

tRNA gene copy number variation (tgCNV) was analyzed in a set of four Schizosaccharomyces species and in Saccharomyces cerevisiae using available whole-genome sequences. Significant variability in tRNA gene numbers in several of the Schizosaccharomyces was well correlated with codon usage, but surprisingly not in Schizosaccharomyces japonicus. Further analysis suggested the existence of japonicus-specific wobble usage that can rationalize the variation in tRNA content in this organism. Additional whole-genome sequencing of different laboratory strains of Saccharomyces pombe also showed tgCNV.
Keywords: copy number variation, tRNA
Abstract
tRNA genes are interspersed throughout eukaryotic DNA, contributing to genome architecture and evolution in addition to translation of the transcriptome. Codon use correlates with tRNA gene copy number in noncomplex organisms including yeasts. Synonymous codons impact translation with various outcomes, dependent on relative tRNA abundances. Availability of whole-genome sequences allowed us to examine tRNA gene copy number variation (tgCNV) and codon use in four Schizosaccharomyces species and Saccharomyces cerevisiae. tRNA gene numbers vary from 171 to 322 in the four Schizosaccharomyces despite very high similarity in other features of their genomes. In addition, we performed whole-genome sequencing of several related laboratory strains of Schizosaccharomyces pombe and found tgCNV at a cluster of tRNA genes. We examined for the first time effects of wobble rules on correlation of tRNA gene number and codon use and showed improvement for S. cerevisiae and three of the Schizosaccharomyces species. In contrast, correlation in Schizosaccharomyces japonicus is poor due to markedly divergent tRNA gene content, and much worsened by the wobble rules. In japonicus, some tRNA iso-acceptor genes are absent and others are greatly reduced relative to the other yeasts, while genes for synonymous wobble iso-acceptors are amplified, indicating wobble use not apparent in any other eukaryote. We identified a subset of japonicus-specific wobbles that improves correlation of codon use and tRNA gene content in japonicus. We conclude that tgCNV is high among Schizo species and occurs in related laboratory strains of S. pombe (and expectedly other species), and tRNAome-codon analyses can provide insight into species-specific wobble decoding.
INTRODUCTION
tRNA gene copy number is of interest to genome biologists (Wood et al. 2002; Bowen et al. 2003; Dujon et al. 2004; Marck et al. 2006; Higgs and Ran 2008; Rogers et al. 2010; Lowe 2011) because of potential match to codon usage by the transcriptome (Higgs and Ran 2008) and because tRNA genes affect nuclear and genome organization (Wang et al. 2005; for review, see McFarlane and Whitehall 2009; Iwasaki et al. 2010; Nguyen et al. 2010). tRNA gene content can vary much among species, both in number and distribution of iso-acceptors (Sharp et al. 1988; Chan and Lowe 2009). It is common for several highly used codons to have no tRNA that can engage in exact Watson:Crick codon:anticodon pairing (Chan and Lowe 2009). Instead, near-cognate tRNAs are used, involving a complex set of determinants that depend on a variety of tRNA modifications that occur mostly on wobble base U34 (Agris et al. 2007; Johansson et al. 2008).
tRNA gene copy number correlates generally well with codon use in some species (Dong et al. 1996; Berg and Kurland 1997; Percudani et al. 1997; Duret 2000; see Plotkin and Kudla 2011). Although codon bias may reflect differently in different species, that which is most clearly supported is correlation with translation efficiency of mRNAs (Sharp et al. 1986; Sorensen et al. 1989; Plotkin and Kudla 2011). Synonymous codon use can have other outcomes; rare codons can create a translational pause that allows for nascent protein folding (Purvis et al. 1987; Cortazzo et al. 2002), in some cases with a phenotype (Kimchi-Sarfaty et al. 2007), and abundance of rare codons can negatively affect expression (Jansen et al. 2003; Letzring et al. 2010). These observations indicate that the genetic code contains information beyond polypeptide sequence, dependent on match of codon use and cognate tRNA levels. Links between tRNA levels and phenotype attributed to codon-matched mRNAs have emerged (Begley et al. 2007; Pavon-Eternod et al. 2009).
tRNAs may be dynamic in specific activity as well as accumulation levels. An advance in this area was the discovery that the wobble U34 modification enzyme Trm9 controls codon-specific decoding of functionally related mRNAs (Begley et al. 2007). These mRNAs are “keyed” to Trm9 because they are enriched in codons decoded by tRNA substrates of Trm9 but specifically lack synonymous codons whose tRNAs are not substrates of Trm9 (Begley et al. 2007; Maraia et al. 2008). This suggests an additional layer of information in the genetic code that can be deciphered through coordination of synonymous codon use and wobble decoding (Begley et al. 2007). However, while advances in understanding wobble decoding have been forthcoming, deciphering the rules is ongoing (Munz et al. 1981; Agris et al. 2007), and the catalog of biologically used wobble pairs remains incomplete. Johansson et al. (2008) advanced this by working out wobble pairs in Saccharomyces cerevisiae for tRNAs whose U34 is modified by one or more modification enzymes.
By developing algorithms to derive gene copy number information from whole-genome Illumina sequencing, we found a tRNA gene duplication as the phenotypic mutation in spontaneous mutants of Schizosaccharomyces pombe (Iben et al. 2011). Ninety percent of these mutants had gained one or more copies of the tRNA gene (Iben et al. 2011). Recent genome assemblies from Illumina sequence data for four Schizosaccharomyces (Schizo) species were analyzed for protein-coding sequences, transposons, mating, and metabolism as well as genome and chromosome structure (Rhind et al. 2011), but not their tRNAomes. Here we report analysis of the data for tRNA gene copy number variation (tgCNV) and codon use including S. cerevisiae for comparison. tRNA gene numbers vary from 171 to 322 among the Schizosaccharomyces, despite similar genome size.
In addition, we subjected multiple laboratory strains of S. pombe to whole-genome sequencing and compared their tRNA gene content. Copy number variation (CNV) was detected for a tRNA gene cluster in laboratory strains separated for 15 yr, indicating that tRNA gene amplification may be more prevalent than appreciated.
We generated codon use tables and analyzed them for correlation with tRNA gene number for each of the four Schizosaccharomyces yeast species as well as S. cerevisiae sequenced the same way. We show that applying wobble rules significantly improves correlation of tRNA gene number and codon use in S. cerevisiae and three of the Schizosaccharomyces species but severely worsens it for the other, Schizosaccharomyces japonicus. Close examination reveals that the tRNAAlaAGC iso-acceptor gene is absent in japonicus but numerous (nine to 15 copies) in all of the other yeast as well as all of the 77 other eukaryotic species for which tRNA gene content had been predicted by the same algorithm used here, tRNAscan-SE (Lowe and Eddy 1997). The absence of tRNAAlaAGC in japonicus is not accompanied by lower usage of the cognate GCU codon but is accompanied by multifold amplification of tRNAAlaUGC relative to the other yeasts. This led to the proposal that GCU codons must rely on wobble decoding by tRNAAlaUGC in japonicus, an unnecessary wobble in S. cerevisiae and the other Schizosaccharomyces species. This tRNA anticodon:codon pair had not been previously proposed for any eukaryote. Further examination also suggested additional anticodon:codon wobble pairs in japonicus that are not used by S. cerevisiae or the other Schizosaccharomyces yeasts.
RESULTS
tRNA gene copy numbers vary markedly among Schizosaccharomyces species
Genomic sequence from each of the four Schizo genome constructions was obtained from the Broad Institute (Rhind et al. 2011) in addition to S. cerevisiae. tRNA gene counts were predicted using tRNAscan-SE (Lowe and Eddy 1997) to be 171, 249, 322, and 278 for Sz. pombe, Schizosaccharomyces cryophilus, Sz. japonicus, and Schizosaccharomyces octosporus, respectively, and compared with existing data from other eukaryotes, reflecting tgCNV with no correlation to genome size (Table 1). All of the tRNA genes in the five yeasts analyzed here are shown in Table 2. Despite variation in number, each tRNA gene family showed a very high degree of sequence identity within each yeast species (data not shown) (Amstutz et al. 1985). An initial survey revealed japonicus as unusual because some of its tRNA iso-acceptors were at relatively low or null copy number (Table 2, tRNASerAGA, tRNAAlaAGC), while others, low copy in other yeasts, were inordinately high copy in japonicus (Table 2, e.g., tRNASerUGA, tRNAAlaUGC).
TABLE 1.
Genome sizes alongside predicted tRNA counts for yeasts and other species demonstrate wide variability
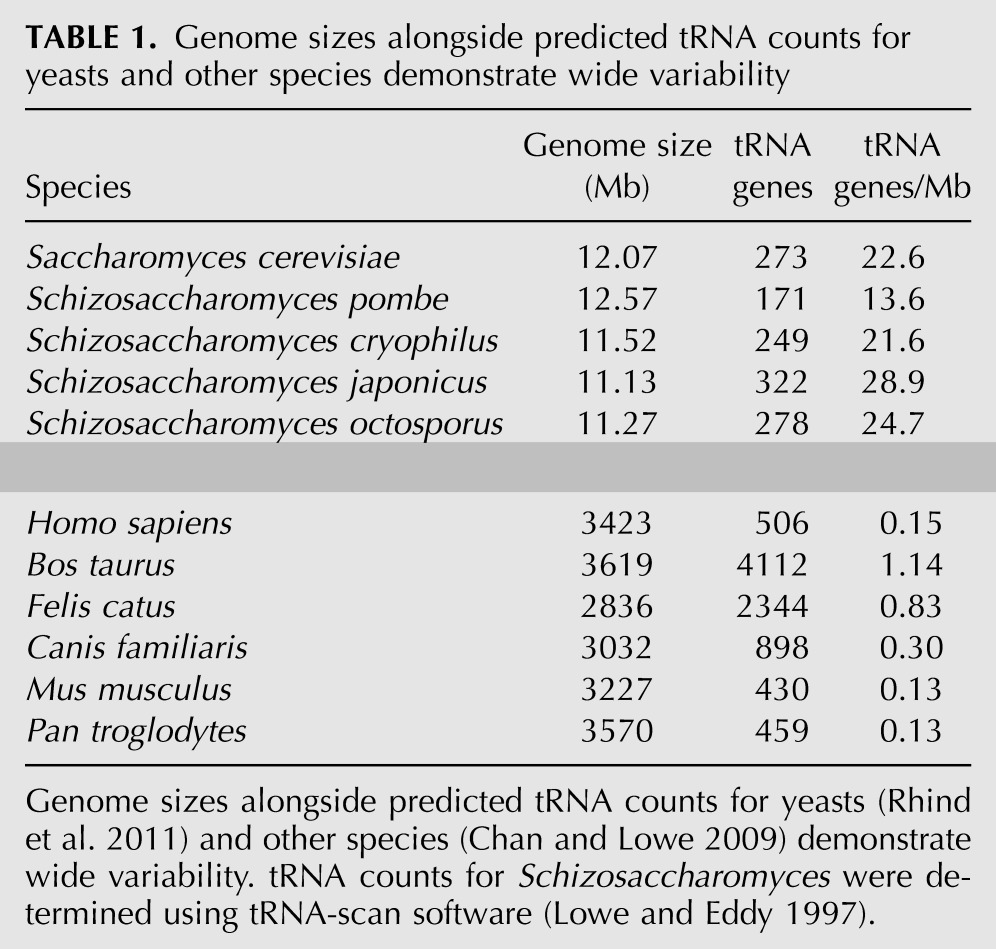
TABLE 2.
Predicted tRNA genes corresponding to different anticodons were tabulated for each species and displayed with calculated genome-wide codon usage rates

tRNA gene CNV occurs in separated laboratory strains of Sz. pombe
We previously found twofold to fourfold increases in the copy number of a suppressor-tRNASerUCA gene in spontaneous mutants of Sz. pombe selected via a genetic screen (Iben et al. 2011). The selected phenotype required that mutants overcome an insufficiency of suppressor-tRNASerUCA in cells bearing a debilitated allele of La (sla1+), which normally protects nascent precursor-tRNAs from 3′-mediated decay, thereby promoting pre-tRNA stability, processing, and folding. Although we expected to obtain mutants in a pre-tRNA decay pathway, instead the tRNASerUCA insufficiency was overcome by increases in the number of tRNASerUCA genes (Iben et al. 2011). tRNA mutants had been isolated from La-deleted S. cerevisiae (lhp1-Δ) cells previously, although as point mutations that impair structure (Yoo and Wolin 1997; Chakshusmathi et al. 2003) rather than tRNA gene duplications.
The high degree of tgCNV observed among the four Schizo species suggested that tgCNV may be apparent in different strains of a species even in the absence of apparent selection. We therefore subjected several strains of Sz. pombe to whole-genome sequencing in parallel, including the reference strain 972h- designated yFS101, which was separated from our oldest laboratory strain, yAS99, by ∼12 yr. We found a significant 2× difference between FS101 and our strains in the number of sequence reads for six of the 171 tRNA genes (Fig. 1).
FIGURE 1.

tRNA gene copy number variation among laboratory strains of S. pombe. Normalized read depth from parallel preparations and sequencing of genomic DNA from pombe strains compared with (A) yAS99 representing the oldest laboratory strain in our laboratory derived collection (Intine et al. 2000) or (B) yFS101. The 171 tRNA gene loci are indicated along the x-axis. (A) Little variation in coverage occurs per tRNA gene among our five laboratory strains. (B) When compared with yFS101, our laboratory strains demonstrate consistent variation, showing increased read depth coverage at the loci containing the highlighted tRNA genes. For each strain, the average read depth was ∼200-fold with genome-wide uniform coverage (data not shown). The Arg(ACG), Glu(TTC), Ile(AAT), Asp(GTC), Ala(AGC), and Val(AAC) each show consistent increased read density, in some cases distributed across multiple identical loci (asterisks), which sum up to be +1×–2× average. (C) tRNA gene copy number in our laboratory strains relative to FS101 calculated from sequence read coverage in a manner different from and confirming B above, displayed for each of the 45 anticodon families in S. pombe.
The five strains from our laboratory are represented in Figure 1A, derived from each other beginning from ∼14 yr ago; they show no significant tgCNV at the loci containing the 171 annotated tRNA genes in the reference Sz. pombe genome and as predicted by tRNAscan-SE. Additional whole-genome sequencing of seven other strains isolated in our laboratory as mutants from different screens also showed no significant CNV in any of the 171 tRNA genes (data not shown). In contrast, parallel sequencing of yFS101 revealed several tRNA loci with 1×–2× increase in tRNA sequence read density in our strains relative to yFS101 (Fig. 1B).
Initial mapping was performed such that reads were allowed to map randomly among multiple copies of identical tRNA genes. Arg(ACG), Glu(TTC), Ile(AAT), Asp(GTC), Ala(AGC), and Val(AAC) each showed increased read density, in some cases distributed unequally across multiple identical tRNA loci (asterisks in Fig. 1B). The sums of these read densities (Fig. 1B, asterisks) produced additional increases for Ala(AGC) and Val(AAC) genes. This suggested amplification of specific Ala(AGC) and Val(AAC) loci even though the mapping randomly distributed over other identical tRNA sequences. To address this, we performed tRNA-independent read scanning that identified specific regions of increased read density that was then cross-referenced with our map of increased tRNAs. This produced an additive total increased density of +2× for each of six tRNAs (Fig. 1C) and also informed us of the loci that had undergone amplification. These data indicate that our strains contain 12 more tRNAs genes than yFS101, and as described below, these appear as two amplifications of a cluster of six tRNA genes.
Furthermore, analysis of the paired-end reads that mapped to the tRNAs indicated that some extended into unique flanking DNA. From these and the context-insensitive increased read densities was formed a simple assembly that identified a contiguous DNA of 2.6 kb containing all six of the amplified tRNA genes and no protein-coding gene on chromosome 2 (position 1617800–1619300). Querying the nonrandom nucleotide sequence database using BLAST revealed a 2.3-kb fragment with 99% identity to “centromere cen2 tRNA cluster, B-repeat region-5” as reported by Carbon and colleagues (Kuhn et al. 1991). Thus, we believe that the amplification of these six tRNA genes likely occurred as an event reflecting the recombination potential of tRNA genes (Pratt-Hyatt et al. 2006; for review, see McFarlane and Whitehall 2009; Iwasaki et al. 2010).
tRNA anticodon distribution is similar in the genomes of the Schizo species except for japonicus but all share similar codon usage
Sz. pombe has the lowest and Sz. japonicus the highest number of tRNA genes among the Schizo species. It is thus notable that for the tRNA genes amplified in our Sz. pombe strains relative to yFS101, their increase trends toward the higher copy numbers in Sz. cryophilus and Sz. octosporus, while their copy numbers are markedly lower in Sz. japonicus (Table 3). This further suggested that japonicus is distinct among the other Schizos in the copy number distribution of its tRNA genes.
TABLE 3.
tRNA gene numbers for the six tRNA genes that exhibit copy number variation among pombe strains yAS99 and yFS101

The fractions of tRNA genes with the same anticodon shows good correlation in all comparisons of Sz. pombe, Sz. cryophilus, Sz. octosporus, and S. cerevisiae, whereas Sz. japonicus shows poor correlation with all others (Fig. 2A; Table 4A). However, the codon use rates predicted from the proteomes (Table 2; Rhind et al. 2011) were similar for all of the yeasts examined including Sz. japonicus (Table 4B).
FIGURE 2.

Correlation of tRNA gene counts per anticodon across yeast species. (A) A high degree of correlation between the distribution of tRNA genes among the anticodon identities exists between most Schizosaccharomyces species with the exception of japonicus. Pearson correlation coefficients are noted and summarized in Table 4. (B,C) Phylogenetic trees derived for the four Schizo species based on their 18S rRNAs (B) and tRNA (C), as indicated.
TABLE 4.
Pearson correlation coefficients for tRNA gene copy number among the different anticodons and for the correlation of tRNA gene number and codon usage rate

We derived a phylogenetic tree based on the 18S rRNA sequences of the four Schizo species (Fig. 2B) that bears a general resemblance to the tree based on 440 core protein-coding genes (see Fig. 1A in Rhind et al. 2011). A phylogenetic tree based on the fractional content of tRNA genes (Fig. 2A; Table 4A) places Sz. japonicus significantly more distant from the other three Schizos (Fig. 2C). This supports the idea that the japonicus tRNAome has diverged from the other Schizo species more so than other components of their genomes.
Codon use correlates with tRNA gene number in the Schizo species except japonicus
It was expected that codon use by the proteome of each species would correlate with tRNA gene counts (Grosjean and Fiers 1982; Sharp et al. 1988). Correlation was lowest for japonicus (Table 5; Fig. 3A). Constitutive highly expressed yeast genes reflect optimization of codon use, whereas conditionally active genes, notably transcription factors, exhibit less optimized codon use (Sharp et al. 1986; Percudani et al. 1997). We found that the correlations were higher for the ribosomal proteins than the transcription factors in all of the Schizo species except japonicus, which showed a striking opposite trend (Fig. 3A–C; Table 5A).
TABLE 5.
Pearson correlations of codon usage rate and tRNA gene count for ribosomal proteins or transcription factor subsets of genes
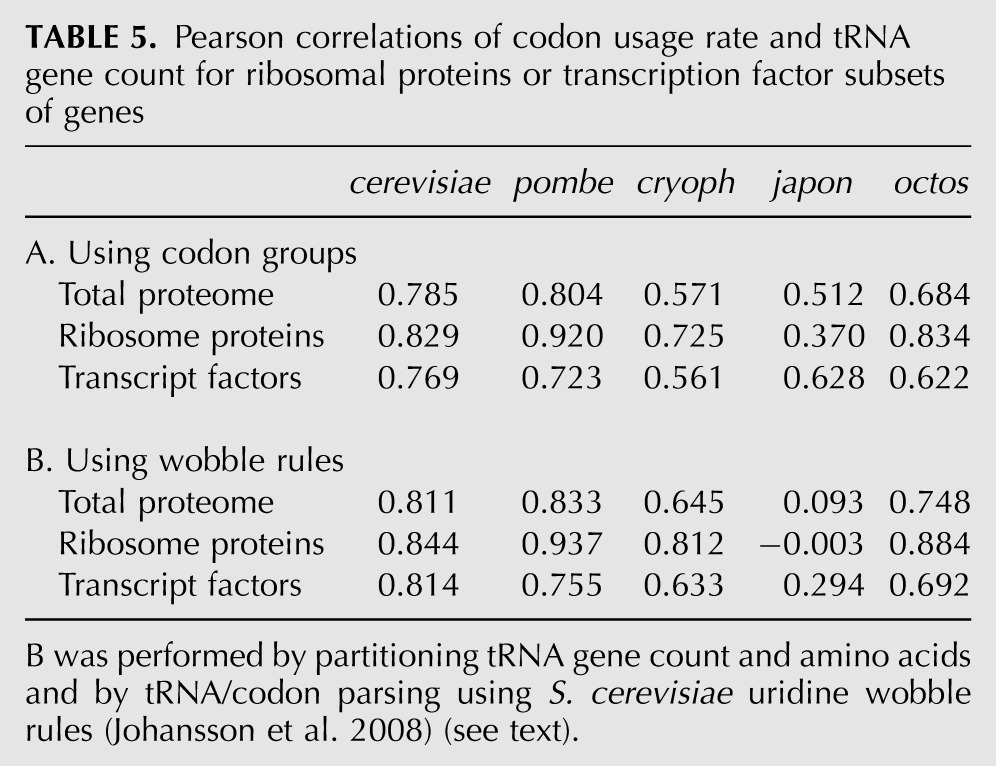
FIGURE 3.

Correlation of tRNA gene number and codon usage improves by partitioning highly expressed genes (ribosomal proteins) in the yeasts except Schizosaccharomyces japonicus. The number of tRNA genes represented as a fraction of the total for each amino acid was plotted against usage rate calculated for (A) the total proteome, (B) ribosomal proteins, and (C) transcription factors for the three yeast species shown, with Pearson correlation coefficients shown. Pearson correlation coefficients were calculated for each of the five yeasts in Table 4. Strong correlations are seen with the exception of Sz. japonicus.
Toward attempts to gain insight into this apparent discrepancy in japonicus, codon use rates were recalculated after elimination of highly codon-biased genes (statistical outliers) (Materials and Methods). By this approach, variance would be reduced and the resulting rates would more closely reflect baseline average; this yielded 90% of the original proteome, from which a set of bias-minimized codon use rates was derived. However, these rates did not improve the correlation of tRNA gene count and codon use for japonicus (data not shown).
Incorporation of S. cerevisiae wobble use increases correlation of tRNA gene count and codon usage in all of the yeasts examined except japonicus in which it decreases
To our knowledge wobble rules had not been applied to correlations of tRNA gene number and codon use. By previous approaches, codons and cognate iso-acceptors were grouped without parsing wobble pairs (Percudani et al. 1997; Duret 2000). We used the wobble pairs determined for S. cerevisiae (Johansson et al. 2008) to partition synonymous codons (Fig. 4; Table 5B). Reassuringly, parsing the wobble pairs (Johansson et al. 2008) improved correlation in S. cerevisiae for total proteome, ribosome proteins, and transcription factors (cf. Fig. 3 with Fig. 4A–C; Table 5). The wobbles also increased correlation for Sz. pombe, Sz. cryophilus, and Sz. octosporus with robustness similar to cerevisiae, but not japonicus, in which it was much decreased in all three protein groups (Fig. 4; Table 5).
FIGURE 4.

Correlation of tRNA gene numbers and codon usage improves by parsing wobble pairs in the yeasts except Sz. japonicus. The number of tRNA genes represented as a fraction of the total was plotted against usage rate but parsing codons and isoacceptors for tRNA wobble, calculated for (A) the total proteome, (B) ribosomal proteins, and (C) transcription factors for the three yeast species shown, with Pearson correlation coefficients shown. Pearson correlation coefficients were calculated for each of the five yeasts in Table 4. Strong correlations are seen with the exception of Sz. japonicus.
Negative effects of wobble rules on the correlation of codon use and tRNA gene numbers might be from two sources: (1) japonicus relies to a significant degree on wobbles not apparent in S. cerevisiae; and (2) a significant fraction of japonicus tRNA genes may be hypoactive for producing functional tRNA. The latter led us to examine the data for sequence evidence of low promoter and/or terminator activity, and/or poor secondary structure in a subset(s) of tRNAs that would contribute to the discordance of tRNA gene counts and codon use in japonicus.
Sz. japonicus tRNA promoter sequences do not reveal an inactive subset(s)
We examined all of the predicted tRNA sequences in japonicus, in comparison with all of those in the other species, both by sequence alignment (data not shown) and sequence Logo (Fig. 5A,B), the latter of which reflects the information content of each consensus position (Schneider and Stephens 1990). While the tRNA genes of many eukaryotes rely only on internal A- and B-box promoter elements for transcription by RNA polymerase (Pol) III, those of Sz. pombe and some other species including plants also use an upstream TATA-box (Hamada et al. 2001). Widespread presence of the Sz. pombe TATA-box was correlated with decreased occurrence of an otherwise conserved G at position 3 of the A-box, as compared with S. cerevisiae and human (Hamada et al. 2001). Sz. japonicus tRNAs exhibit less prevalence at A-box position 3 than Sz. pombe and the other Schizo species, and this was not accompanied by higher prevalence of TATA (Fig. 5A,B). Parsing of promoter sequences did not lead to a tRNA set that when removed from the gene counts increased correlation with codon use (data not shown).
FIGURE 5.

Sequence analyses of RNA polymerase III control elements. (A) Sequences of all of the tRNA genes from each species were aligned and then subjected to Sequence Logo (Schneider and Stephens 1990). Position 1 reflects the first nucleotide of mature tRNA. The A and B boxes are indicated as are the positions of the anticodon (ac) and intron plus variable stem (i/v). (*) The third position of the A box (see text), which shows variable conservation. (B) The sequences upstream of the first nucleotide in all of the tRNA genes are aligned and then subjected to Sequence Logo to evaluate conservation of TATA-box sequences in the different yeasts. (C) Plot of fractional abundance of the oligo(dT) length of the first encountered transcription terminator relative to the total tRNA gene pool for each yeast and for human tRNAs predicted by the same method.
We also considered the possibility that tRNA-ScanSE may have somehow failed to detect tRNAAlaAGC genes in japonicus. We performed two BLAST searches using tRNAAlaAGC sequence in the other Schizos, which are all identical (data not shown). Our searches used sequence positions 1–38 and 39–72, each significantly longer than the A- and B-box promoters that are searched for by tRNA-ScanSE. Our searches found no homologous sequences in the japonicus sequence database (data not shown).
Sz. japonicus tRNA terminators exhibit distinct oligo(dT) profile but this does not contribute to discordance of tRNA gene number and codon use
RNA polymerase III terminates transcription upon encountering an oligo(dT) terminator found at the 3′ ends of tRNA genes. The 3′ oligo(dT) is represented in nascent pre-tRNAs as 3′ terminal oligo(U), which serves as a sequence-specific and length-dependent binding site for the La protein. Since 3′ terminal oligo(U) length can be a determinant of pre-tRNA stability and maturation (Huang et al. 2005; for review, see Maraia and Lamichhane 2011), terminator length could conceivably affect tRNA levels. We examined tRNA terminator lengths, comparing the four Schizo species, as well as S. cerevisiae and Homo sapiens (Fig. 5C). It is important to note for this purpose that four, five, and six consecutive dTs represent minimal efficient Pol III terminators in human, Sz. pombe, and S. cerevisiae, respectively (Hamada et al. 2000), as also reflected by genome profiles (Braglia et al. 2005). Notably, the tRNAome profile of oligo(dT) length in japonicus was distinct from the other Schizo species in three characteristics—width, position, and a relatively high fraction of genes with four T’s (Fig. 5C). However, while the Sz. japonicus profile is interesting in its own right, attempts to partition japonicus tRNA genes according to oligo(dT) length did not improve correlation of codon use and gene number, and in some cases eliminated entire iso-acceptor groups (data not shown).
Predicted secondary structure does not reveal potential unstable subset of japonicus tRNAs
Another possible explanation of the apparent discordance of tRNA gene number and codon use in japonicus was that some tRNAs may be unstable due to poor secondary structure. To address this, the Cove scores from tRNAscan-SE, which incorporate secondary structure information, were filtered using a sliding cutoff. However, correlation of tRNA gene number and codon use was not improved regardless of the stringency (data not shown). Thus, the possibility that a significant subset(s) of japonicus tRNA genes is deficient for transcription or accumulation could not be supported by analyses of the tRNA sequences themselves.
Conservation of tRNA wobble base modification enzymes among the Schizo species
Worsening of the correlation of codon use and tRNA gene number by incorporation of wobble rules suggested that japonicus uses wobbles not used by other yeasts and/or vice versa. We compared the known yeast tRNA wobble base modification enzymes of S. cerevisiae for homologs in the Schizo species (Chan et al. 2010; Phizicky and Hopper 2010). We found homologs in all of the species with no obvious variance in homology (data not shown). We next examined the distribution of tRNA iso-acceptors for evidence of wobble use by japonicus that would not be apparent from the known S. cerevisiae wobbles (Johansson et al. 2008).
Informatics evidence that S. japonicus uses tRNAAlaUGC to wobble decode GCU
Alanine is encoded by four codons, of which the most abundant in yeasts is GCU, which is decoded by tRNAAlaAGC, the most numerous of the Ala tRNA genes in the yeasts, with the striking exception of japonicus, which has none (Table 2). Despite this, usage rates among Ala codons is similar in japonicus to the other yeasts, with GCU used most. This raises the question of how GCU codons are decoded in japonicus. Although there are three genes for tRNAAlaCGC, C34 does not decode a wobble U and cannot be used for GCU codons (Agris et al. 2007). As in the other yeasts, there is no tRNAAlaGGC in japonicus (Table 2). This leaves tRNAAlaUGC as the only tRNA to decode GCU codons in japonicus. Consistent with this expectation and absence of tRNAAlaAGC, the tRNAAlaUGC gene was found multifold amplified in japonicus relative to the other yeasts (Table 2).
The second most plausible wobble in japonicus that is not apparent in S. cerevisiae involves tRNASerUGA, which is highly amplified relative to the other yeasts (Table 2). In contrast, tRNASerAGA is normally the most abundant of the tRNASer’s in the other yeasts (seven to 11 copies) but only one copy in japonicus (Table 2). Despite this disparity, UCU is the most abundant serine codon in all of the species examined, including japonicus (Table 1). This leads to the proposal that tRNASerUGA decodes UCU codons in japonicus, while this wobble is unnecessary in S. cerevisiae (Johansson et al. 2008). It is noteworthy that for both tRNASerUGA and tRNAAlaUGC, the proposed wobble involves U34.
japonicus wobbles partially improve correlation of tRNA gene counts and codons
Using the wobble pairs determined for S. cerevisiae led to improvement in the correlation of tRNA gene counts and codon usage in all yeasts examined except japonicus, in which it was drastically worsened (Figs. 3, 4; Table 5). We reasoned that if the wobbles proposed for japonicus were actually used, parsing the appropriate synonymous codons might increase the correlation with tRNA gene count more so than do the S. cerevisiae rules. A more thorough examination of japonicus tRNAs for imbalances of iso-acceptors relative to other yeasts uncovered tRNAValUAC as wobble decoding GUU, and what appears to be tRNAGlyUCC wobble decoding GGU, in addition to the Ala and Ser wobble pairs noted above. The reasoning for the tRNAGlyUCC wobble decoding GGU is as follows: Although tRNAGlyGCC could wobble decode the GGU codon, its gene number of four in japonicus is significantly lower than the eight, 17, and 18 copies in the other Schizo species (Table 1). In contrast, tRNAGlyUCC gene number is higher in japonicus than in the others. Specifically, the ratio of tRNAGlyUCC to tRNAGlyGCC gene numbers is 2.5 in japonicus but 0.38, 0.47, and 0.39 in the others. This suggests that japonicus may prefer to use U:U over U:G wobble.
This information was used to adjust the S. cerevisiae wobble rules for Sz. japonicus (Johansson et al. 2008) (Materials and Methods). Application of these modified rules, referred to as Sj-adjusted rules, significantly increased the correlation of tRNA gene copy number and codon use for all three protein-coding groups in Sz. japonicus but less so for the other yeasts (Fig. 6). In Table 6 are the individual changes to correlation values for each genome for each of the four new tRNA donors (SerTGA, AlaTGC, GlyTCC, and ValTAC) separately. Each of these improved japonicus correlation individually. Table 6 also shows the difference for each of the four donors relative to the total change in correlation from the cerevisiae rules (Sj-rules and Difference, respectively). That the improvement with Sj rules was significant but only partial suggests that there may be other wobble rules that we have not tried to account for or speculate about here. Nonetheless, partial improvement should not detract from the fact that the wobble rules that we have elucidated do, indeed, improve the correlation. The data support the idea that the tRNAome of Sz. japonicus has evolved to rely on a subset of wobbles unnecessary in the other yeasts.
FIGURE 6.

Species-specific improvement of the correlation of tRNA gene count and codon usage upon application of Sz. japonicus–adjusted wobbles. Four additional wobbles of U(NN) anticodons are added to the cerevisiae defined rules (Ala-TGC, Gly-TCC, Ser-TGA, Val-TAC) enabling decoding of the respective four-box through those tRNA genes. Although a significant improvement is seen for japonicus, little change is seen in correlation scores of most yeasts. Graphed data are numerically presented in Table 6.
TABLE 6.
Comparison of correlation of codon use and tRNA gene number with and without S. cerevisiae (Sc) or japonicus-adjusted (Sj) wobble rules

DISCUSSION
Four conclusions can be derived from this work: (1) There has been a significant amount of tRNA gene copy number variation (tgCNV) in the genomes of the four Schizosaccharomyces species investigated here. (2) We found significant tgCNV in different laboratory strains of S. pombe. (3) The tRNAome of Sz. japonicus has been most divergent among the Schizosaccharomyces species in iso-acceptor constitution, discordance with codon use rates, and oligo(dT) terminator length. (4) Comparative tRNAome analyses indicate use of a tRNA anticodon:codon wobble pair in Sz. japonicus for which there has been no prior evidence in a eukaryote. Although wobble decoding of GCU codons by tRNAAlaUGC is consistent with wobble rules involving U34 (Agris et al. 2007), to our knowledge there has not been biological evidence for use of this pair. This wobble is not readily apparent in S. cerevisiae, which has an abundance of an tRNAAlaAGC to decode the GCU codon. A similar argument can be made for tRNASerUGA to decode UCU codons.
The tgCNV found between our Sz. pombe strains and yFS101 involves a cluster of six tRNA genes near the centromere of chromosome 2 (Kuhn et al. 1991), apparently amplified twice, leading to 12 more tRNA genes in our strains relative to yFS101 (Table 3).
Analysis of all of the 77 eukaryotic genomes for which all tRNA genes were predicted using tRNAscan-SE available on the tRNA gene database reveal none that lacks a gene for tRNAAlaAGC, although absence of a tRNAAlaAGC gene is exceedingly common in archaea and Eubacteria (Chan and Lowe 2009; Lowe 2011).
Our strongest and most clear data suggest that U:U wobble is for tRNAAlaUGC in japonicus. For SerUGA and ValUAC, there is a possibility that another tRNA for Ser or Val, with A at position 34, may contribute to decoding following A-to-I editing, although for tRNAAlaUGC and tRNAGlyUCC, A-to-I editing cannot apply since there is no tRNA gene with A in the 34 position.
The wobble base uridines of tRNAs are decorated with a variety of complex modifications whose precise chemical composition can either restrict or extend wobble capability, including pairing with U in the codon wobble position (Agris et al. 2007). The data uncovered lead us to propose that tRNAAlaUGC, tRNASerUGA, tRNAValUAC, and tRNAGlyUCC engage in wobble U:U decoding in Sz. japonicus. We do not know the modification state of any of the U34’s in these tRNAs. However, it was recently determined that the corresponding tRNAs in S. cerevisiae, tRNAAlaUGC, tRNAValUAC, and tRNASerUGA contain 5-carbamoylmethyl-U34 (ncm5U34), while tRNAGlyUCC contains 5-methoxycarbamoylmethyl-U34 (mcm5U34) (Johansson et al. 2008). It is possible for unmodified U:U base pairs to form with significant stability although unmodified U:U wobble pairing in the ribosome had not been observed (for review, see Yokoyama and Nishimura 1995; Agris et al. 2007). It is therefore notable that recent evidence indicates U:U wobble decoding of CCU by tRNAProncm5UGG (Johansson et al. 2008). Moreover, the ability of tRNAProUGG to decode CCU occurred even in the elp3Δ strain, which lacks the ncm5 U34-modification (Johansson et al. 2008). From that study it appears that tRNAProUGG with an unmodified U34 can decode CCU, and furthermore, that the presence of ncm5U in tRNAProUGG does not interfere with decoding CCU. If the same applies to tRNAAlaAGC in japonicus, either an unmodified or a ncm5-modified U34 may support decoding of GCU codons. Unfortunately, Sz. japonicus has only recently attracted attention as a model system with very few tools available for manipulation, and it is beyond our present means to determine which if any modifications affect the ability of tRNAAlaUGC to decode GCU.
The U:U wobble pair of tRNAProUGG and CCU codon observed in S. cerevisiae supports the idea that U:U wobbles also occur in S. japonicus as the most likely mechanism of compensating for loss of tRNAAlaAGC with no change in GCU codon use. We should also consider other compensatory mechanisms, such as RNA editing, although this seems less likely. Since we have used genomic DNA sequence for this analysis, it is possible that some japonicus tRNAs are edited. A-to-I, and C-to-U editing has been observed at wobble positions of tRNAs, but not editing of U (pseudouridylation, considered a modification, would not resolve the U:U wobble issue) (Paris et al. 2011). Thus if amplified tRNAAlaUGC genes in japonicus compensate for loss of tRNAAlaAGC by a mechanism that does not involve U:U wobble, one may imagine U34 editing to A, G, or I, although such editing has not been observed (Paris et al. 2011). Alternatively, RNA editing of GCU codons in mRNA may occur, although more unlikely as GCU codons occur on a transcriptome-wide scale.
It is intriguing that Sz. japonicus has diverged from the other yeasts to the degree that it has in its tRNA gene distribution and apparent reliance on U:U wobble decoding. This suggests that other aspects of decoding may also be divergent in this fission yeast. As such, it may be interesting to examine the exact modification profiles on some of its tRNAs and/or if there is any divergence in its 18S rRNA sequence in the vicinity of the decoding center. In any case, the apparent reliance of Sz. japonicus on U:U wobble pairs suggests this yeast as a possible source of new insights into how eukaryotes deal with wobble decoding.
MATERIALS AND METHODS
Estimation of tgCNV in different strains of S. pombe
yFS101 representing wild-type (h-972) Sz. pombe was kindly provided by Nick Rhind, who obtained it from Paul Russell’s laboratory, who in turn brought it from Paul Nurse’s laboratory (N Rhind, pers. comm.). This strain is also known as ATCC 24843, deposited by A.M. Ali, who obtained it from U. Leupold. Our laboratory strain yAS99 was derived in 1996 after crossing sp1190 (Connolly and Beach 1994) (a.k.a., yAS50 and ATCC 96082: hS-, ade6-704, leu1-32, ura4D) with spDJV1 (diploid; h−/h+, leu1-32, ade6-M216/ade6-M210, ura4-D18/ura+, sla1+/sla1-D) (Van Horn et al. 1997), which was derived from spB67 (McLeod et al. 2000). Genomic DNA was prepared in parallel from various Sz. pombe laboratory strains including yFS101. Paired-end, multiplexed sequencing was performed by Illumina HiSeq at the NIH Intramural Sequencing Center (NISC); for each data set, paired ends were pooled and aligned independently to the pombe reference genome used for tRNA gene prediction. Normalizing for total average read-depth (consistently about 200 for each data set; data not shown), the read coverage at each predicted tRNA gene was compared for each data set against that determined for either yFS101 or yAS99 (Fig. 1A,B).
For Figure 1C, read-depth was combined for each tRNA-anticodon to correct for random distribution of read mapping among multiple identical tRNA sequences (see text). Simply, average read-depth was summed across each tRNA-anticodon family and divided by average mapped read-depth to approximate the number of genomic copies present for a given anticodon. For each anticodon, the copy number for strain FS101 was subtracted from each other strain to yield copy number change for each tRNA-anticodon family. The average difference and standard deviation among strains were plotted.
Determination of codon use and tRNA gene copy numbers
Assemblies of all available Schizosaccharomyces genomes were obtained from the Broad Institute (C Nusbaum, pers. comm.), and genomic sequence for S. cerevisiae was obtained from GenBank. From primary sequence, tRNA genes were predicted blindly using tRNAscan-SE (Lowe and Eddy 1997). From predicted genes, gene counts per iso-acceptor were tabulated, and the genomic sequence corresponding to each was obtained.
The total protein gene content of each strain was obtained from previous predictions (Rhind et al. 2011; C Nusbaum, pers. comm.). For each gene, the occurrence of each codon was counted, excluding stop and initiator methionine codons. To limit bias in overall genome averages due to potential functional enrichment of codons, on a per-codon basis, statistical outlier genes (>3 standard deviations in usage from the mean) were removed from consideration through successive rounds of minimization until no further change was observed. Initiator methionine and stop codons were not considered and were removed from the functional size of the gene. Additionally, only genes that use the codon were considered in tabulating usage rates.
Correlation of tRNA gene copy number and codon usage rates
Pearson correlation coefficients were calculated evaluating codon usage rate as a function of tRNA gene copy number both with and without wobble rules for S. cerevisiae. Calculations were performed using the total proteome or subsets as noted. tRNA gene counts and usage rates were either pooled among all synonymous codons or within subsets using wobble rules previously determined (Johansson et al. 2008) where noted.
Conservation of the distribution of tRNA gene counts among iso-acceptors was evaluated by calculating Pearson correlation coefficients comparing the number of tRNA genes per iso-acceptor across species.
Phylogenetic tree constructions
The 18S rRNA sequence from pombe was used to identify corresponding rRNA sequence from full genomic sequences of the other Schizos using BLAST. Genes were aligned using CLUSTAL and submitted to PHYLIP for calculation of a distance matrix and tree drawing. In parallel, correlation values calculated from tRNA gene content were transformed directly into a distance matrix for tree drawing through PHYLIP by taking (1 − ρ) (Felsenstein 1989, 1997).
Evaluation of tRNA promoter sequences
Flanking sequence for each predicted tRNA gene was compiled and evaluated for the presence of promoter elements. For each species, 100 bases of upstream sequence for each tRNA gene, normalized to the first nucleotide of the mature tRNA, were aligned. The A- and B-box elements within the tRNA sequence were additionally aligned. Sequence logos were generated from each alignment using web logo software (Crooks et al. 2004). The S. cerevisiae A-box and B-box were again determined using MEME software (Bailey et al. 2009) and used in a MAST search of Sz. japonicus to evaluate their composition relative to cerevisiae through the reported p-score of hits.
Evaluation of oligo(dT) terminator lengths
Using the predicted tRNA genes of each yeast genome and also for the human genome, 100 bases downstream from the defined ends of tRNA sequence were clipped from the genomic DNA for each gene. These downstream sequences were evaluated for the first encountered stretch of four or more consecutive T’s. Those lacking a 4T or more sequence in that stretch were reevaluated for an occurrence of 3Ts. Counts for each primary terminator length were tabulated for each strain.
Derivation of japonicus-specific, Sj wobble rules
The initial wobble rules used here (Fig. 3) for codon:tRNA gene correlation allow certain tRNAs to wobble to some but not to other codons in their box according to Johansson et al. (2008). Since the absence of tRNAAlaAGC accompanied by amplification of tRNAAlaUGC led to the worsening of codon:tRNA gene correlation in japonicus using the initial cerevisiae wobble rules, we examined Table 2 for other tRNAs that are significantly imbalanced in their iso-acceptor ratios in japonicus relative to the other Schizos. This identified tRNASerTGA, tRNAAlaTGC, tRNAGlyTCC, and tRNAValTAC. We modified these rules by permitting each of the four japonicus tRNAs (SerTGA, AlaTGC, GlyTCC, ValTAC) to donate to the codons in their box without limiting them to only a subset of codons. No wobbles observed in cerevisiae were disallowed by the Sj wobble rules.
ACKNOWLEDGMENTS
We thank N. Rhind and others at the Broad Institute for making Sz. octosporus, Sz. japonicus, and Sz. cryophilus sequence data available and for providing the wild-type Sz. pombe strain yFS101; and I. Targanski and V. Cherkasova for insightful comments. This work was supported by the Intramural Research Program on Genomics of Differentiation in the Eunice Kennedy Shriver National Institute of Child Health and Human Development.
Footnotes
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.032151.111.
REFERENCES
- Agris PF, Vendeix FA, Graham WD 2007. tRNA’s wobble decoding of the genome: 40 years of modification. J Mol Biol 366: 1–13 [DOI] [PubMed] [Google Scholar]
- Amstutz H, Munz P, Heyer WD, Leupold U, Kohli J 1985. Concerted evolution of tRNA genes: Intergenic conversion among three unlinked serine tRNA genes in S. pombe. Cell 40: 879–886 [DOI] [PubMed] [Google Scholar]
- Bailey TL, Boden M, Buske FA, Frith M, Grant CE, Clementi L, Ren J, Li WW, Noble WS 2009. MEME SUITE: Tools for motif discovery and searching. Nucleic Acids Res 37: W202–W208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begley U, Dyavaiah M, Patil A, Rooney JP, Direnzo D, Young CM, Conklin DS, Zitomer RS, Begley TJ 2007. Trm9-catalyzed tRNA modifications link translation to the DNA damage response. Mol Cell 28: 860–870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg OG, Kurland CG 1997. Growth rate-optimised tRNA abundance and codon usage. J Mol Biol 270: 544–550 [DOI] [PubMed] [Google Scholar]
- Bowen NJ, Jordan IK, Epstein JA, Wood V, Levin HL 2003. Retrotransposons and their recognition of pol II promoters: A comprehensive survey of the transposable elements from the complete genome sequence of Schizosaccharomyces pombe. Genome Res 13: 1984–1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braglia P, Percudani R, Dieci G 2005. Sequence context effects on oligo(dT) termination signal recognition by Saccharomyces cerevisiae RNA polymerase III. J Biol Chem 280: 19551–19562 [DOI] [PubMed] [Google Scholar]
- Chakshusmathi G, Kim SD, Rubinson DA, Wolin SL 2003. A La protein requirement for efficient pre-tRNA folding. EMBO J 22: 6562–6572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan PP, Lowe TM 2009. GtRNAdb: A database of transfer RNA genes detected in genomic sequence. Nucleic Acids Res 37: D93–D97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan CT, Dyavaiah M, DeMott MS, Taghizadeh K, Dedon PC, Begley TJ 2010. A quantitative systems approach reveals dynamic control of tRNA modifications during cellular stress. PLoS Genet 6: e1001247 doi: 10.1371/journal.pgen.1001247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connolly T, Beach D 1994. Interaction between the Cig1 and Cig2 B-type cyclins in the fission yeast cell cycle. Mol Cell Biol 14: 768–776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortazzo P, Cervenansky C, Marin M, Reiss C, Ehrlich R, Deana A 2002. Silent mutations affect in vivo protein folding in Escherichia coli. Biochem Biophys Res Commun 293: 537–541 [DOI] [PubMed] [Google Scholar]
- Crooks GE, Hon G, Chandonia JM, Brenner SE 2004. WebLogo: A sequence logo generator. Genome Res 14: 1188–1190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong H, Nilsson L, Kurland CG 1996. Co-variation of tRNA abundance and codon usage in Escherichia coli at different growth rates. J Mol Biol 260: 649–663 [DOI] [PubMed] [Google Scholar]
- Dujon B, Sherman D, Fischer G, Durrens P, Casaregola S, Lafontaine I, De Montigny J, Marck C, Neuvéglise C, Talla E, et al. 2004. Genome evolution in yeasts. Nature 430: 35–44 [DOI] [PubMed] [Google Scholar]
- Duret L 2000. tRNA gene number and codon usage in the C. elegans genome are co-adapted for optimal translation of highly expressed genes. Trends Genet 16: 287–289 [DOI] [PubMed] [Google Scholar]
- Felsenstein J 1989. PHYLIP—Phylogeny Inference Package (Vers 3.2). Cladistics 5: 164–166 [Google Scholar]
- Felsenstein J 1997. An alternating least squares approach to inferring phylogenies from pairwise distances. Syst Biol 46: 101–111 [DOI] [PubMed] [Google Scholar]
- Grosjean H, Fiers W 1982. Preferential codon usage in prokaryotic genes: The optimal codon–anticodon interaction energy and the selective codon usage in efficiently expressed genes. Gene 18: 199–209 [DOI] [PubMed] [Google Scholar]
- Hamada M, Sakulich AL, Koduru SB, Maraia R 2000. Transcription termination by RNA polymerase III in fission yeast: A genetic and biochemical model system. J Biol Chem 275: 29076–29081 [DOI] [PubMed] [Google Scholar]
- Hamada M, Huang Y, Lowe TM, Maraia RJ 2001. Widespread use of TATA elements in the core promoters for RNA polymerases III, II, and I in fission yeast. Mol Cell Biol 21: 6870–6881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgs PG, Ran W 2008. Coevolution of codon usage and tRNA genes leads to alternative stable states of biased codon usage. Mol Biol Evol 25: 2279–2291 [DOI] [PubMed] [Google Scholar]
- Huang Y, Intine RV, Mozlin A, Hasson S, Maraia RJ 2005. Mutations in the RNA polymerase III subunit Rpc11p that decrease RNA 3′ cleavage activity increase 3′-terminal oligo(U) length and La-dependent tRNA processing. Mol Cell Biol 25: 621–636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iben JR, Epstein JA, Bayfield MA, Bruinsma MW, Hasson S, Bacikova D, Ahmad D, Rockwell D, Kittler EL, Zapp ML, et al. 2011. Comparative whole genome sequencing reveals phenotypic tRNA gene duplication in spontaneous Schizosaccharomyces pombe La mutants. Nucleic Acids Res 39: 4728–4742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Intine RVA, Sakulich AL, Koduru SB, Huang Y, Pierstorrf E, Goodier JL, Phan L, Maraia RJ 2000. Control of transfer RNA maturation by phosphorylation of the human La antigen on serine 366. Mol Cell 6: 339–348 [DOI] [PubMed] [Google Scholar]
- Iwasaki O, Tanaka A, Tanizawa H, Grewal SI, Noma K 2010. Centromeric localization of dispersed Pol III genes in fission yeast. Mol Biol Cell 21: 254–265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen R, Bussemaker HJ, Gerstein M 2003. Revisiting the codon adaptation index from a whole-genome perspective: Analyzing the relationship between gene expression and codon occurrence in yeast using a variety of models. Nucleic Acids Res 31: 2242–2251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansson MJ, Esberg A, Huang B, Bjork GR, Bystrom AS 2008. Eukaryotic wobble uridine modifications promote a functionally redundant decoding system. Mol Cell Biol 28: 3301–3312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimchi-Sarfaty C, Oh JM, Kim IW, Sauna ZE, Calcagno AM, Ambudkar SV, Gottesman MM 2007. A “silent” polymorphism in the MDR1 gene changes substrate specificity. Science 315: 525–528 [DOI] [PubMed] [Google Scholar]
- Kuhn RM, Clarke L, Carbon J 1991. Clustered tRNA genes in Schizosaccharomyces pombe centromeric DNA sequence repeats. Proc Natl Acad Sci 88: 1306–1310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letzring DP, Dean KM, Grayhack EJ 2010. Control of translation efficiency in yeast by codon–anticodon interactions. RNA 16: 2516–2528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe TM. 2011. A genomic tRNA database. http://gtrnadbucscedu/
- Lowe TM, Eddy SR 1997. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res 25: 955–964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maraia RJ, Lamichhane TN 2011. 3′ processing of eukaryotic precursor tRNAs. Wiley Interdiscip Rev RNA 2: 362–375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maraia RJ, Blewett NH, Bayfield MA 2008. It’s a mod mod tRNA world. Nat Chem Biol 4: 162–164 [DOI] [PubMed] [Google Scholar]
- Marck C, Kachouri-Lafond R, Lafontaine I, Westhof E, Dujon B, Grosjean H 2006. The RNA polymerase III-dependent family of genes in hemiascomycetes: Comparative RNomics, decoding strategies, transcription and evolutionary implications. Nucleic Acids Res 34: 1816–1835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McFarlane RJ, Whitehall SK 2009. tRNA genes in eukaryotic genome organization and reorganization. Cell Cycle 8: 3102–3106 [DOI] [PubMed] [Google Scholar]
- McLeod M, Shor B, Caporaso A, Wang W, Chen H, Hu L 2000. Cpc2, a fission yeast homologue of mammalian RACK1 protein, interacts with Ran1 (Pat1) kinase to regulate cell cycle progression and meiotic development. Mol Cell Biol 20: 4016–4027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munz P, Leupold U, Agris P, Kohli J 1981. In vivo decoding rules in Schizosaccharomyces pombe are at variance with in vitro data. Nature 294: 187–188 [DOI] [PubMed] [Google Scholar]
- Nguyen VC, Clelland BW, Hockman DJ, Kujat-Choy SL, Mewhort HE, Schultz MC 2010. Replication stress checkpoint signaling controls tRNA gene transcription. Nat Struct Mol Biol 17: 976–981 [DOI] [PubMed] [Google Scholar]
- Paris Z, Fleming IM, Alfonzo JD 2011. Determinants of tRNA editing and modification: Avoiding conundrums, affecting function. Semin Cell Dev Biol doi: org/10.1016/j.semcdb.2011.10.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavon-Eternod M, Gomes S, Geslain R, Dai Q, Rosner MR, Pan T 2009. tRNA over-expression in breast cancer and functional consequences. Nucleic Acids Res 37: 7268–7280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Percudani R, Pavesi A, Ottonello S 1997. Transfer RNA gene redundancy and translational selection in Saccharomyces cerevisiae. J Mol Biol 268: 322–330 [DOI] [PubMed] [Google Scholar]
- Phizicky EM, Hopper AK 2010. tRNA biology charges to the front. Genes Dev 24: 1832–1860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plotkin JB, Kudla G 2011. Synonymous but not the same: The causes and consequences of codon bias. Nat Rev Genet 12: 32–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pratt-Hyatt MJ, Kapadia KM, Wilson TE, Engelke DR 2006. Increased recombination between active tRNA genes. DNA Cell Biol 25: 359–364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purvis IJ, Bettany AJ, Santiago TC, Coggins JR, Duncan K, Eason R, Brown AJ 1987. The efficiency of folding of some proteins is increased by controlled rates of translation in vivo. A hypothesis. J Mol Biol 193: 413–417 [DOI] [PubMed] [Google Scholar]
- Rhind N, Chen Z, Yassour M, Thompson DA, Haas BJ, Habib N, Wapinski I, Roy S, Lin MF, Heiman DI, et al. 2011. Comparative functional genomics of the fission yeasts. Science 332: 930–936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers HH, Bergman CM, Griffiths-Jones S 2010. The evolution of tRNA genes in Drosophila. Genome Biol Evol 2: 467–477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider TD, Stephens RM 1990. Sequence logos: A new way to display consensus sequences. Nucleic Acids Res 18: 6097–6100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp PM, Tuohy TM, Mosurski KR 1986. Codon usage in yeast: Cluster analysis clearly differentiates highly and lowly expressed genes. Nucleic Acids Res 14: 5125–5143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp PM, Cowe E, Higgins DG, Shields DC, Wolfe KH, Wright F 1988. Codon usage patterns in Escherichia coli, Bacillus subtilis, Saccharomyces cerevisiae, Schizosaccharomyces pombe, Drosophila melanogaster and Homo sapiens; a review of the considerable within-species diversity. Nucleic Acids Res 16: 8207–8211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorensen MA, Kurland CG, Pedersen S 1989. Codon usage determines translation rate in Escherichia coli. J Mol Biol 207: 365–377 [DOI] [PubMed] [Google Scholar]
- Van Horn DJ, Yoo CJ, Xue D, Shi H, Wolin SL 1997. The La protein in Schizosaccharomyces pombe: A conserved yet dispensable phosphoprotein that functions in tRNA maturation. RNA 3: 1434–1443 [PMC free article] [PubMed] [Google Scholar]
- Wang L, Haeusler RA, Good PD, Thompson M, Nagar S, Engelke DR 2005. Silencing near tRNA genes requires nucleolar localization. J Biol Chem 280: 8637–8639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood V, Gwilliam R, Rajandream MA, Lyne M, Lyne R, Stewart A, Sgouros J, Peat N, Hayles J, Baker S, et al. 2002. The genome sequence of Schizosaccharomyces pombe. Nature 415: 871–880 [DOI] [PubMed] [Google Scholar]
- Yokoyama S, Nishimura S 1995. Modified nucleosides and codon recognition. In tRNA: Structure, biosynthesis and function (ed. D Soll, UL RajBhandary), pp. 207–223. ASM Press, Washington, DC [Google Scholar]
- Yoo CJ, Wolin SL 1997. The yeast La protein is required for the 3′ endonucleolytic cleavage that matures tRNA precursors. Cell 89: 393–402 [DOI] [PubMed] [Google Scholar]