

Alternative splicing and trans-splicing events have not been systematically studied in the silkworm Bombyx mori. Here, the silkworm transcriptome was analyzed by RNA-seq. The authors identified 320 novel genes, modified 1140 gene models, and found thousands of alternative splicing and 58 trans-splicing events. Studies of three SR proteins show that both their alternative splicing patterns and mRNA products are conserved from insect to human, and one isoform of Srsf6 with a retained intron is expressed sex-specifically in silkworm gonads.
Keywords: RNA-seq, alternative splicing, trans-splicing, Bombyx mori, SR protein
Abstract
Alternative splicing and trans-splicing events have not been systematically studied in the silkworm Bombyx mori. Here, the silkworm transcriptome was analyzed by RNA-seq. We identified 320 novel genes, modified 1140 gene models, and found thousands of alternative splicing and 58 trans-splicing events. Studies of three SR proteins show that both their alternative splicing patterns and mRNA products are conserved from insect to human, and one isoform of Srsf6 with a retained intron is expressed sex-specifically in silkworm gonads. Trans-splicing of mod(mdg4) in silkworm was experimentally confirmed. We identified integrations from a common 5′-gene with 46 newly identified alternative 3′-exons that are located on both DNA strands over a 500-kb region. Other trans-splicing events in B. mori were predicted by bioinformatic analysis, in which 12 events were confirmed by RT-PCR, six events were further validated by chimeric SNPs, and two events were confirmed by allele-specific RT-PCR in F1 hybrids from distinct silkworm lines of JS and L10, indicating that trans-splicing is more widespread in insects than previously thought. Analysis of the B. mori transcriptome by RNA-seq provides valuable information of regulatory alternative splicing events. The conservation of splicing events across species and newly identified trans-splicing events suggest that B. mori is a good model for future studies.
INTRODUCTION
Thousands to hundreds of thousands of introns must be removed from new transcripts by pre-mRNA splicing in metazoans. As an essential step of RNA processing in all eukaryotes, splicing is catalyzed by the spliceosome through two transesterification reactions to generate mature mRNA (Wahl et al. 2009). Exons and introns are defined by spliceosomal factors as well as by cis-RNA elements and trans-protein regulators (Wang and Burge 2008; Long and Caceres 2009). Alternative splicing and trans-splicing enormously increase gene complexity and allow delicate regulation of gene expression, both of which are necessary for differentiation and development (Horiuchi and Aigaki 2006; Pan et al. 2008; Gingeras 2009; Nilsen and Graveley 2010; Witten and Ule 2011).
Genes typically contain multiple introns, with an average of 7.8–9.0 introns per gene in vertebrates and 2.4–5.4 in invertebrates (Mourier and Jeffares 2003; Roy 2006). By using different splice sites (SS), alternative splicing can generate multiple mRNA products and thus multiple protein isoforms from a single transcript. One well-characterized example of regulation by alternative splicing is the sex-determination cascade in Drosophila melanogaster (Dm). Several genes, including Sex lethal (Sxl), transformer (tra), and doublesex (dsx), are spliced differently in male and female flies, and these alternative splicing events are critical for the sexual differentiation pathway (Black 2003).
Trans-splicing integrates two distinct nascent transcripts into a single mRNA transcript, and it is widespread in lower eukaryotes, such as trypanosomes and nematodes (Zorio et al. 1994; Haile and Papadopoulou 2007). Nearly all transcripts in Trypanosoma brucei and ∼70% of transcripts in Caenorhabditis elegans are trans-spliced, with a short spliced leader (SL RNA) being added to the 5′ end of transcripts by a reaction similar to that of cis-splicing, except that a Y-structured intermediate is formed instead of a lariat intermediate (Allen et al. 2011; Michaeli 2011). Compared with cis-splicing, trans-splicing has been observed much less often in higher eukaryotes (McManus et al. 2010; Zhang et al. 2010). The best-characterized trans-spliced gene in higher eukaryotes is mod(mdg4) in Drosophila, which contains common 5′ exons from one chromosomal locus and 31 alternative 3′ exons from multiple loci, that are located on both DNA strands (Dorn and Krauss 2003; Gabler et al. 2005; McManus et al. 2010). It has been reported that the trans-splicing of mod(mdg4) is conserved in silkworm, based on evidence from one public EST sequence (Krauss and Dorn 2004).
Investigations in D. melanogaster, the best-studied insect model system, have provided deep insights into alternative splicing and trans-splicing, including the examples mentioned above. However, insects are a large and diverse clade, with more than 1 million identified species, and many of the metabolic and developmental pathways in D. melanogaster are distinct from those found in other insects (Borst 2009); for example, insects exhibit many different mechanisms for sexual development and reproduction (Shukla and Nagaraju 2010). Because the silkworm is economically important for silk production, hundreds of mutant strains and dozens of cell lines have been generated for this species during its long history of domestication (Goldsmith et al. 2005). The genome sequence of Bombyx mori (Bm) was also recently released and updated (Xia et al. 2004, 2009). Furthermore, the silkworm belongs to the order Lepidoptera, the second largest order of Insecta, which includes many agricultural pests, such as the cotton bollworm and corn borer, with poorly understood genomic backgrounds and fewer available strains. Therefore, B. mori has the potential to become another important insect model system. To date, 316,223 ESTs and 4918 mRNA sequences from B. mori have been deposited in the NCBI database (http://www.ncbi.nlm.nih.gov/nuccore). However, these are insufficient to provide a global view of alternative splicing and trans-splicing in the silkworm. Only 277 alternative splicing events were identified in a previous study based on the available EST sequences (Zha et al. 2005). Recently, powerful high-throughput sequencing technology (RNA-seq) has been widely applied to transcriptome profiling and has been shown to be an effective and accurate method for the identification of novel transcripts and alternative splicing and trans-splicing events in many organisms, including D. melanogaster, C. elegans, rice, and human (Wang et al. 2008; Zhang et al. 2010; Daines et al. 2011; Graveley et al. 2011).
Here, we applied RNA-seq to samples collected throughout the lifetime of B. mori to analyze its transcriptome, leading to the identification of novel genes, alternative splicing, and trans-splicing events, and to the correction of predicted gene models. We further investigated alternative splicing isoforms of three SR proteins across species, and their expression profiles in developmental stages and tissues of silkworm. Trans-splicing of mod(mdg4) in B. mori was experimentally confirmed with multiple alternative 3′ exons originating from genes on both DNA strands. Another 12 trans-splicing events were identified, in which six events were further confirmed by chimeric SNPs analysis and two events were validated by allele-specific RT-PCR/sequencing assay using F1 hybrids from two distinct lines of silkworm.
RESULTS
RNA-seq data and gene modification in B. mori
To obtain a global view of the silkworm transcriptome, we collected 77 samples from B. mori at various developmental stages, including 18 from embryos, 29 from larvae, 26 from pupae, and four from adults (Fig. 1A). Females and males were equally represented in the larval, pupae, and adult samples (see Materials and Methods). Equal amounts of total RNA from each sample were mixed together, and ribosomal RNA was removed by the Ribominus method. The RNA sample was then applied to an Illumina Genome Analyzer (II) for sequencing. We collected a total of 16,172,896 reads with an average length of 75 bp. After quality filtering, the remaining 14,034,606 reads were mapped to the B. mori genome (release_2.0) using TopHat (Trapnell et al. 2009; Duan et al. 2010). Only 0.70% of reads matched ribosomal RNA sequences. There were 9,373,785 reads that uniquely mapped to the silkworm genome (Table 1), covering 12,441 of the 14,623 genes predicted in the Silkworm Genome Database (SilkDB; http://silkworm.genomics.org.cn/).
FIGURE 1.

RNA-seq and the identification of novel genes in B. mori. (A) Strategy for RNA-seq in the silkworm. Total RNAs were isolated from samples at different stages throughout the lifetime of the silkworm as indicated and mixed together in equal parts. After depletion of ribosomal RNAs, reverse transcription, and fragmentation, double-stranded cDNAs were sequenced on an Illumina Genome Analyzer. (B) Validation of the predicted novel genes by RT-PCR. Twenty novel genes were randomly selected and amplified from genomic DNA and cDNA templates. Migration differences between two templates indicate that the gene contains one or more introns. Validation failure could be caused by inaccurate genome information or poor PCR amplification. (C) Distribution of the RNA-seq reads that support exon and intron boundaries for novel genes. The positions of the start and stop codons and the depth of supported reads are indicated. RT-PCR analysis, shown in panel B, confirmed that these two novel genes contained intron(s).
TABLE 1.
Analysis of RNA-seq data from Bombyx mori
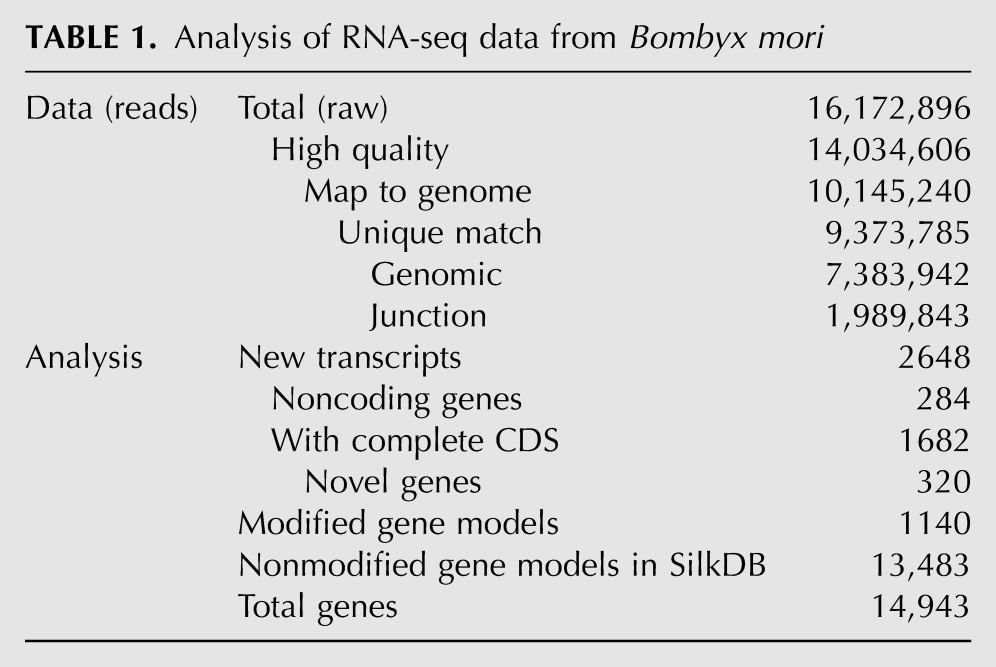
Gene prediction was performed with AUGUSTUS (Stanke et al. 2004, 2008) using our RNA-seq data combined with publicly available mRNA sequences. In comparison to the predicted gene models from SilkDB, we identified 2648 new transcripts, of which 1682 have complete coding sequences, and 284 are noncoding genes (Table 1). Using stringent criteria, we found 320 high-confidence novel genes that were designated SIBSBM000001–SIBSBM000320, most of which have homologs in other organisms (Supplemental Table S1). We also analyzed the Gene Ontology classifications of all of the genes identified in this study (Supplemental Fig. S1). To validate our predictions, 20 of the previously unidentified genes were chosen randomly and tested by RT-PCR. Among these, 19 were confirmed by amplification products from DNA and cDNA that matched our predictions (Fig. 1B); the only one that failed to yield the predicted amplification product from cDNA is not listed here. Locations of exons and introns from each novel gene are also defined (Supplemental Table S2). For example, there was one intron in the novel gene SIBSBM000044 and two introns in SIBSBM000166 (Fig. 1C), which were supported by both the RNA-seq and validating RT-PCR results (Fig. 1B). Moreover, 1140 silkworm gene models were modified based on exon–exon junction reads. Although most of these modifications were minor, they will allow more accurate prediction of protein products. The corrected genes were named SIBSBM000321–SIBSBM001460 and are listed with the previous SilkDB models to show the modifications (Supplemental Table S3).
Alternative splicing events in the silkworm
Alternative splicing often produces protein isoforms with functional domain changes or introduces a premature stop codon that would trigger the nonsense-mediated decay surveillance pathway (Chang et al. 2007; McGlincy and Smith 2008). To further investigate alternative splicing events in B. mori, we searched the 1,989,843 uniquely mapped exon–exon junction reads that contained potential splice sites, along with nonjunction reads, and identified 1923 alternative splicing events (Supplemental Table S4). These events were classified into five groups: skipped exons (8.6%), retained introns (46.1%), alternative 5′SS (27.6%), alternative 3′SS (16.9%), and mutually exclusive exons (0.8%). Three individual events from each group were randomly chosen and validated by RT-PCR. All of the RT-PCR products were of a size and sequence consistent with the results of the bioinformatic analysis. The ratio between the two isoforms amplified by RT-PCR was similar to that between the two isoforms identified by the RNA-seq analysis (Fig. 2A–E; data not shown).
FIGURE 2.

Identification of alternative splicing events in B. mori. The alternative splicing events in the silkworm are classified as (A) skipped exons, (B) retained introns, (C) alternative 5′SS, (D) alternative 3′SS, or (E) mutually exclusive exons. The numbers of each type of event are indicated, and there were 1923 events in total. (F) As a subtype of alternative 3′SS, events of tandem 3′ splice site separated by 3 nt were specifically analyzed. Supporting RNA-seq reads and RT-PCR amplifications are shown for randomly selected events from each type. The positions of all primers used for PCR are indicated, and the amplified products were confirmed by sequencing. Specifically for the case of tandem 3′SS in panel F, the two alternative splicing products are 3 nt different in length, and were detected as 2:2 after sequencing of four clones. This is reasonable with the abundance of RNA-seq reads that support the prediction of the two isoforms, which was 105 to 59.
Tandem splice sites separated by 2–12 nt are common at both the 5′SS and 3′SS and comprise approximately one-third of alternative splicing events (Kuhn et al. 2007). The use of tandem splice sites is considered to be a subtle form of alternative splicing because the resulting mRNAs differ by only a few nucleotides (Sinha et al. 2010). Here, we studied tandem 3′SS separated by 3 nt (represented by NAG ↓NAG ↓), which would either change the protein product by in-frame variation of one or two amino acids or introduce a UAG stop codon at a low frequency. We captured 2658 NAGNAG hexamers from the entire collection of B. mori intron–exon boundaries. Based on the RNA-seq data, alternative splicing with tandem 3′SS had occurred at 66 of such hexamers (Fig. 2F; Supplemental Table S5), with only one case introducing a UAG stop codon (#19 in Supplemental Table S5). This result demonstrated that alternative splicing of tandem 3′SS separated by 3 nt is a common event and mostly produces subtle changes at the protein level.
SR proteins and their alternative splicing in B. mori
Regulation of alternative splicing is a complicated and delicate process, which not only depends on the sequence of pre-mRNA itself, but also is determined by upstream regulators, such as SR proteins and hnRNPs. Members of the SR protein family usually function as activators in pre-mRNA splicing (Long and Caceres 2009). Evidence shows that alternative splicing of many SR proteins affects downstream genes' alternative splicing to establish a regulatory network (Black 2003). For example, alternative splicing of tra, encoding an SR-like protein, plays an important role in the sex determination pathway in D. melanogaster by sex-specific regulation of alternative splicing of dsx gene (Inoue et al. 1992). Therefore, as an example of a group of potentially alternatively spliced isoforms, we next focused on the alternative splicing of SR proteins in the silkworm. To detect SR proteins in B. mori, we generated a database of all silkworm proteins and then searched for proteins with more than 10 SR/RS di-peptides plus at least one SRSRSR/RSRSRS triple di-peptide repeat, based on the amino acid composition that is characteristic of the SR protein family (Fig. 3A). We identified 37 SR or SR-like proteins, 23 of which had homologs that have been described as splicing factors in other species (Supplemental Table S6). For further analysis, we selected three SR proteins: SRSF6 (B52), U1-70K, and U2AF2 (U2AF65), for which alternatively spliced mRNA isoforms were captured by RNA-seq.
FIGURE 3.

Alternative splicing of three SR proteins are conserved from silkworm to human. (A) Strategy for searching for SR proteins from the silkworm. (B) Alternative splicing of Srsf6 is highly conserved across species from silkworms to humans. Three alternatively spliced isoforms of Bm Srsf6 were identified by RNA-seq analysis. Early stop codons in the intron-retention and exon-inclusion isoforms of Srsf6 are indicated by asterisks (upper). Both alternative splicing types and the resulting SRSF6 protein isoforms are conserved in the silkworm, D. melanogaster, C. elegans, and humans (bottom). (C) mRNA profiles of Srsf6, U1-70K, and U2AF2 in different developmental stages and tissues. Each isoform amplified by RT-PCR was isolated and confirmed by sequencing. Actin3 was used as a loading control. Alternatively spliced isoforms of (D) U1-70K and (E) U2AF2 are conserved in the silkworm and human. The absence of related isoforms in D. melanogaster and C. elegans might be due to incomplete investigation. All of the mRNA sequences from D. melanogaster, C. elegans, and human were retrieved from the NCBI database. The SR domain and RNA recognition motif (RRM) in SRSF6 are indicated.
Based on analysis of the RNA-seq data, there were three isoforms of Srsf6 and two isoforms each of U1-70K and U2AF2 in the silkworm (Fig. 3B,D,E, upper part of each). Isoform A of Srsf6 would produce a full-length protein with two RRMs and one RS domain, similar to the domain structure of other classic SR proteins (Shepard and Hertel 2009). However, both isoforms B and C of Srsf6 would generate truncated proteins lacking an RS domain and exhibiting an incomplete RRM2 due to early stop codons being introduced either by the retention of intron 4 or by the inclusion of an additional exon derived from the middle part of intron 4 (Fig. 3B). To characterize these isoforms, we investigated their mRNA levels during different silkworm developmental stages and tissues by RT-PCR. The full-length (A) and exon inclusion (C) isoforms of Srsf6 were consistently expressed in all tested stages and tissues, and the intron retention isoform (B) was expressed at low levels throughout the whole body during all developmental stages. However, this form was more highly expressed in some of the tissues tested, such as the silk gland and fat body. Importantly, this isoform was differentially expressed in male and female gonads, with no detectable mRNA being observed in the testis, whereas high expression was seen in the ovary, suggesting that this truncated Srsf6 isoform might play a role in the sexual differentiation of B. mori (Fig. 3C). We also searched for human, D. melanogaster, and C. elegans mRNA isoforms of Srsf6 from the NCBI database. Strikingly, both intron retention and exon inclusion isoforms with early stop codons at similar positions to those in the silkworm were also identified in D. melanogaster and C. elegans (Fig. 3B). A similar exon inclusion isoform of Srsf6 was found in humans. The absence of the intron retention isoform in humans might be due to the lower frequency of intron retention in mammals (Zhou et al. 2009) or incomplete analysis of human Srsf6 (Fig. 3B). The existence and conservation of alternative splicing of Srsf6 across species strongly suggest that either the mRNA isoforms with early stop codons or the truncated Srsf6 protein products could have similar regulatory functions in most animals.
The two isoforms of U1-70K were present in all of the tested silkworm samples, but the ratios between these two isoforms were variable in stages and tissues (Fig. 3C). In contrast, expression of the two isoforms of U2AF2 were relatively stable in stages and tissues (Fig. 3C), consistent with its predicted function in constitutive splicing (Sickmier et al. 2006). Similar conservation of alternative splicing patterns for U1-70K and U2AF2 is also observed in human. The exon inclusion forms of U1-70K (Fig. 3D) and alternatively spliced forms of U2AF2 (Fig. 3E) are conserved in both silkworm and human. However, it is unclear whether these isoforms are conserved in D. melanogaster or C. elegans due to the absence of such isoforms from the NCBI database (Fig. 3D,E).
Identification of trans-splicing events in silkworm
Trans-splicing integrates nascent RNAs from two transcripts into a single mRNA; this occurs frequently in trypanosomes and nematodes but much less often in higher animals. To identify trans-splicing events in the silkworm, a library of all possible hybrid exon–exon sequences except from the same gene locus was generated; all RNA-seq reads that could not be mapped to a single gene locus were aligned to this library. Two groups of trans-splicing events were identified. The first group contained 46 trans-splicing events from the homolog of D. melanogaster mod(mdg4); the second group included 80 non-mod(mdg4) trans-splicing events. To reduce the possibilities of false events and provide a reliable prediction for the second group, we further strengthened the prediction with three more criteria: (1) reads mapped to the hybrid exon–exon sequence should have at least 15 nt on the two exons; (2) each event should be supported by at least two nonredundant reads; (3) considering the possibility of gene fusion, predicted events were removed when the two genes are neighboring genes and located within 20 kb. Finally, we found 19 events for the second group with high confidence.
Trans- and alternative trans-splicing of mod(mdg4) in B. mori
The mod(mdg4) locus in the silkworm has previously been reported based on one EST sequence and, therefore, had been proposed to be expressed through a trans-splicing mechanism (Krauss and Dorn 2004). However, this was predicted before the release of the silkworm genome, and trans-splicing of Bm mod(mdg4) has not been experimentally and completely investigated. Analysis of our RNA-seq data revealed nine products of Bm mod(mdg4) that share common 5′ exons from the BGIBMGA006426 gene and alternative 3′ exons from eight genes located on the antiparallel strand in the same scaffold of B. mori (Fig. 4A). Similar to what is seen for Dm mod(mdg4), trans-splicing of Bm mod(mdg4) yielded an mRNA product encoding 317 amino acids with a BTB domain at the N terminus from the BGIBMGA006426 gene alone (including exon1 through exon4) and conserved FLYWCH motifs in the C termini of eight variable loci from the opposite DNA strand. Moreover, we also identified 37 cases of alternative 3′ exons from the same DNA strand joined to the common 5′-exon gene (Fig. 4A; Supplemental Table S7). Most of these alternative 3′ exons encoded the conserved FLYWCH motif, similar to the 3′ exons on the opposite strand (Supplemental Table S7). Because the same-strand alternative 3′ exons were distributed over a chromosomal locus of nearly 440 kb, we propose that their mRNA products were derived through trans-splicing.
FIGURE 4.

Trans- and alternative trans-splicing of mod(mdg4) in the silkworm. (A) Trans-spliced forms of mod(mdg4) in B. mori encode a common BTB domain at the N terminus from exon1 to exon4 of BGIBMGA006426 and variable FLYWCH domains at the C terminus from eight genes on the opposite DNA strand in the same scaffold (nscaf2853). RT-PCR analysis and sequencing confirmed all nine trans-splicing events with precise splice sites. (B) mRNA profiles of the nine opposite-strand trans-splicing events in different developmental stages and tissues. RT-PCR analysis from different B. mori samples indicated that some trans-splicing events were specific to one or several stages or tissues. Alternative trans-splicing was observed for one of the 3′-exon genes, BGIBMGA006451, which can be trans-spliced into two isoforms of mod(mdg4) to produce trans-P51.1 and P51.2.
To investigate the expression profiles of the nine trans-spliced products that came from the opposite strand, we analyzed the mRNA levels of all of the forms in samples from silkworm developmental stages and tissues. Each trans-spliced product was named using the last two digits of its 3′-exon gene. The expression profiles of these nine mRNA products showed diverse patterns. Most of the trans-spliced products were expressed at high levels during the embryonic stage and at low levels during the larval stage. Trans-P54, P53, P51.2, P51.1, P46, and P45 were consistently expressed at similar levels throughout all tested stages and tissues, with the exception of their low expression in larvae; trans-P52 was highly expressed in the ovary as well as in the early embryo; trans-P50 was found at high levels in all tested tissues but was not detectable in the whole body of larva, pupa, and adult samples; trans-P49 was highly expressed in the silk gland and in the adult stage (Fig. 4B). Interestingly, one 3′-exon gene, BGIBMGA006451, was ligated to the common 5′ exons in two different ways, thus generating two alternative trans-spliced products, trans-P51.1 and P51.2 (Fig. 4A,B). Taking these findings together and considering the known function of mod(mdg4) in chromatin structure rearrangement in the fly, these data strongly imply that the distinct isoforms of mod(mdg4) act separately and/or coordinately in the process of insect development and tissue differentiation.
Validation of novel trans-splicing events
To validate the trans-splicing events, we first performed individual RT-PCR/sequencing for the predicted 19 trans-splicing events, in which 12 events generated the predicted amplification products (Table 2). All of the ligated trans-spliced products were generated by intron removal at precise splice sites (data not shown). According to the data of chromosomal locations of the loci from the SilkDB, three events were between interchromosomal genes; two were between intrachromosomal genes; and the origins of the other seven events were unclear due to insufficient information (Table 2).
TABLE 2.
The second group of newly identified trans-splicing events in the silkworm
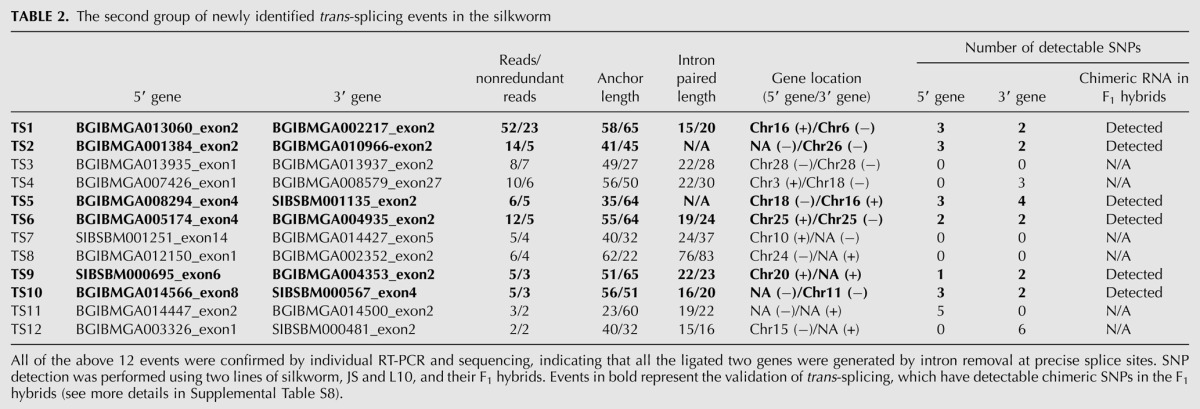
To rule out the possibility of artifacts generated during the library construction or genome misassembly in the tested strain, we further analyzed these 12 trans-splicing events in two distinct silkworm lines, JS and L10, as well as in their F1 hybrids (Fig. 5A). JS is an agricultural line that produces high-quality silk; L10 is a line with poor silk production but strong stress resistance (Zhan et al. 2009). We first searched for SNPs in both the 5′ and 3′ genes by sequencing in JS and L10. For six events, available SNPs were found in exons of both the 5′ and 3′ genes (Fig. 5B; Table 2, events in bold). Another six events either have SNPs only in one gene or have no SNP at all in the two trans-spliced genes, which are thereby unsuitable for further analysis for validation (Table 2 and Supplemental Table S8). Then, cDNAs of the six events with available SNPs were amplified from F1 hybrids. Clones having exchanged SNPs were observed in all these six cases, suggesting that their mRNA products were generated by trans-splicing (Fig. 5B).
FIGURE 5.

Validation of newly identified non-mod(mdg4) trans-splicing events in the silkworm. (A) Schematics of two silkworm lines and their F1 hybrids. (Red) mRNA and SNPs from JS line; (green) mRNA and SNPs from L10 line. (B) Exchanged SNPs from JS and L10 were observed in their F1 hybrids to produce a chimeric RNA for six events, indicating that these events were through trans-splicing. Other events cannot be validated due to insufficient SNPs in the two trans-spliced genes. (C) Two trans-splicing events were further validated by using allele-specific RT-PCR and sequencing. cDNAs from JS, L10, and their mixtures were used as controls. Species-specific primers for TS5 and TS6 events were indicated in B. Sequences of products from each template were listed, and the SNPs were colored to indicate their origins.
Furthermore, to rule out the possibility of strand switching from templates, one of the current methods for validation of trans-splicing is allele-specific RT-PCR/sequencing assay by using species-specific primers based on SNP information (McManus et al. 2010). Design of the specific primers and final PCR amplification were difficult due to either insufficient SNPs or sequence similarities around SNP sites. In these six events, five (TS1, 2, 5, 6, and 10, but not the TS9 event) had at least two SNPs on each side of genes (Fig. 5B). We tested the five events by allele-specific RT-PCR and found two events could be validated. For the events of TS5 and TS6, primers from the same species amplified the anticipated mRNA products only in the samples from their own species (Fig. 5C, lanes 1,2,4,5). However, the combined primers from two species amplified the anticipated mRNA products only from F1 hybrids (Fig. 5C, lanes 3,6,7), but not from either parent species or their mixtures. All of the mRNA products were confirmed by DNA sequencing, arguing that TS5 and TS6 were produced by trans-splicing, not due to errors of strand switching from templates by RT-PCR.
DISCUSSION
RNA-seq analyses have shown that >80% of mammalian genes include introns, and >95% of transcripts in humans are alternatively spliced (Pan et al. 2008; Wang et al. 2008); furthermore, ∼40% of genes exhibit introns, and 31% of transcripts are alternatively spliced in D. melanogaster (Daines et al. 2011; Graveley et al. 2011). This powerful emerging technique has stimulated the study of transcriptomes and revealed an unexpected complexity of gene expression in higher eukaryotes. To obtain detailed information about gene expression and regulation in the silkworm, it is necessary to analyze pre-mRNA splicing events systematically by RNA-seq. Here, our analysis of RNA-seq data revealed valuable information about gene structure and alternative splicing and trans-splicing events in the silkworm.
Gene annotations in the silkworm
The current gene annotations of B. mori in the SilkDB, which include 14,623 predicted gene models, are based on an insufficient number of EST sequences from the NCBI database and genomic structures from other species. In this RNA-seq study of the silkworm transcriptome, nearly 3000 new mRNA transcripts were identified; 320 of these were high-confidence predictions of novel genes, most of which have homologs in other organisms. We also modified 1140 gene annotations based on the captured junction reads and subsequent analyses, which provide more accurate coding sequences. Further statistical validation by RT-PCR proved that these new identifications are mostly correct and will benefit future studies in the silkworm. Some rare mRNA products might be diluted in the mixed sample used here, such that they cannot be detected by RNA-seq. In the future, deeper sequencing of individual samples of tissues and developmental stages will be helpful for defining additional gene annotations precisely in B. mori.
Alternative splicing of SR proteins
A great deal of evidence has demonstrated that alternative splicing is an important regulatory step in cell differentiation and tissue development (Chen and Manley 2009; Yeo et al. 2009). Based on nearly 2 million junction reads, we identified almost 2000 alternative splicing events under strict criteria. Retained introns comprised almost half of all identified events, and intron retention was the major type of alternative splicing in B. mori, consistent with transcriptomal analyses in D. melanogaster and C. elegans (Daines et al. 2011; Ramani et al. 2011). We also showed that alternative splicing at tandem 3′ splice sites in the silkworm mostly introduced subtle changes to the primary structure of proteins.
SR proteins contribute to multiple steps of pre-mRNA splicing that affect splice site recognition, spliceosome assembly, and catalysis, as well as cotranscriptional splicing and RNA export (Graveley 2000; Sanford et al. 2005; Long and Caceres 2009 and references therein). Alternative splicing of SR proteins plays critical roles in the regulation of gene expression. Here, we found that one of the three Srsf6 isoforms was differentially expressed in the gonads of B. mori. This isoform encodes an SRSF6 protein with an absent RS-domain and an incomplete RRM2 that might have regulatory functions in the sexual development of B. mori. Consistent with this finding, studies from D. melanogaster have shown that Srsf6(B52) interacts genetically with sex determination regulators, such as doublesex, Tra, and Tra2 (Peng and Mount 1995; Lynch and Maniatis 1996) and that the intron-retention isoform is sex-related (Telonis-Scott et al. 2009). Importantly, the Srsf6 isoforms and their alternative splicing pattern are conserved across organisms. Taken together, these data indicate that analyses of alternative splicing based on RNA-seq data could help to identify regulatory factors in certain biological pathways.
Conservation of trans-splicing of the mod(mdg4) gene
mod(mdg4) is the best-characterized trans-spliced gene in D. melanogaster, producing multiple protein isoforms that are involved in chromatin structure rearrangements (Dorn and Krauss 2003). Here, we demonstrated that mod(mdg4) is also trans-spliced in the silkworm. Several lines of evidence strongly suggest that the trans-splicing of mod(mdg4) is highly conserved between the fly and the silkworm. (1) Previous studies have found a mod (mdg4) locus in the silkworm (Krauss and Dorn 2004). (2) Both Bm mod (mdg4) and Dm mod (mdg4) encode hybrid BTB and FLYWCH domains that come from the 5′ exons and 3′ exons, respectively. (3) Both genes are characterized by more than 30 alternative 3′ exons that are transcribed from genes on two DNA strands. (4) One observed alternative trans-splicing event of mod(mdg4) in the silkworm, in which one 3′-exon transcript is trans-spliced with the common 5′ exons to produce two isoforms, trans-P51.1 and trans-P51.2 (Fig. 4B), the similar alternative trans-splicing of Dm mod(mdg4) is also observed in the recently updated FlyBase.
Our detailed analysis of the nine opposite-strand trans-splicing events of mod(mdg4) genes revealed interesting specificities of these hybrid mRNA products in different developmental stages and tissues in the silkworm, which strongly suggest that their expression is regulated by splicing factors. The similarity of the mRNA profiles of the multiple mod(mdg4) genes between the silkworm and Drosophila implies that the mechanism regulating the trans-splicing of mod(mdg4) is conserved in these two insects. One difference between the mod(mdg4) genes in the silkworm and the fly is in the length of the region in which the alternative 3′-exon genes are distributed. In Drosophila, all of the 3′-exon genes are concentrated within <25 kb of the common 5′-exon gene (Krauss and Dorn 2004). However, this distance is expanded to >500 kb in the silkworm (Fig. 4A). The reason for such difference between the fly and the silkworm is not clear yet. In addition, based on information from SilkDB and analysis of our RNA-seq data, there is no obvious silkworm homolog of lola, another well-characterized trans-spliced gene in D. melanogaster.
Identification of trans-splicing events in the silkworm
Trans-splicing is rare in higher eukaryotes and can be falsely predicted from RNA-seq data by bioinformatic analysis. To exclude false predictions, several steps of controls were performed in this study of trans-splicing events in B. mori. Firstly, stringent criteria were used for bioinformatics analysis and prediction, which resulted in 19 predicted events. Secondly, individual experimental RT-PCR/sequencing analyses were performed, which reduced the events to 12. Lastly, two lines of silkworm and their F1 hybrids were used for further validation based on SNP information and followed by allele-specific RT-PCR/sequencing assays. Due to insufficient SNPs and difficulties of allele-specific RT-PCR, we only can validate part of the 12 events. However, we believe that further studies, including DNA/RNA fluorescent in situ hybridization and silkworm genetic analysis, would likely validate more of them.
Most of the identified 12 trans-splicing events would encode proteins with ORFs derived from two transcripts, including novel domains or domains that would be completely functional only after trans-splicing. For example, both the 5′ gene (BGIBMGA013060) and 3′ gene (BGIBMGA002217) only encode part of a RhoGAP domain, which cannot be functional. However, the trans-spliced product from these two genes would encode a protein with an intact, and presumably functional, RhoGAP domain like the Rho family of GTPases found in other organisms (Supplemental Fig. S2A). Furthermore, it has been revealed that RNA base-pairing is critical for the trans-splicing (Fischer et al. 2008; Kamikawa et al. 2011; Roy et al. 2011). We assembled each set of the 5′ and 3′ trans-introns to reveal potential RNA base-pairings. Most of these intron sets were able to form relatively stable RNA secondary structures by base-pairing (Table 2). Two examples were selected for further analysis. In the trans-splicing event between genes BGIBMGA012150 and BGIBMGA002352, trans-introns were able to form a stable RNA secondary structure with 76 bp over an 83-nt region (Supplemental Fig. S2B). Similarly, intron 6 of gene SIBSBM000695 and intron 2 of gene BGIBMGA004353 were observed to form an RNA structure with 22 bp over a 23-nt-long RNA region (Supplemental Fig. S2C). RT-PCR and sequencing results confirmed that the cleavage and ligation of these trans-splicing events occurred at conventional splice sites.
Taken together, we have provided evidence of the existence of trans-splicing events in the silkworm that suggest B. mori is a good system for further studies.
MATERIALS AND METHODS
Silkworm culture and sample collection
B. mori strains, including P50 (Dazao), JS, L10, and their F1 hybrid, were cultured by standard methods (Zhou et al. 2010). Samples of P50 were collected for RNA-seq. In the embryo stage, 100 embryos were collected at the times of 0, 2, 4, 6, 8, 10, 12, and 24 h, and then once a day in the next 9 d. In the stages of larva, pupa, and adult, samples were collected once each day, in which the female and male were equally collected when the sex of silkworm could be distinguished.
RNA-seq and read alignment
Total RNA from 77 samples was isolated using TRIzol (Invitrogen) and mixed together in equal parts. Ribosomal RNA was depleted using the Ribominus kit (Invitrogen) according to the manufacturer's protocol. The RNA sample was subsequently fragmented by Covaris and reverse-transcribed by random priming, followed by second-strand synthesis to create double-stranded cDNA fragments with blunt ends. These cDNA fragments were ligated to Illumina Single-End Sequencing adapters for PCR and sequenced on an Illumina Genome Analyzer.
Reads that did not pass the Illumina chastity filter or did not contain at least 25 Q20 bases (a base with a Q-value ≥20, which is defined as an error probability of ≤1%) among the first 35 cycles were removed. Any 3′-end sequences of low quality were trimmed off using a custom Perl script. Quality-filtered reads were then aligned to the genome of B. mori (release_2.0, ftp://silkdb.org/pub/current/Genome/) by TopHat (version 1.1.4) (Trapnell et al. 2009) with the parameters “-F 0 -solexa1.3-quals -g 1”; thus, de novo splice junction reads that uniquely mapped to the genome were retained. To avoid possible artifacts from the PCR amplification, nonredundant reads were taken into consideration for the subsequent analysis.
Identification of novel genes and modification of existing gene models
Gene prediction at the genome level was performed with AUGUSTUS (Stanke et al. 2004, 2008) using both the RNA-seq reads and retrieved mRNAs from NCBI (currently 4918 for B. mori). A “Drosophila melanogaster”–trained version of AUGUSTUS was used in this study. A predicted gene model was considered to be of high confidence when more than half of its exons or introns were supported by the RNA-seq or mRNA data. The obtained high-confidence gene models were compared with those previously predicted in SilkDB. Novel genes were defined when they emerged in our high-confidence models and were located in intergenic regions of SilkDB. Modified gene models were evaluated when there were different exon–exon boundaries between our gene models and the SilkDB gene models and were accepted only when the RNA-seq data or retrieved mRNA sequences supported our models, but not the SilkDB models.
Alternative splicing analysis
All RNA-seq reads were mapped to the reference genome from SilkDB by TopHat, and then a “junction.bed” file was generated to obtain potential exon–exon junction sites. RNA-seq reads that support a junction site must meet two conditions: (1) one read mapped to both flanking regions of a potential junction site with at least 8-nt perfect matches; and (2) there were at least two nonredundant reads covering a junction. Then, we applied custom Perl scripts to distinguish five types of alternative splicing events: alternative 5′ splicing sites, alternative 3′ splicing sites, mutually exclusive exons, skipped exon, and intron retention.
Trans-splicing analysis
To identify trans-splicing events, a hybrid sequence library based on our new gene model data set was generated to include exon–exon junctions of all possible combinations, except junctions from the same gene. Each junction sequence consisted of the last 60 nt from the 5′ exon and the first 60 nt from the 3′ exon. RNA-seq reads that failed to map to the silkworm genome, known mRNAs, or annotated genes were aligned to this library using Bowtie (version 0.12.7) (Langmead et al. 2009) with up to two mismatches allowed. The anchor length from the retained reads was parsed to evaluate accuracy. To remove false-positive events, trans-splicing events were required to meet the following criteria: (1) reads mapped to the chimeric sequence should have at least 15-nt perfect matches on one side of the flanking region, (2) supported by at least two nonredundant reads, (3) events from neighboring genes <20 kb in distance were removed due to the possibility of gene fusion.
RT-PCR and sequencing validation
RT-PCR was performed using reverse transcriptase (Toyobo) and ExTaq (Takara) using the total RNA isolated from either mixed or individual samples, as indicated. Actin3 was used as a loading control. PCR products were excised from agarose gels and sequenced.
DATA DEPOSITION
Deep sequencing data can be found at the SRA database, accession code SRP007541.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
ACKNOWLEDGMENTS
We thank the Center for Epigenomics at the Albert Einstein College of Medicine for the Illumina RNA-seq runs; C.J. McManus for communications on the allele-specific RT-PCR assay; and A. Moldón and C. Query for discussions and critical reading of the manuscript. This work was supported by the National Basic Research Program of China (973, 2012CB114101 to Y.-Z.X. and 2012CB316501 to X.L.), the Science and Technology Commission of Shanghai Municipality (10JC1416200), and the Chinese Academy of Sciences (KSCX2-EW-J-12) to Y.-Z.X.
Footnotes
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.029751.111.
REFERENCES
- Allen MA, Hillier LW, Waterston RH, Blumenthal T 2011. A global analysis of C. elegans trans-splicing. Genome Res 21: 255–264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black DL 2003. Mechanisms of alternative pre-messenger RNA splicing. Annu Rev Biochem 72: 291–336 [DOI] [PubMed] [Google Scholar]
- Borst A 2009. Drosophila's view on insect vision. Curr Biol 19: R36–R47 [DOI] [PubMed] [Google Scholar]
- Chang YF, Imam JS, Wilkinson MF 2007. The nonsense-mediated decay RNA surveillance pathway. Annu Rev Biochem 76: 51–74 [DOI] [PubMed] [Google Scholar]
- Chen M, Manley JL 2009. Mechanisms of alternative splicing regulation: insights from molecular and genomics approaches. Nat Rev Mol Cell Biol 10: 741–754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daines B, Wang H, Wang L, Li Y, Han Y, Emmert D, Gelbart W, Wang X, Li W, Gibbs R, et al. 2011. The Drosophila melanogaster transcriptome by paired-end RNA sequencing. Genome Res 21: 315–324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorn R, Krauss V 2003. The modifier of mdg4 locus in Drosophila: Functional complexity is resolved by trans splicing. Genetica 117: 165–177 [DOI] [PubMed] [Google Scholar]
- Duan J, Li R, Cheng D, Fan W, Zha X, Cheng T, Wu Y, Wang J, Mita K, Xiang Z, et al. 2010. SilkDB v2.0: A platform for silkworm (Bombyx mori) genome biology. Nucleic Acids Res 38: D453–D456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer SE, Butler MD, Pan Q, Ruvkun G 2008. Trans-splicing in C. elegans generates the negative RNAi regulator ERI-6/7. Nature 455: 491–496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabler M, Volkmar M, Weinlich S, Herbst A, Dobberthien P, Sklarss S, Fanti L, Pimpinelli S, Kress H, Reuter G, et al. 2005. Trans-splicing of the mod(mdg4) complex locus is conserved between the distantly related species Drosophila melanogaster and D. virilis. Genetics 169: 723–736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gingeras TR 2009. Implications of chimaeric non-co-linear transcripts. Nature 461: 206–211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldsmith MR, Shimada T, Abe H 2005. The genetics and genomics of the silkworm, Bombyx mori. Annu Rev Entomol 50: 71–100 [DOI] [PubMed] [Google Scholar]
- Graveley BR 2000. Sorting out the complexity of SR protein functions. RNA 6: 1197–1211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graveley BR, Brooks AN, Carlson JW, Duff MO, Landolin JM, Yang L, Artieri CG, van Baren MJ, Boley N, Booth BW, et al. 2011. The developmental transcriptome of Drosophila melanogaster. Nature 471: 473–479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haile S, Papadopoulou B 2007. Developmental regulation of gene expression in trypanosomatid parasitic protozoa. Curr Opin Microbiol 10: 569–577 [DOI] [PubMed] [Google Scholar]
- Horiuchi T, Aigaki T 2006. Alternative trans-splicing: A novel mode of pre-mRNA processing. Biol Cell 98: 135–140 [DOI] [PubMed] [Google Scholar]
- Inoue K, Hoshijima K, Higuchi I, Sakamoto H, Shimura Y 1992. Binding of the Drosophila transformer and transformer-2 proteins to the regulatory elements of doublesex primary transcript for sex-specific RNA processing. Proc Natl Acad Sci 89: 8092–8096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamikawa R, Inagaki Y, Tokoro M, Roger AJ, Hashimoto T 2011. Split introns in the genome of Giardia intestinalis are excised by spliceosome-mediated trans-splicing. Curr Biol 21: 311–315 [DOI] [PubMed] [Google Scholar]
- Krauss V, Dorn R 2004. Evolution of the trans-splicing Drosophila locus mod(mdg4) in several species of Diptera and Lepidoptera. Gene 331: 165–176 [DOI] [PubMed] [Google Scholar]
- Kuhn RM, Karolchik D, Zweig AS, Trumbower H, Thomas DJ, Thakkapallayil A, Sugnet CW, Stanke M, Smith KE, Siepel A, et al. 2007. The UCSC Genome Browser Database: Update 2007. Nucleic Acids Res 35: D668–D673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL 2009. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10: R25 doi: 10.1186/gb-2009-10-3-r25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long JC, Caceres JF 2009. The SR protein family of splicing factors: Master regulators of gene expression. Biochem J 417: 15–27 [DOI] [PubMed] [Google Scholar]
- Lynch KW, Maniatis T 1996. Assembly of specific SR protein complexes on distinct regulatory elements of the Drosophila doublesex splicing enhancer. Genes Dev 10: 2089–2101 [DOI] [PubMed] [Google Scholar]
- McGlincy NJ, Smith CW 2008. Alternative splicing resulting in nonsense-mediated mRNA decay: What is the meaning of nonsense? Trends Biochem Sci 33: 385–393 [DOI] [PubMed] [Google Scholar]
- McManus CJ, Duff MO, Eipper-Mains J, Graveley BR 2010. Global analysis of trans-splicing in Drosophila. Proc Natl Acad Sci 107: 12975–12979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaeli S 2011. Trans-splicing in trypanosomes: Machinery and its impact on the parasite transcriptome. Future Microbiol 6: 459–474 [DOI] [PubMed] [Google Scholar]
- Mourier T, Jeffares DC 2003. Eukaryotic intron loss. Science 300: 1393 doi: 10.1126/science.1080559 [DOI] [PubMed] [Google Scholar]
- Nilsen TW, Graveley BR 2010. Expansion of the eukaryotic proteome by alternative splicing. Nature 463: 457–463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan Q, Shai O, Lee LJ, Frey BJ, Blencowe BJ 2008. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat Genet 40: 1413–1415 [DOI] [PubMed] [Google Scholar]
- Peng X, Mount SM 1995. Genetic enhancement of RNA-processing defects by a dominant mutation in B52, the Drosophila gene for an SR protein splicing factor. Mol Cell Biol 15: 6273–6282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramani AK, Calarco JA, Pan Q, Mavandadi S, Wang Y, Nelson AC, Lee LJ, Morris Q, Blencowe BJ, Zhen M, et al. 2011. Genome-wide analysis of alternative splicing in Caenorhabditis elegans. Genome Res 21: 342–348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy SW 2006. Intron-rich ancestors. Trends Genet 22: 468–471 [DOI] [PubMed] [Google Scholar]
- Roy SW, Hudson AJ, Joseph J, Yee J, Russell AG 2011. Numerous fragmented spliceosomal introns, AT–AC splicing, and an unusual dynein gene expression pathway in Giardia lamblia. Mol Biol Evol 29: 43–49 [DOI] [PubMed] [Google Scholar]
- Sanford JR, Ellis J, Caceres JF 2005. Multiple roles of arginine/serine-rich splicing factors in RNA processing. Biochem Soc Trans 33: 443–446 [DOI] [PubMed] [Google Scholar]
- Shepard PJ, Hertel KJ 2009. The SR protein family. Genome Biol 10: 242 doi: 10.1186/gb-2009-10-10-242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shukla JN, Nagaraju J 2010. Doublesex: A conserved downstream gene controlled by diverse upstream regulators. J Genet 89: 341–356 [DOI] [PubMed] [Google Scholar]
- Sickmier EA, Frato KE, Shen H, Paranawithana SR, Green MR, Kielkopf CL 2006. Structural basis for polypyrimidine tract recognition by the essential pre-mRNA splicing factor U2AF65. Mol Cell 23: 49–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha R, Lenser T, Jahn N, Gausmann U, Friedel S, Szafranski K, Huse K, Rosenstiel P, Hampe J, Schuster S, et al. 2010. TassDB2—A comprehensive database of subtle alternative splicing events. BMC Bioinformatics 11: 216 doi: 10.1186/1471-2105-11-216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanke M, Steinkamp R, Waack S, Morgenstern B 2004. AUGUSTUS: A web server for gene finding in eukaryotes. Nucleic Acids Res 32: W309–W312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanke M, Diekhans M, Baertsch R, Haussler D 2008. Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics 24: 637–644 [DOI] [PubMed] [Google Scholar]
- Telonis-Scott M, Kopp A, Wayne ML, Nuzhdin SV, McIntyre LM 2009. Sex-specific splicing in Drosophila: Widespread occurrence, tissue specificity and evolutionary conservation. Genetics 181: 421–434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Pachter L, Salzberg SL 2009. TopHat: Discovering splice junctions with RNA-Seq. Bioinformatics 25: 1105–1111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahl MC, Will CL, Luhrmann R 2009. The spliceosome: Design principles of a dynamic RNP machine. Cell 136: 701–718 [DOI] [PubMed] [Google Scholar]
- Wang Z, Burge CB 2008. Splicing regulation: From a parts list of regulatory elements to an integrated splicing code. RNA 14: 802–813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang ET, Sandberg R, Luo S, Khrebtukova I, Zhang L, Mayr C, Kingsmore SF, Schroth GP, Burge CB 2008. Alternative isoform regulation in human tissue transcriptomes. Nature 456: 470–476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witten JT, Ule J 2011. Understanding splicing regulation through RNA splicing maps. Trends Genet 27: 89–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia Q, Zhou Z, Lu C, Cheng D, Dai F, Li B, Zhao P, Zha X, Cheng T, Chai C, et al. 2004. A draft sequence for the genome of the domesticated silkworm (Bombyx mori). Science 306: 1937–1940 [DOI] [PubMed] [Google Scholar]
- Xia Q, Guo Y, Zhang Z, Li D, Xuan Z, Li Z, Dai F, Li Y, Cheng D, Li R, et al. 2009. Complete resequencing of 40 genomes reveals domestication events and genes in silkworm (Bombyx). Science 326: 433–436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeo GW, Coufal NG, Liang TY, Peng GE, Fu XD, Gage FH 2009. An RNA code for the FOX2 splicing regulator revealed by mapping RNA–protein interactions in stem cells. Nat Struct Mol Biol 16: 130–137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zha XF, Xia QY, Zhao P, Li J, Duan J, Wang ZL, Qian JF, Xiang ZH 2005. Detection and analysis of alternative splicing in the silkworm by aligning expressed sequence tags with the genomic sequence. Insect Mol Biol 14: 113–119 [DOI] [PubMed] [Google Scholar]
- Zhan S, Huang J, Guo Q, Zhao Y, Li W, Miao X, Goldsmith MR, Li M, Huang Y 2009. An integrated genetic linkage map for silkworms with three parental combinations and its application to the mapping of single genes and QTL. BMC Genomics 10: 389 doi: 10.1186/1471-2164-10-389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang G, Guo G, Hu X, Zhang Y, Li Q, Li R, Zhuang R, Lu Z, He Z, Fang X, et al. 2010. Deep RNA sequencing at single base-pair resolution reveals high complexity of the rice transcriptome. Genome Res 20: 646–654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou W, Calciano MA, Jordan H, Brenner M, Johnson S, Wu D, Lei L, Pallares D, Beurdeley P, Rouet F, et al. 2009. High resolution analysis of the human transcriptome: detection of extensive alternative splicing independent of transcriptional activity. BMC Genet 10: 63 doi: 10.1186/1471-2156-10-63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou B, Yang X, Jiang J, Wang Y, Li M, Miao X, Huang Y 2010. Silkworm (Bombyx mori) BmLid is a histone lysine demethylase with a broader specificity than its homolog in Drosophila and mammals. Cell Res 20: 1079–1082 [DOI] [PubMed] [Google Scholar]
- Zorio DA, Cheng NN, Blumenthal T, Spieth J 1994. Operons as a common form of chromosomal organization in C. elegans. Nature 372: 270–272 [DOI] [PubMed] [Google Scholar]