression of DN-PKCα or ILK-DM significantly inhibited the Shh-induced RGC growth cone collapse (Fig. 4B, B′). Expression of DN-PKCα appeared to completely abolish the Shh-induced growth cone collapse, whereas expression of ILK-DM resulted in a significant inhibition of axonal response to Shh but to a lesser extent than that of DN-PKCα (Fig. 4B′). These results demonstrate that disruption of PKCα and ILK function diminished the Shh-induced macropinocytosis in RGC axons, and support the notion that negative factor-induced macropinocytosis is closely associated with growth cone collapse elicited by these factors.
Figure 4.

Expression of DN-PKCα or ILK-DM inhibited Shh-induced dextran uptake. A, A′, RGC axons infected with various RCAS viruses were treated with vehicle control or Shh for 2 mins with 10K rhodamine dextran. Representative fluorescent images of GFP-positive axons are shown. Arrowheads indicate dextran labeled macropinosomes. Percentages of dextran+ axons were quantified in the infected GFP-positive axons and the data were normalized to control. B, B′, Growth cone collapse was also scored in the RCAS virus-infected RGC axons in response to 5 min treatment of vehicle control or Shh. Data are represented as mean±SEM. * p<0.05, ** p< 0.01, Student’s t test. Numbers in parentheses indicate the total number of axons scored from three independent experiments. Scale bar, 5μm.
We next set out to test whether PKCα and its downstream ILK play a role in Shh-induced axonal turning. As in our previous report (Kolpak et al., 2009), Shh (2.5–3.0μg/ml) pulsed from a micropipette positioned at a 45° angle with respect to the original direction of axon extension caused repulsive turning of the RGC axons. The majority of growth cones turned away from the Shh gradients, with an average turning angle of − 32.89° compared to the control (< 5°) (Fig. 5A–D). Pre-treatment of Gö6976, but not PKCβ inhibitor, abolished Shh-induced repulsive turning, yielding an average turning angle of 4.74° and −26.43°, respectively (Fig. 5B–D). Random turning of RGC axons was observed in the Gö6976-treated cultures, indicating that inhibition of PKCα did not affect the ability of the axon to turn but affects their turning response to Shh.
Figure 5.

Activation of PKCα and ILK is required for the repulsive axon turning induced by Shh. A, Dark field images of RGC growth cones before (0 min) and after (10 min and 20 min) exposure to a vehicle or Shh gradient delivered by a micropipette positioned at 45° angle to the direction of axon growth (arrows). B, Superimposed traces depict the trajectory of wild type RGC axon extension in the presence or absence of PKC signaling pathway inhibitors in the culture media (upper row). For some experiments, RGC axon cultures were derived from retinas infected with retroviruses, including RCAS-GFP, RCAS-DN-PKCα or RCAS-ILK-DM (lower row). Infected axons were selected based on GFP fusion protein expression. The origin is the centre of the growth cone at the beginning of the recording and the original direction of axon extension coincides with the y-axis. Arrows indicate the direction of the Shh protein gradient. Tick marks on the x- and y-axis represent 5μm. C, Average axonal turning angles and extension under various experimental conditions. Data are represented as mean±SEM. *p<0.001, **p<0.05, Student’s t test. D, Cumulative distribution plots of axonal turning angles of each condition. Each point represents the turning angle of growth cone at the end of 20 min exposure to vehicle control or Shh. The percentage represents the percentage of growth cone bearing turning angle ≤ the value indicated on the x axis. Positive angles represent axon turning toward the pipette, whereas negative angles represent axon turning away from the pipette. Gö, Gö6976, β inhibitor, PKCβ inhibitor.
To further confirm the results, RGC axons infected with the RCAS-DN-PKCα, RCAS-ILK-DM or control RCAS-GFP viruses were tested in the axon turning assays. Identified by expression of GFP, the infected axons were selected for turning assays. Similar repulsive turning was observed in the RCAS-GFP-infected axons in response to Shh, compared to uninfected controls (−23.04° vs − 32.89°, respectively) (Fig. 5B–D). A significant inhibition of Shh-induced turning was observed by expression of RCAS-DN-PKCα or RCAS-ILK-DM, with an average turning angle of −3.14° and −5.89°, respectively (Fig. 5B–D). In all cases, the rates of axon extension were not significantly affected by the viral infection (Fig. 5C). These results demonstrate that PKCα and ILK activities are required for Shh-induced repulsive axon turning.
PKCα-ILK pathway is also required for proper RGC axon guidance at the optic chiasm
Finally, we analyzed the roles of PKCα and ILK in Shh-mediated RGC axonal guidance in vivo. It has been shown that a high level of Shh is present at the anterior and posterior borders of the developing chick optic chiasm to confine the RGC axon projection within the borders at the chiasm (Trousse et al., 2001) (Fig. 8). In mouse, injection of hybridoma producing an antibody to neutralize Shh protein at the chiasm region resulted in an increase of RGC axonal projections into ipsilateral tract and contralateral optic nerve (Sanchez-Camacho and Bovolenta, 2008). However, the signaling pathway that mediates the effect of Shh on RGC pathfinding at the chiasm remains unclear. To determine whether PKCα-ILK pathway may be involved in the effect of Shh on guiding the RGC axon at the chiasm, optic vesicles were injected with the replication-competent RCAS viruses expressing GFP, or the GFP fusion with the DN-PKCα or ILK-DM at E1.5. At E7, fluorescent lipophilic dye DiI was injected unilaterally into the right eye cups to label the RGC axons.
Figure 8.

A model of PKCα-ILK signaling in the RGC axon guidance. A, In chick RGC axons, Shh decreases [cAMP]I (Trousse et al., 2001) and increases [Ca2+]i leading to phosphorylation and translocation of PKCα to the plasma membrane. Activated PKCαphosphorylates ILK. The PKCα-ILK and the Rho GTPase pathways (Kolpak et al., 2009) are critical for Shh-induced negative axon guidance effects. B, In wild-type chick embryos, RGC axons that originate from optic disc cross at the optic chiasm to the contralateral optic tract. B′, Expression of DN-PKCα or ILK-DM results in a portion of axons that fail to respond to Shh, leading to aberrant axon pathfinding at the chiasm. PM, plasma membrane; V, Ventral; D, dorsal; N, nasal; T, temporal; on, optic nerve; ot, optic tract.
In chicken, 100% of RGC axons cross at the optic chiasm to the contralateral side. In the control group expressing GFP, 1 out of 10 embryos showed minor projection abnormality. A small number of errors of axon projection are known to occur during embryonic development which is corrected at later stages (O’Leary et al., 1983). In the embryos with expression of DN-PKCα, the optic nerve appeared loose at the chiasm, and RGC axon misprojection at the chiasm was observed in 100% of the injected embryos (n=10 embryos). In 2 out of 10 embryos injected with RCAS-DN-PKCα, severe misprojection of RGC axons was observed at the chiasm; a large fraction of axons failed to cross the diencephalon midline and projected ipsilaterally, or crossed the midline but deviated into the contralateral optic nerve of the un-labeled eye (Fig. 6B, B′). For the axons managed to project to the correct contralateral optic tract, the axon bundles appeared disorganized at the midline and some splayed out from the bundle (Figure. 6B′). In the rest of the embryos (n=8), a less severe phenotype was observed; small bundles of axons were observed to erroneously traverse into the opposite optic nerve or ipsilateral optic tract (Fig. 6C–D). 100% of embryos injected with RCAS-ILK-DM (n=10) also showed axon misprojection at the chiasm, similar to those less severe phenotypes in the RCAS-DN-PKCα injected samples (Fig. 6E–E′). Axons appeared disorganized, splayed out from the bundle, and erroneously projected to the opposite optic nerve or ipsilateral optic tract. We previously reported that a low concentration of Shh acted as a positive factor in guiding the RGC axon toward the optic disc (Kolpak et al., 2005). No obvious defect of intraretinal projection of RGC axons towards the optic disc was observed by expression of DN-PKCα or ILK-DM (Fig. 7). This is consistent with the in vitro experimental results that the positive effect of low concentrations of Shh on RGC axons was not inhibited by pre-treatment of PKCα inhibitor Gö6976 (data not shown). Therefore, PKCα-ILK pathway along with other signaling events in the axons specifically mediates the negative guidance effect of high concentration of Shh on RGC axons (Fig. 8).
Figure 6.

Expression of DN-PKCα or ILK-DM resulted in misguidance of chick RGC axons at the optic chiasm. The optic vesicles of the chick embryos were injected with RCAS viruses at E1.5 and DiI was injected into the eye cups of the right eyes at E7. DiI was allowed to diffuse for 2 to 3 weeks to label the RGC axon pathway. A, In RCAS-GFP infected retinas, DiI-labeled RGC axons exhibited normal pathfinding at the chiasm projecting into the contralateral optic tract (asterisk). B–E′, Misprojection of RGC axons was found at the chiasm in the samples infected with RCAS-DN-PKCα (B–D) or RCAS-ILK-DM (E, E′). Axons were misrouted into the ipsilateral optic tract (arrowheads) and contralateral optic nerve (arrows). A′, B′, E′ are higher magnification views of the regions boxed in A, B, E, respectively. C, D, Confocal images of two consecutive sections of a RCAS-DN-PKCα infected sample. Vertical dash line indicates the midline. * indicates the contralateral optic tract.
DISCUSSION
In this paper, by both in vitro and in vivo experiments, we demonstrate that a novel signaling pathway consisting of PKCα and ILK specifically mediates the negative guidance effects of high concentration of Shh on RGC axons. Despite the reports of direct actions of Shh on growth cones of various axons, noncanonical signaling pathways of Shh mediating its effects in axon guidance have just begun to be elucidated. Shh rapidly increased Ca2+ concentration, and activated PKCα and ILK in the growth cones of RGC axons. By in vitro kinase assay, we found that PKCα directly phosphorylated ILK, and identified two phosphorylation sites by PKCα on ILK. Inhibition of PKCα or expression of a mutant form of ILK (T173A/T181A) that eliminates the phosphorylation by PKCα significantly inhibited the negative guidance effects of Shh, both in vitro and in vivo.
Expressed in the dorsal and posterior borders of the chiasm in chick and mouse embryos, Shh has been shown to play important roles in promotion of RGC axon fasciculation and defining a constrained pathway at the chiasm (Sanchez-Camacho and Bovolenta, 2008; Trousse et al., 2001). Injection of E13.5 mouse embryos with a hybridoma producing a Shh-blocking antibody resulted in RGC axon guidance defects at the chiasm; the RGC axon bundles were disorganized and expanded at the chiasm region (Sanchez-Camacho and Bovolenta, 2008). In another study, Shh has been implicated to prevent ipsilateral RGC axons from crossing the optic chiasm in mouse through interaction with the Shh receptor Boc (Fabre et al., 2010). As 100% of RGC axons cross to the contralateral side in the chick embryos unlike in mouse, we observed consistent negative effects of high concentration of Shh on RGC axons. Expression of dominant negative PKCα and the double mutant of ILK (T173A/T181A) resulted in similar RGC axonal phenotypes as those by injection of anti-Shh antibody in mouse embryos (Sanchez-Camacho and Bovolenta, 2008). These results support the previous notion that Shh likely act directly on the RGC axons as a guidance factor rather than indirectly in patterning the neural tube (Sanchez-Camacho and Bovolenta, 2008; Trousse et al., 2001). The presence of other guidance factors such as Slit1 and Slit2 in the area may help confine the misprojected axons to existing axonal tracks including ipsilateral optic tract and the contralateral optic nerve rather than entering the preoptic or hypothalamic area, when the response to Shh was interfered at the chiasm (Erskine et al., 2000; Marcus and Mason, 1995). Our growth cone collapse results indicate that the conventional PKC signaling is not involved in the Slit2-induced negative effects on RGC axons.
A novel PKC isoform, PKCδ, was shown to be essential for GLI-dependent reporter transcription in the canonical signaling pathway in the mouse LIGHT2 cells (Riobo et al., 2006). However, the involvement of PKCα in Shh signaling has not been previously demonstrated. Translocation experiments and immunocytochemical staining with antibodies specific for anti-phospho-PKCα and anti-phospho-PKC (pan) confirmed that PKCα was rapidly and most predominantly activated by high-concentration of Shh. The increase of phospho-PKCα appeared to be concentrated in the growth cone but also span the distal axon shaft, suggesting that PKCαis predominantly activated inside the growth cones. This is consistent with the pattern of Ca2+ increase in the growth cone in response to the Shh protein. When Shh was applied at 45° angle to the direction of axon extention, asymmetrical rise of Ca2+ concentration in the growth cones was observed in 3 of 7 axons examined; Ca2+ elevation first occurred at the side of the growth cone facing the Shh source, and then quickly spread to the entire growth cone (data not shown). This transient Ca2+ gradient in response to Shh in the growth cones was similar to a previous report of transient Ca2+ gradient in response to netrin-1 (Hong et al., 2000). We cannot rule out whether the rapid dissipation of asymmetrical Ca2+ increase is due to a technical reason that infusion of Ca2+-sensing dye, such as fluo-3, facilitates the diffusion of Ca2+, eliminating local Ca2+ gradient that would normally persist longer without the dye. In addition, it was necessary to wash out fluo-3 before the initiation of Ca2+ imaging which could disturb the axons resulting in axon stalling or retraction, thus no asymmetrical Ca2+ response could be observed. Previous study has demonstrated that PKCα and PKCγ, can rapidly respond to Ca2+ concentration changes, shuttling between the plasma membrane and cytosol nearly synchronously with the repetitive calcium spikes (Mogami et al., 2003; Oancea and Meyer, 1998). Short-lived Ca2+ signals through PKC activation can be further transduced into long-lived phosphorylation of downstream target genes, modulating long term physiological phenomena (Mogami et al., 2003). Therefore, transient Ca2+ increase on one side of the growth cone may be sufficient to trigger local activation of PKCα and ILK, triggering signaling cascades that lead to repulsive axon turning. However, right now, we don’t know if the transient Ca2+ increase is the result of calcium influx from the medium or calcium release from intracellular store. It will be interesting in future study to detect whether inositol trisphosphate (IP3), which binds to Ca2+ channel on the endoplasmic reticulum(ER) and thus releases Ca2+ from the ER, is involved in Shh-induced Ca2+ elevation in growth cone.
By using a combined approach of pharmacological inhibitors and a dominant negative construct, we were able to inhibit PKCα activity in RGC axons for both the in vitro and in vivo experiments. Widely used in many studies, Gö6976 is reported to inhibit PKCα and β1 at 100 nM, and only PKCα at 5 nM (Martiny-Baron et al., 1993). In contrast to the PKCβ-specific inhibitor, Gö6976 potently inhibited Shh-induced negative effects on RGC axons including growth cone turning, axon retraction, and repulsive turning. Consistent with the acute inhibition of PKCα by Gö6976, longer-term inhibition of PKCα by expression of a well characterized dominant-negative PKCα construct similarly abolished the Shh-induced negative effects in in vitro experiments and resulted in aberrant axon pathfinding at the optic chiasm. Increasing evidence indicates that PKC signaling pathways play important roles in axon guidance. Thrombin and protein tyrosine phosphatase μ (PTPμ)-induced growth cone collapse of rat dorsal root ganglion axon and chick RGC axon were shown to result from selective activation of novel PKCε and PKCδ, respectively (Ensslen and Brady-Kalnay, 2004); while Wnt-mediated attractive guidance of rat commissural axons requires atypical PKCζ (Wolf et al., 2008). The distinct roles of individual PKCs may be attributed to the differences in their molecular structures, mechanisms that regulate their activation, and isoform-selective interacting proteins targeting them to specific substrates (Steinberg, 2008).
Our previous results suggest that Rho GTPase is activated by high concentration of Shh and inhibition of Rho activity blocks the repulsive axon turning induced by Shh (Kolpak et al., 2009). In addition, high concentration of Shh has been shown to decrease cAMP level in RGC axons (Trousse et al., 2001). As growth cone turning involves complex coordinated reorganization of cytoskeleton, regulated membrane retrieval and addition, and local protein translation and degradation (Dent and Gertler, 2003; Lowery and Van Vactor, 2009), it is not surprising that more than one signaling pathways are activated by guidance molecules. In addition, several lines of evidence suggest that cross talk between PKCα and Rho GTPase pathways allow them to coordinate their activities in a number of biological processes. PKCα has been shown to directly interact with Rho GTPase (Pang and Bitar, 2005; Slater et al., 2001) and PKCα can regulate the activity of Rho through regulation of GDI and RND proteins (Madigan et al., 2009; Mehta et al., 2001). On the other hand, Rho GTPase potentiates the activity of PKCαand the activity of PKCα depends on the GTP- or GDP-bound state of the Rho GTPases. Cross talks among various signaling pathways ensure orchestrated and consistent axonal responses to the signaling of extracellular factors.
Our results demonstrate that ILK can be directly phosphorylated by PKCα on T173 and T181 in vitro and the expression of the ILK-DM significantly abolished the negative effects of Shh on RGC axons, suggesting a new role for ILK in axon guidance. ILK has been shown to play important roles as a signaling and scaffolding protein in connecting integrins to the actin cytoskeleton and regulating actin dynamics (Hannigan et al., 2005). The mechanism underlying the effect of ILK-DM on blocking Shh signaling in RGC axons is currently unclear, although interference of interaction of ILK with other proteins is a possibility, as phosphorylation of T173/T181 likely alters the regional surface electrostatic potential of ILK based on molecular modeling (supplemental Figure 4). Previous studies demonstrated that the ILK can increase phosphorylation of AKT or GSK3β, two effectors of PI3K (Guo et al., 2007; Wu and Dedhar, 2001). However, as PI3K pathway is not involved in the effect of high concentration of Shh on RGC axon growth and guidance (Kolpak et al., 2005), ILK appears to play a distinct role in this process. ILK is also directly phosphorylated by p21-activated kinase 1 (PAK1) on T173 and S246 (Acconcia et al., 2007). By in vitro kinase assay, we found that T173 and T181 but not S246 was directly phosphorylated by PKCα (Fig. 3D). We also made retrovirus expressing the single mutant of ILK T173A and expression of the single mutant ILK T173A did not inhibit the Shh-induced RGC axon growth cone collapse (data not shown). Therefore, eliminating the other phosphorylation site by PKCα(T181A) is necessary for blocking the Shh effects on RGC axons.
ILK-DM is less effective than DN-PKCα in inhibition of Shh-induced negative axon guidance effects, suggesting that other effector(s) downstream of PKCα could potentially mediate the negative effects of Shh. Although increased phosphorylation level of β1 integrin on threonine 788 and 789 by Shh treatment was not observed (Hannigan et al., 2005; Hannigan et al., 1996), other sites may be phosphorylated by ILK. The exact role of ILK in Shh-mediated negative axon guidance is currently unclear. The effects of ILK were thought to result from the putative kinase activity of ILK (Guo et al., 2007). However, whether ILK possesses kinase activity has been challenged, especially by the recently available crystal structure of ILK (Fukuda et al., 2009; Wickstrom et al., 2010). Up to present, no report has shown direct interaction between calcium and ILK. Our data support the view that PKCα functions downstream of calcium but upstream of ILK. ILK may function as an adaptor coupling integrin and growth factor signaling through interactions with many proteins including PINCH, parvin, paxillin, and ILKAP (Legate et al., 2006).
We found that inhibition of PKCα-ILK signaling pathway significantly reduced Shh-induced macropinocytosis as well as antagonized the negative effects of Shh on RGC axons including growth cone collapse and repulsive axon turning, supporting that the macropinosome-mediated membrane removal is associated with the negative axon guidance effects of Shh. Although the exact mechanism was not completely understood, previous studies have shown the importance of PKC isoforms in regulating macropinocytosis in various cell types. PMA, an activator of conventional and novel PKCs, was shown to induce membrane ruffling and macropinocytosis in A431 cells (Grimmer et al., 2002). PKCβ along with PKCδ mediate cholesterol accumulation when macropinocytosis of LDL is stimulated in PMA-activated monocyte-derived macrophages (Ma et al., 2006). Furthermore, adenovirus-triggered macropinocytosis in epithelial cells (Meier et al., 2002) and constitutive fluid-phase pinocytosis in dendritic cells (Sarkar et al., 2005) have been shown to require conventional and novel PKCs, respectively. These studies indicate that the specific members of PKC family proteins may regulate macropinocytosis in different cellular contexts and possibly through distinct downstream events. Since the formation of macropinosomes is initiated by F-actin reorganization around the membrane ruffles which subsequently fold and fuse to form macropinosomes (Swanson, 2008), the involvement of ILK in Shh-induced macropinocytosis is not surprising. Given that ILK can regulate F-actin dynamics through a group of actin-associated binding partners (Legate et al., 2006), it is possible that ILK may play a role in the ruffle formation, but the exact mechanism is open to future research.
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health Grant EY014980 (Z.-Z.B.). We thank Dr. E. Campeau and P. Kaufman (UMass Med. Sch.) for providing pENTR vectors, Dr. Chuanyue Wu (Univ. of Pittsburgh) for providing pGEX-ILK-WT construct, Dr. Yi Rao (Beijing University) and Dr. Jane Wu (Northwestern University) for providing Slit2 construct, Dr. Stephen Doxsey (UMass Med. Sch.) for anti-Na+/K+ ATPase antibody, Dong Han and Chenjian Li for technical help on in vitro kinase assay, and Adrianne Kolpak for critical reading of the manuscript.
Footnotes
The authors declare no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Acconcia F, Barnes CJ, Singh RR, Talukder AH, Kumar R. Phosphorylation-dependent regulation of nuclear localization and functions of integrin-linked kinase. Proc Natl Acad Sci U S A. 2007;104:6782–6787. doi: 10.1073/pnas.0701999104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao ZZ. Intraretinal projection of retinal ganglion cell axons as a model system for studying axon navigation. Brain Research. 2008;1192:165–177. doi: 10.1016/j.brainres.2007.01.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourikas D, Pekarik V, Baeriswyl T, Grunditz A, Sadhu R, Nardo M, Stoeckli ET. Sonic hedgehog guides commissural axons along the longitudinal axis of the spinal cord. Nat Neurosci. 2005;8:297–304. doi: 10.1038/nn1396. [DOI] [PubMed] [Google Scholar]
- Campeau E, Ruhl VE, Rodier F, Smith CL, Rahmberg BL, Fuss JO, Campisi J, Yaswen P, Cooper PK, Kaufman PD. A versatile viral system for expression and depletion of proteins in mammalian cells. PLoS One. 2009;4:e6529. doi: 10.1371/journal.pone.0006529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charron F, Stein E, Jeong J, McMahon AP, Tessier-Lavigne M. The morphogen Sonic Hedgehog is an axonal chemoattractant that collaborates with Netrin-1 in midline axon guidance. Cell. 2003;113:11–23. doi: 10.1016/s0092-8674(03)00199-5. [DOI] [PubMed] [Google Scholar]
- Chau MD, Tuft R, Fogarty K, Bao ZZ. Notch signaling plays a key role in cardiac cell differentiation. Mech Dev. 2006;123:626–640. doi: 10.1016/j.mod.2006.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dempsey EC, Newton AC, Mochly-Rosen D, Fields AP, Reyland ME, Insel PA, Messing RO. Protein kinase C isozymes and the regulation of diverse cell responses. Am J Physiol Lung Cell Mol Physiol. 2000;279:L429–438. doi: 10.1152/ajplung.2000.279.3.L429. [DOI] [PubMed] [Google Scholar]
- Dent EW, Gertler FB. Cytoskeletal dynamics and transport in growth cone motility and axon guidance. Neuron. 2003;40:209–227. doi: 10.1016/s0896-6273(03)00633-0. [DOI] [PubMed] [Google Scholar]
- Ensslen SE, Brady-Kalnay SM. PTPmu signaling via PKCdelta is instructive for retinal ganglion cell guidance. Mol Cell Neurosci. 2004;25:558–571. doi: 10.1016/j.mcn.2003.12.003. [DOI] [PubMed] [Google Scholar]
- Erskine L, Williams SE, Brose K, Kidd T, Rachel RA, Goodman CS, Tessier-Lavigne M, Mason CA. Retinal ganglion cell axon guidance in the mouse optic chiasm: expression and function of robos and slits. Journal of Neuroscience. 2000;20:4975–4982. doi: 10.1523/JNEUROSCI.20-13-04975.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabre PJ, Shimogori T, Charron F. Segregation of ipsilateral retinal ganglion cell axons at the optic chiasm requires the Shh receptor Boc. J Neurosci. 2010;30:266–275. doi: 10.1523/JNEUROSCI.3778-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuda K, Gupta S, Chen K, Wu C, Qin J. The pseudoactive site of ILK is essential for its binding to alpha-Parvin and localization to focal adhesions. Mol Cell. 2009;36:819–830. doi: 10.1016/j.molcel.2009.11.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginnan R, Pfleiderer PJ, Pumiglia K, Singer HA. PKC-delta and CaMKII-delta 2 mediate ATP-dependent activation of ERK1/2 in vascular smooth muscle. Am J Physiol Cell Physiol. 2004;286:C1281–1289. doi: 10.1152/ajpcell.00202.2003. [DOI] [PubMed] [Google Scholar]
- Grimmer S, van Deurs B, Sandvig K. Membrane ruffling and macropinocytosis in A431 cells require cholesterol. J Cell Sci. 2002;115:2953–2962. doi: 10.1242/jcs.115.14.2953. [DOI] [PubMed] [Google Scholar]
- Guo W, Jiang H, Gray V, Dedhar S, Rao Y. Role of the integrin-linked kinase (ILK) in determining neuronal polarity. Dev Biol. 2007;306:457–468. doi: 10.1016/j.ydbio.2007.03.019. [DOI] [PubMed] [Google Scholar]
- Hannigan G, Troussard AA, Dedhar S. Integrin-linked kinase: a cancer therapeutic target unique among its ILK. Nat Rev Cancer. 2005;5:51–63. doi: 10.1038/nrc1524. [DOI] [PubMed] [Google Scholar]
- Hannigan GE, Leung-Hagesteijn C, Fitz-Gibbon L, Coppolino MG, Radeva G, Filmus J, Bell JC, Dedhar S. Regulation of cell adhesion and anchorage-dependent growth by a new beta 1-integrin-linked protein kinase. Nature. 1996;379:91–96. doi: 10.1038/379091a0. [DOI] [PubMed] [Google Scholar]
- Hines JH, Abu-Rub M, Henley JR. Asymmetric endocytosis and remodeling of beta1-integrin adhesions during growth cone chemorepulsion by MAG. Nat Neurosci. 2010;13:829–837. doi: 10.1038/nn.2554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong K, Nishiyama M, Henley J, Tessier-Lavigne M, Poo M. Calcium signalling in the guidance of nerve growth by netrin-1. Nature. 2000;403:93–98. doi: 10.1038/47507. [DOI] [PubMed] [Google Scholar]
- Ishii T, Satoh E, Nishimura M. Integrin-linked kinase controls neurite outgrowth in N1E-115 neuroblastoma cells. Journal of Biological Chemistry. 2001;276:42994–43003. doi: 10.1074/jbc.M105198200. [DOI] [PubMed] [Google Scholar]
- Jin Z, Zhang J, Klar A, Chedotal A, Rao Y, Cepko CL, Bao ZZ. Irx4-mediated regulation of Slit1 expression contributes to the definition of early axonal paths inside the retina. Development. 2003;130:1037–1048. doi: 10.1242/dev.00326. [DOI] [PubMed] [Google Scholar]
- Kolpak A, Zhang J, Bao ZZ. Sonic hedgehog has a dual effect on the growth of retinal ganglion axons depending on its concentration. Journal of Neuroscience. 2005;25:3432–3441. doi: 10.1523/JNEUROSCI.4938-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolpak AL, Jiang J, Guo D, Standley C, Bellve K, Fogarty K, Bao ZZ. Negative guidance factor-induced macropinocytosis in the growth cone plays a critical role in repulsive axon turning. J Neurosci. 2009;29:10488–10498. doi: 10.1523/JNEUROSCI.2355-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legate KR, Montanez E, Kudlacek O, Fassler R. ILK, PINCH and parvin: the tIPP of integrin signalling. Nat Rev Mol Cell Biol. 2006;7:20–31. doi: 10.1038/nrm1789. [DOI] [PubMed] [Google Scholar]
- Lin YF, Leu SJ, Huang HM, Tsai YH. Selective activation of specific PKC isoforms dictating the fate of CD14(+) monocytes towards differentiation or apoptosis. J Cell Physiol. 2011;226:122–131. doi: 10.1002/jcp.22312. [DOI] [PubMed] [Google Scholar]
- Linding R, Jensen LJ, Ostheimer GJ, van Vugt MA, Jorgensen C, Miron IM, Diella F, Colwill K, Taylor L, Elder K, Metalnikov P, Nguyen V, Pasculescu A, Jin J, Park JG, Samson LD, Woodgett JR, Russell RB, Bork P, Yaffe MB, Pawson T. Systematic discovery of in vivo phosphorylation networks. Cell. 2007;129:1415–1426. doi: 10.1016/j.cell.2007.05.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowery LA, Van Vactor D. The trip of the tip: understanding the growth cone machinery. Nat Rev Mol Cell Biol. 2009;10:332–343. doi: 10.1038/nrm2679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma HT, Lin WW, Zhao B, Wu WT, Huang W, Li Y, Jones NL, Kruth HS. Protein kinase C beta and delta isoenzymes mediate cholesterol accumulation in PMA-activated macrophages. Biochem Biophys Res Commun. 2006;349:214–220. doi: 10.1016/j.bbrc.2006.08.018. [DOI] [PubMed] [Google Scholar]
- Madigan JP, Bodemann BO, Brady DC, Dewar BJ, Keller PJ, Leitges M, Philips MR, Ridley AJ, Der CJ, Cox AD. Regulation of Rnd3 localization and function by protein kinase C alpha-mediated phosphorylation. Biochem J. 2009;424:153–161. doi: 10.1042/BJ20082377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcus RC, Mason CA. The first retinal axon growth in the mouse optic chiasm: axon patterning and the cellular environment. J Neurosci. 1995;15:6389–6402. doi: 10.1523/JNEUROSCI.15-10-06389.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martiny-Baron G, Kazanietz MG, Mischak H, Blumberg PM, Kochs G, Hug H, Marme D, Schachtele C. Selective inhibition of protein kinase C isozymes by the indolocarbazole Go 6976. J Biol Chem. 1993;268:9194–9197. [PubMed] [Google Scholar]
- Mehta D, Rahman A, Malik AB. Protein kinase C-alpha signals rho-guanine nucleotide dissociation inhibitor phosphorylation and rho activation and regulates the endothelial cell barrier function. J Biol Chem. 2001;276:22614–22620. doi: 10.1074/jbc.M101927200. [DOI] [PubMed] [Google Scholar]
- Meier O, Boucke K, Hammer SV, Keller S, Stidwill RP, Hemmi S, Greber UF. Adenovirus triggers macropinocytosis and endosomal leakage together with its clathrin-mediated uptake. J Cell Biol. 2002;158:1119–1131. doi: 10.1083/jcb.200112067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills J, Digicaylioglu M, Legg AT, Young CE, Young SS, Barr AM, Fletcher L, O’Connor TP, Dedhar S. Role of integrin-linked kinase in nerve growth factor-stimulated neurite outgrowth. Journal of Neuroscience. 2003a;23:1638–1648. doi: 10.1523/JNEUROSCI.23-05-01638.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills J, Digicaylioglu M, Legg AT, Young CE, Young SS, Barr AM, Fletcher L, O’Connor TP, Dedhar S. Role of integrin-linked kinase in nerve growth factor-stimulated neurite outgrowth. J Neurosci. 2003b;23:1638–1648. doi: 10.1523/JNEUROSCI.23-05-01638.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mogami H, Zhang H, Suzuki Y, Urano T, Saito N, Kojima I, Petersen OH. Decoding of short-lived Ca2+ influx signals into long term substrate phosphorylation through activation of two distinct classes of protein kinase C. J Biol Chem. 2003;278:9896–9904. doi: 10.1074/jbc.M210653200. [DOI] [PubMed] [Google Scholar]
- O’Leary DM, Gerfen CR, Cowan WM. The development and restriction of the ipsilateral retinofugal projection in the chick. Brain Research. 1983;312:93–109. doi: 10.1016/0165-3806(83)90124-4. [DOI] [PubMed] [Google Scholar]
- Oancea E, Meyer T. Protein kinase C as a molecular machine for decoding calcium and diacylglycerol signals. Cell. 1998;95:307–318. doi: 10.1016/s0092-8674(00)81763-8. [DOI] [PubMed] [Google Scholar]
- Ohno S, Konno Y, Akita Y, Yano A, Suzuki K. A point mutation at the putative ATP-binding site of protein kinase C alpha abolishes the kinase activity and renders it down-regulation-insensitive. A molecular link between autophosphorylation and down-regulation. J Biol Chem. 1990;265:6296–6300. [PubMed] [Google Scholar]
- Pang H, Bitar KN. Direct association of RhoA with specific domains of PKC-alpha. Am J Physiol Cell Physiol. 2005;289:C982–993. doi: 10.1152/ajpcell.00364.2004. [DOI] [PubMed] [Google Scholar]
- Prada C, Puga J, Perez-Mendez L, Lopez, Ramirez G. Spatial and Temporal Patterns of Neurogenesis in the Chick Retina. Eur J Neurosci. 1991;3:1187. doi: 10.1111/j.1460-9568.1991.tb00053.x. [DOI] [PubMed] [Google Scholar]
- Riobo NA, Haines GM, Emerson CP., Jr Protein kinase C-delta and mitogen-activated protein/extracellular signal-regulated kinase-1 control GLI activation in hedgehog signaling. Cancer Res. 2006;66:839–845. doi: 10.1158/0008-5472.CAN-05-2539. [DOI] [PubMed] [Google Scholar]
- Rosse C, Linch M, Kermorgant S, Cameron AJ, Boeckeler K, Parker PJ. PKC and the control of localized signal dynamics. Nat Rev Mol Cell Biol. 2010;11:103–112. doi: 10.1038/nrm2847. [DOI] [PubMed] [Google Scholar]
- Sanchez-Camacho C, Bovolenta P. Autonomous and non-autonomous Shh signalling mediate the in vivo growth and guidance of mouse retinal ganglion cell axons. Development. 2008;135:3531–3541. doi: 10.1242/dev.023663. [DOI] [PubMed] [Google Scholar]
- Sarkar K, Kruhlak MJ, Erlandsen SL, Shaw S. Selective inhibition by rottlerin of macropinocytosis in monocyte-derived dendritic cells. Immunology. 2005;116:513–524. doi: 10.1111/j.1365-2567.2005.02253.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirai Y, Saito N. Activation mechanisms of protein kinase C: maturation, catalytic activation, and targeting. J Biochem. 2002;132:663–668. doi: 10.1093/oxfordjournals.jbchem.a003271. [DOI] [PubMed] [Google Scholar]
- Slater SJ, Seiz JL, Stagliano BA, Stubbs CD. Interaction of protein kinase C isozymes with Rho GTPases. Biochemistry. 2001;40:4437–4445. doi: 10.1021/bi001654n. [DOI] [PubMed] [Google Scholar]
- Soh JW, Weinstein IB. Roles of specific isoforms of protein kinase C in the transcriptional control of cyclin D1 and related genes. J Biol Chem. 2003;278:34709–34716. doi: 10.1074/jbc.M302016200. [DOI] [PubMed] [Google Scholar]
- Steinberg SF. Structural basis of protein kinase C isoform function. Physiol Rev. 2008;88:1341–1378. doi: 10.1152/physrev.00034.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson JA. Shaping cups into phagosomes and macropinosomes. Nat Rev Mol Cell Biol. 2008;9:639–649. doi: 10.1038/nrm2447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka M, Sagawa S, Hoshi J, Shimoma F, Matsuda I, Sakoda K, Sasase T, Shindo M, Inaba T. Synthesis of anilino-monoindolylmaleimides as potent and selective PKCbeta inhibitors. Bioorg Med Chem Lett. 2004;14:5171–5174. doi: 10.1016/j.bmcl.2004.07.061. [DOI] [PubMed] [Google Scholar]
- Tojima T, Akiyama H, Itofusa R, Li Y, Katayama H, Miyawaki A, Kamiguchi H. Attractive axon guidance involves asymmetric membrane transport and exocytosis in the growth cone. Nat Neurosci. 2006 doi: 10.1038/nn1814. [DOI] [PubMed] [Google Scholar]
- Tojima T, Itofusa R, Kamiguchi H. Asymmetric clathrin-mediated endocytosis drives repulsive growth cone guidance. Neuron. 2010;66:370–377. doi: 10.1016/j.neuron.2010.04.007. [DOI] [PubMed] [Google Scholar]
- Trousse F, Marti E, Gruss P, Torres M, Bovolenta P. Control of retinal ganglion cell axon growth: a new role for Sonic hedgehog. Development - Supplement. 2001;128:3927–3936. doi: 10.1242/dev.128.20.3927. [DOI] [PubMed] [Google Scholar]
- Wickstrom SA, Lange A, Montanez E, Fassler R. The ILK/PINCH/parvin complex: the kinase is dead, long live the pseudokinase! EMBO J. 2010;29:281–291. doi: 10.1038/emboj.2009.376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf AM, Lyuksyutova AI, Fenstermaker AG, Shafer B, Lo CG, Zou Y. Phosphatidylinositol-3-kinase-atypical protein kinase C signaling is required for Wnt attraction and anterior-posterior axon guidance. J Neurosci. 2008;28:3456–3467. doi: 10.1523/JNEUROSCI.0029-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong EV, Kerner JA, Jay DG. Convergent and divergent signaling mechanisms of growth cone collapse by ephrinA5 and slit2. J Neurobiol. 2004;59:66–81. doi: 10.1002/neu.10342. [DOI] [PubMed] [Google Scholar]
- Wu C, Dedhar S. Integrin-linked kinase (ILK) and its interactors: a new paradigm for the coupling of extracellular matrix to actin cytoskeleton and signaling complexes. J Cell Biol. 2001;155:505–510. doi: 10.1083/jcb.200108077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu DY, Zheng JQ, McDonald MA, Chang B, Twiss JL. PKC isozymes in the enhanced regrowth of retinal neurites after optic nerve injury. Invest Ophthalmol Vis Sci. 2003;44:2783–2790. doi: 10.1167/iovs.02-0715. [DOI] [PubMed] [Google Scholar]
- Xiang Y, Li Y, Zhang Z, Cui K, Wang S, Yuan XB, Wu CP, Poo MM, Duan S. Nerve growth cone guidance mediated by G protein-coupled receptors. Nat Neurosci. 2002;5:843–848. doi: 10.1038/nn899. [DOI] [PubMed] [Google Scholar]
- Yam PT, Langlois SD, Morin S, Charron F. Sonic hedgehog guides axons through a noncanonical, Src-family-kinase-dependent signaling pathway. Neuron. 2009;62:349–362. doi: 10.1016/j.neuron.2009.03.022. [DOI] [PubMed] [Google Scholar]
- Zhou, Cohan Growth cone collapse through coincident loss of actin bundles and leading edge actin without actin depolymerization. J Cell Biol. 2001;153:1071–1083. doi: 10.1083/jcb.153.5.1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.