

Abstract
Toxin–antitoxin (TA) loci are common in archaea and prokaryotes and allow cells to rapidly adapt to changing environmental conditions through release of active regulators of metabolism. Many toxins are endonucleases that target cellular mRNA and tRNAs, while the antitoxins tightly wrap around the toxins to inhibit them under normal circumstances. The antitoxins also bind to operators in the promoter regions of the cognate TA operon and thereby regulate transcription. For enteric vapBC TA loci, the VapC toxins specifically cleave tRNAfMet and thus down-regulate protein synthesis. Here, we describe the crystal structure of the intact Shigella flexneri VapBC TA complex, determined to 2.7 Å resolution. Both in solution and in the crystal structure, four molecules of each protein combine to form a large and globular hetero-octameric assembly with SpoVT/AbrB-type DNA-binding domains at each end and a total molecular mass of about 100 kDa. The structure gives new insights into the inhibition of VapC toxins by VapB and provides the molecular basis for understanding transcriptional regulation through VapB dimerization.
Abbreviations: TA, toxin–antitoxin; PIN, PilT N-terminal; SIRAS, single isomorphous replacement with anomalous scattering; FEN-1, flap endonuclease-1; PDB, Protein Data Bank; DMSI, dimethyl superimidate; EMSA, electrophoretic mobility shift assay
Keywords: RNA interferase, tRNA, protein–DNA interaction
Graphical Abstract
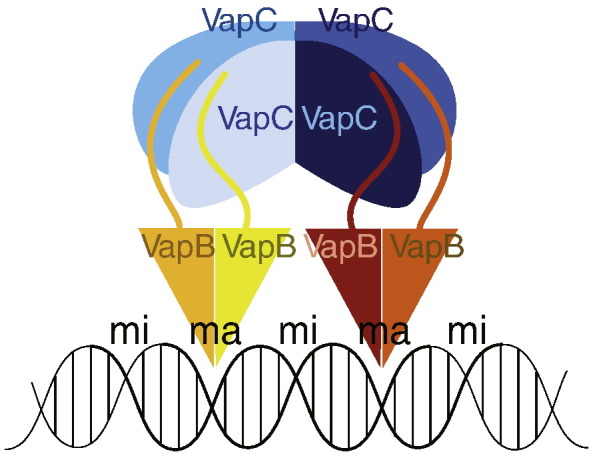
Highlights
► S. flexneri VapBC form a large, 100-kDa hetero-octamer assembly. ► We observe two DNA-binding domains of the AbrB/SpoVT type. ► The complex binds tightly to operator DNA at two distinct sites. ► The structure suggests that induced fit takes place upon DNA binding.
Introduction
Toxin–antitoxin (TA) loci are widespread in prokaryotes and code for an active “toxin” molecule, typically, a translational regulator, and an “antitoxin” that forms a tight complex with the toxin and thus inhibits it1-3. Upon changes in the surrounding environment, such as during nutritional stress, the antitoxin is degraded and the toxin is released intracellularly. Functions have not been ascribed to all types of toxins, but many possess “RNA interferase” activity, that is, they are able to cleave mRNA or tRNA to regulate overall rates of translation.4 At the genomic level, TA loci are organized in a tightly controlled operon with the toxin downstream of the antitoxin, transcriptionally regulated through a DNA-binding domain on the antitoxin.5 Type II TA loci, for which both toxin and antitoxin are proteins, have been subdivided into six evolutionarily independent families: ccdAB, mazEF, phd/doc, parDE, higAB, and relBE.5 VapC, HigA, and MazF toxins are active ribonucleases in isolation3,6,7 while the RelE-type toxins require the ribosome in order to cleave mRNA during translation.8,9 CcdB and ParE toxins, on the other hand, target DNA gyrase and thus inhibit DNA replication,10,11 while Doc has been proposed to inhibit translation through static binding to the ribosome in a way akin to antibiotics and thus does not induce RNA cleavage.12
In the largest TA family, vapBC, the VapC toxin contains a PilT N-terminal (PIN) domain, which is a compact ribonuclease domain consisting of an αβ-fold harboring four highly conserved acidic residues required for catalysis.13 PIN domains have only been functionally characterized in a few cases but are believed to cleave single-stranded RNA in a sequence and divalent metal-ion-dependent manner.14 They are found in all domains of life but are in bacteria mainly encoded by vapBC loci, where they are active on a range of substrates including mRNA and tRNA14 and often in a sequence-specific manner like their eukaryotic counterparts.6 Interestingly, vapBC loci are common among pathogenic bacteria, such as Mycobacterium tuberculosis, which contains an astounding 47 vapBC loci.1 The evolutionary benefit of having such an extreme number of similar genetic loci is not known, but recent results suggest that the loci are involved in the formation of persister cells, which are critical to pathogeniticy.15 Crystal structures of VapBC complexes and isolated VapC toxins currently exist from both archaea and the pathogenic bacteria M. tuberculosis and Neisseria gonorrhoeae.16–18 Like other TA systems, VapBC regulate their own transcription through binding of VapBC complexes to operators in its own promoter region.19–21 The structure of the N. gonorrhoeae FitAB complex, which is a VapBC-type TA system, bound to its operator site on DNA showed that the complex forms relatively loose hetero-octamer structure that interacts with DNA through two ribbon–helix–helix motifs.18 However, in the structure of unbound VapBC-5 from M. tuberculosis, the DNA-binding region of VapB was disordered and could not be resolved (Table 2).17
Table 2.
Comparison of S. flexneri VapBC with similar structures
| Structural similarity (RMSD Cα positions) (Å) | ||
| VapC (core only) | M. jannaschii FEN-1 nuclease (PDB 1A7623) | 3.44 |
| VapC | N. gonorrhoeae FitB (PDB 2H1O18) | 1.78 |
| VapC | M. tuberculosis Rv0301 (VapC) (PDB 3H87) | 2.15 |
| VapC | M. tuberculosis VapC-5 (PDB 3DBO17) | 0.97 |
| VapB (N-domain) | B. subtilis AbrB (N domain, PDB 2K1N26) | 3.66 |
| VapB (β3 + 4 only) | B. subtilis AbrB (β3+4 only, PDB 2K1N26) | 0.76 |
Root-mean-square deviation values (RMSD; measured in angstroms) are calculated by superpositioning of the indicated VapBC components (left column) onto other known structures (middle column). RMSD values are for Cα atoms only.
Recently, it was found that VapC (MvpT) from the Gram-negative pathogen Shigella flexneri 2a virulence plasmid pMYSH6000 functions by specifically cleaving initiator tRNAfMet in the anticodon region, thus globally down-regulating translation.22 This showed that VapC toxins are capable of very specifically recognizing molecular targets and open up entirely new ways of fine-tuning cell metabolism. In order to understand the activity, mode of inhibition, and DNA-binding properties of the VapBC family, we have determined the crystal structure of the VapBC complex from S. flexneri. The structure reveals a compact hetero-octameric assembly with two unique DNA-binding domains of the SpoVT/AbrB type that have not been previously observed in VapBC complexes. Gel permeation chromatography and in vitro cross-linking experiments confirm that the octamer is present in solution and in the crystal, thus strongly suggesting that S. flexneri VapBC interacts with the promoter through interaction with adjacent major grooves.
Results and Discussion
Overall structure of the S. flexneri VapBC complex
His6-VapB:VapC was expressed in Escherichia coli from a bicistronic construct encoding genes optimized for expression, purified by Ni-NTA and gel-filtration chromatography, and concentrated to 7 mg/ml before crystallization. Large hexagonal crystals containing both components appeared in 1.0 M ammonium sulfate and 0.5% (v/v) polyethylene glycol 3350 at pH 5.5 and diffracted to about 2.7 Å. Following unsuccessful attempts at structure determination by molecular replacement using existing VapBC structures, the structure was eventually determined by single isomorphous replacement with anomalous scattering (SIRAS) using a uranyl acetate data set to 2.9 Å and refined to a final R (Rfree) of 18.2% (23.7%) using iterative rebuilding and refinement in Coot and PHENIX (see Table 1 for crystallographic data statistics). The asymmetric unit contains four copies of both VapB and VapC, for which the VapC structures are complete (residues 1–132), while the VapB structures cover residues 2–67 of the complete sequence (1–75). The C-terminal residues 68–75 of VapB extend into the solvent region and are not visible.
Table 1.
Crystallographic data statistics
| Native | U (SIRAS) | |
|---|---|---|
| Data collection | ||
| Wavelength (Å) | 0.94645 | 1.04002 |
| Space group | P6122 | P6122 |
| Cell dimensionsa | ||
| a, b, c (Å) | 91.4, 91.4, 549.1 | 92.4, 92.4, 548.9 |
| α, β, γ (°) | 90, 90, 120 | 90, 90, 120 |
| Resolution (Å) | 39.6–2.7 | 39.5–2.9 |
| I/σ(I)b | 20.5 (2.2) | 28.76 (8.79) |
| Completeness (%) | 99.6 (96.9) | 99.5 (95.6) |
| Redundancy | 22.4 (21.0) | 6.1 (5.5) |
| Mosaicity (°) | 0.1 | 0.09 |
| Rsym (%) | 16.6 (164.8) | 4.3 (19.2) |
| Rmrdg,F (%)c | 9.0 (70.7) | 3.5 (18.1) |
| Refinementd | ||
| Resolution (Å) | 39.6–2.7 | |
| No. of reflections | 38,932 | |
| Rwork/Rfree (%) | 18.2/23.7 (28.9/39.6) | |
| Total protein atoms | 6280 | |
| Total water molecules | 205 | |
| B-factor (Å2) | 65.9 | |
| RMSD bonds (Å) | 0.0052 | |
| RMSD angles (°) | 0.838 | |
| Ramachandran statisticsd (%) | ||
| Favored | 95.6 | |
| Allowed | 4.1 | |
| Outliers | 0.3 | |
Crystallographic data collection and refinement statistics for VapBC native and uranium derivative crystals.
Values given by XSCALE.
Figures in parentheses represent the outermost-resolution shell (2.76–2.69 Å).
Redundancy-independent R-factor.
Values given by PHENIX.
S. flexneri VapC contains a typical PIN domain structure consisting of a small and central five-stranded β-sheet (β1–β5) surrounded by seven α-helices, denoted α1 through α7 (Fig. 1a, blue). VapB has an N-terminal domain consisting of four β-strands (residues 1–45), which dimerizes with a neighboring VapB to form a complete DNA-binding domain, and an extended C-terminal tail (residues 46–67) that wraps around VapC (Fig. 1a, orange). In the crystal, the four copies of VapBC in the asymmetric unit form a large and globular VapB4C4 hetero-octameric assembly, consisting of two VapB2C2 complexes related to each other by a dyad axis (Fig. 1b, top view; the dyad is indicated by the black lens shape at the center). The VapB2C2 complexes are strongly stabilized through interaction of the VapB N-terminal domains to form two DNA-binding domains (Fig. 1b, side view). The VapB2 dimer itself has an interface area of 1510 Å2 (Fig. 1b, orange/yellow chains) and consists of a layer of a four-stranded and a three-stranded antiparallel β-sheets. Interactions between the two VapC molecules in the VapB2C2 assembly are weak and water mediated (Fig. 1b, e.g., VapC2/VapC1, the dark-blue chains); however, there are strong interactions to the two VapC molecules of the adjacent VapB2C2 hetero-tetramer mediated through helices α4, α5, and α6 with a total interface area of 1050 Å2 (Fig. 1b, e.g., VapC2/VapC3, the dark-blue and white chains). Overall, these interactions give rise to a ring-shaped structure with weaker water-mediated contacts at the center. Interestingly, the DNA-bound conformation of the N. gonorrhoeae FitAB hetero-octamer shows a ring-shaped structure similar to but much more loose and open than that observed for VapBC (Table 2 and Supplementary Fig. 1).
Fig. 1.

Structure overview. (a) The VapBC hetero-dimer shown in ribbon representation labeled with termini and secondary structure elements. The VapC toxin is in blue, and the VapB antitoxin is in orange. (b) Overview on the hetero-octameric assembly of four VapBC hetero-dimers found in the asymmetric unit, shown in top and side views with VapC in shades of blue and with VapB in shades of orange/yellow. The two DNA-binding domains formed upon dimerization of adjacent VapB molecules are orange and yellow, and on the top view, the dyad axis is indicated in a black lens shape. (c) Interaction of conserved hydrophobic residues in VapB (yellow) with the hydrophobic core of VapC. VapC is shown as a semitransparent surface colored from white (hydrophobic) to blue (hydrophilic) with the cartoon inside. This figure and subsequent structure figures were produced with PyMOL (Schrödinger, LLC).
The VapB C-terminal domain inhibits VapC by a conserved mechanism
The interaction between the extended C-terminal region of VapB and the VapC PIN domain shows two main features, which are replicated in all four copies in the asymmetric unit. Firstly, four aromatic residues in VapB (Trp47, Trp50, Phe51, and Phe60) point into the hydrophobic interface between helices α1, α2, α3, and α4 of VapC where they interact tightly with the hydrophobic core of the protein (Fig. 1c). Secondly, near the C-terminus of VapB, the side chains of Arg64 and Gln66 of VapB point directly into the active site of VapC, where they make close, charged interactions to the conserved acidic active residues Asp7, Glu42, and Asp98 (Fig. 2a). In this interaction, the guadinium group of Arg64 interacts strongly with the carboxylic oxygen atoms of Glu42 and Asp98 in VapC through two hydrogen bonds to each residue. VapB Arg64 makes additional contacts to a water molecule located in the active site, which is also coordinated by Asp7, Asp98, and the main-chain nitrogen of Thr8 in VapC (the latter is not shown). In the FitAB complex, Arg68 in Fit B engages in a similar close interaction with the active-site residues of FitA, confirming the generality of this observation.18 In addition, a BLAST search using only the C-terminus of VapB (residues 46–75) identified 110 VapB proteins with a sequence similarity of 54–95%, and a multiple sequence alignment reveals a high degree of conservation for both interaction regions (i.e., the intercalating aromatic residues, as well as Arg64 and Gln66; Supplementary Fig. 2, marked in red). The high level of conservation strongly suggests that these are general mechanisms of inhibition within the VapBC group. Interestingly, however, in the structure of the M. tuberculosis VapBC-5 complex, the arginine was found in a different orientation, which may indicate that not all interactions are simultaneously required for inhibition to take place.17
Fig. 2.

Comparison of the active site of VapC to FEN-1 nuclease. (a) Close-up of the active site of VapC shown in blue with yellow sticks and with relevant residues of VapB in orange sticks. Residues shown with yellow sticks are conserved in PIN domains. Water molecules in the active site are shown with red small spheres. (b) The active site of the M. jannaschii flap endonuclease (FEN-1; PDB 1A7618) shown in the same orientation as in (a). Conserved active-site residues are shown as yellow sticks; non-conserved residues, in green; and the two Mn2+ ions, as violet big spheres.
In order to understand the functional implications of the close interactions at the active site, we superimposed the structure of VapB-inhibited VapC on the PIN domain from Methanococcus jannaschii flap endonuclease-1 [FEN-1; Protein Data Bank (PDB) ID: 1A76], which is more well characterized functionally.23 Overall, the superposition gives a good fit of secondary structure elements surrounding the active site and places all four conserved acidic residues in reasonable positions (Table 2 and Fig. 2b). Although the cleavage mechanism employed by PIN domains is still debated,14 there is growing evidence that it involves two divalent metal ions at the active site, coordinated by the acidic residues.13,14 Mutant studies implicate metal ion 1 (Fig. 2b, right) directly in the cleavage mechanism, whereas metal ion 2 (left) is thought to stabilize the conformation of the active site and enhance substrate binding.23 Both ions are present in the FEN-1 structure, and comparison to VapC reveals that the positively charged guadinium group of Arg64 in VapB appears to take up the position of metal ion 1 (Fig. 2b), while Gln66 may substitute metal ion 2. Together, this suggests that the nuclease activity of VapC is at least in part inhibited through displacement of the natural divalent metal ions from the active site.
S. flexneri VapBC forms a higher-order structure in solution
As the higher-order octameric structure observed in the crystal structure of S. flexneri VapBC could arise due to crystal packing interactions, we asked if this VapBC complex also forms a higher-order structure in solution. Analysis of purified VapBC by high-resolution gel permeation chromatography using a 24-ml Superdex 200 10/300 GL column showed a major early peak containing both VapB and VapC around 12.7 ml. Consistently, this elution volume corresponds to a molecular mass of approximately 105 kDa (Fig. 3a), closely fitting the predicted size of a hetero-octameric VapB4C4 complex (VapC = 15 kDa, VapB = 10 kDa, and VapB4C4 = 100 kDa). We also analyzed the purified VapBC complex by chemical cross-linking using dimethyl superimidate (DMSI), which shows an increasingly complex pattern of higher-order complexes appearing over time (Fig. 3b). Early cross-linked complexes indicate formation of VapB2 (20 kDa) and VapBC (25 kDa) dimers (10 min), while later on, presumably due to variations in accessibility of cross-linking sites, VapB4, VapB2C2, and VapC4 species appear. The observation of a molecular species heavier than 50 kDa strongly indicates that a tetramer of VapC molecules is present in solution (molecular mass, 15 kDa) and, consequently, that the hetero-octamer is the prevalent oligomeric assembly due to the strong interaction between VapB and VapC. The lower abundance of the higher-order complexes on the gel is likely due to the statistical chance of cross-linking all four molecules in the same complex combined with accessibility of cross-linking sites. Thus, based on combination of gel permeation chromatography and chemical cross-linking, we therefore conclude that VapB and VapC assemble as an octameric complex in solution, consistent with the crystal structure.
Fig. 3.

S. flexneri VapBC forms a hetero-octamer in solution. (a) Chromatogram from gel permeation chromatography using a 24-ml Superdex 200 10/300 GL column showing a major early peak containing both VapB and VapC at an elution volume of 12.7 ml, corresponding to a molecular mass of 105 kDa. Vo is the void volume (approximately 8 ml). OD280 is shown in blue, and OD260 is shown in red. (b) DMSI cross-linking time-course experiment analyzed by 15% Coomassie-stained SDS-PAGE with tentative assignment of the cross-linked species. MW is a molecular mass standard with the indicated sizes in kilodaltons. Monomeric VapC weighs 15 kDa, and VapB, 10 kDa.
VapB dimerizes to form a SpoVT/AbrB-like DNA-binding domain
A BLAST search using the N-terminal sequence of S. flexneri VapB (residues 1–45) identifies the region as a putative SpoVT/AbrB-like DNA-binding domain (Pfam accession number PF04014). AbrB is a transcriptional regulator found in Bacillus subtilis, which is involved in the regulation of more than 60 genes.24 The solution structure of the DNA-binding domain of AbrB revealed the so-called “swapped β-hairpin” fold, in which two molecules each containing two β-hairpins dimerize through interweaving of the hairpins, forming a layered β-sandwich (Fig. 4a, right).24 Analogously, in the octamer structure of VapBC, the four N-terminal VapB domains come together to form two DNA-binding domains each consisting of a three-stranded antiparallel β-sheet (β1′ + β2′ and β2″; Fig. 4a, left, top layer) and a four-stranded antiparallel β-sheet (β3′ + β4″ and β3″ + β4″; left, bottom layer). The two β-sheets pack tightly against each other in a layered fashion to form the complete DNA-binding domain. Because the three-stranded β-sheets are formed asymmetrically (i.e., with one strand from one molecule and two from the other), two of four VapB molecules have an N-terminal domain consisting of two long and two short β-strands, while the other VapB molecules in our structure contain two long and a single short β-strand. The N-termini that only contribute a single short β-strand are extended and engage in crystal contacts with other symmetry-related octamers in the crystal. We speculate that the complete β-sandwich is formed upon binding to DNA. Interestingly, the DNA-binding domain is reminiscent of the β-barrel found in the MazEF TA complex but without the continuous interstrand hydrogen bonds required to form a proper β-barrel.25 This is therefore, to our knowledge, the first observation of a layered DNA-binding domain in bacterial TA systems.
Fig. 4.

VapBC contains a SpoVT/AbrB-type DNA-binding domain. (a) Left: The DNA-binding domain in VapB resulting from homodimerization of two N-terminal domains (shown in red and green). Right: The homologous DNA-binding domain found in B. subtilis AbrB (light red/light green; PDB 2K1N26). The annotation of secondary structure elements for VapB follows the standard used for AbrB. (b) A model for the interaction between the VapB DNA-binding domain and linear B-form DNA with putative interacting residues in green. Both Lys18 residues from the VapB dimer interact with the DNA backbone, while residues 8–11 are proposed to interact with the DNA major groove. (c) An overview model of how DNA binding to adjacent major grooves might be accomplished by the VapBC hetero-octamer. Color scheme as in Fig. 1b. ma, major groove; mi, minor groove.
In the DNA-binding domain of AbrB, the layers of β-sheets are connected by short two-turn α-helices. In comparison to VapB, we find that these are much shorter (Table 2 and Fig. 4a). In addition, in AbrB, the loops connecting β1′ and β2′ corresponding to residues 8–11 in VapB have been shown to be involved directly in sequence readout in the major groove of DNA, while the loops connecting β2 and β3 were predicted to be involved in DNA backbone recognition.26 Due to the aforementioned asymmetry in the present VapB structure, we only observe one of the major groove-binding loops at each end of the octamer, again suggesting that this region may undergo induced fit upon binding DNA. However, using the structure of the N-terminal domain of AbrB bound to its operator (PDB entry 2K1N), we can get some idea of how VapB binds DNA.26 In this model, the two Lys18 of both VapB molecules in the DNA-binding dimer are located close to the DNA backbone and probably contribute general affinity through charge–charge interactions (Fig. 4b). Inside the major groove, Ser8, Asn9, Arg10, and Ser11 from the β-loop come very close to the DNA bases and are therefore likely involved in sequence readout. We predict that an analogous interaction mediated by the other VapB molecule takes place in the same major groove on the other side of the double helix. The residues involved in direct contacts are only partially conserved across species, but similar functional groups are found close by in the related sequences, suggesting that the loops are used to adapt the domain to varying DNA sequences (Supplementary Fig. 3). Considering the entire octamer structure and the orientation of the two DNA-binding domains relative to each other, we find that the distance does not match exactly an integer multiple of helical turns and further that the domains are rotated 56° with respect to one another. This supports the idea that induced fit takes place upon DNA binding, either through deformation (bending) of the DNA or through conformational changes in the protein (Fig. 4c). Furthermore, the relative orientation of the two domains suggests that the DNA is approached from two sides and thus clamped by the VapBC complex.
VapBC binds to two operator sites
To functionally investigate the binding of VapBC to promoter DNA, we first inspected the vapBC promoter region on the S. flexneri pMYSH6000 genome, which revealed two putative VapBC binding sites overlapping with the − 10 Pribnow box and the − 35 sequence (Fig. 5a, shown with broken lines). The specificity of VapBC toward these putative operator sites was then investigated by electrophoretic mobility shift assays (EMSA) using three different DNA fragments spanning either one or both of the regions. Incubation of the VapBC complex with the entire vapBC promoter region (DNA I) results in formation of two distinct complexes (Fig. 5b, lanes 3 and 4, and Supplementary Fig. 4a, lanes 7–12). VapBC is specific to these promoter sequences as no shifts are observed when the complex is incubated with an unrelated control DNA amplified from the pUC plasmid (Fig. 5b, lanes 1 and 2, and Supplementary Fig. 4a, lanes 1–6). This indicates that VapBC specifically binds to two sites in the vapBC promoter region. To further delineate the observed mode of binding, we incubated VapBC with two smaller DNA fragments containing the isolated operator sites (DNA II and DNA III; Fig. 5a and b). In each case, incubation produces single shifts (Fig. 5b, lanes 5–8). Since the VapB4C4 octamer is the stable form in solution, this strongly suggests that two VapBC octamers bind in the promoter region. Both DNA II and DNA III have identical lengths, and the shifts observed are of equal size, which indicates that the complexes formed with DNA II and DNA III are identical. However, the band shift observed for DNA III is of higher intensity than that for DNA II, which further suggests that VapBC has the highest affinity for the operator site overlapping with − 35 sequences (compare Fig. 5b, lanes 6 and 8, and Supplementary Fig. 4b, lanes 1–6 and 7–12). Both operator sites overlap with RNA polymerase binding sites; thus, VapBC binding to either site most likely results in transcriptional repression.
Fig. 5.

VapBC specifically binds the promoter DNA at two operator sites. (a) The sequence of the S. flexneri vapBC promoter region with the − 10 Pribnow box and the − 35 sequences is indicated in boldface. The transcription start site is shown with a bent arrow, and putative VapB binding sites in adjacent major grooves are indicated by broken lines. The position and extent of the DNA oligos (DNA I, DNA II, and DNA III) analyzed in (b) are indicated with continuous lines. (b) EMSA showing 32P-labeled DNA fragments binding to VapBC. Control DNA (lanes 1 and 2), DNA I (lanes 3 and 4), DNA II (lanes 5 and 6), or DNA III (lanes 7 and 8) were incubated with (+) or without (−) 2.5 ng/μl VapBC, and DNA and protein–DNA complexes were separated by 6% native PAGE. Arrows indicate shifted protein–DNA complexes.
Conclusion
In this paper, we have shown that the S. flexneri VapBC complex forms a large hetero-octameric assembly both in solution and in the crystal structure. This assembly most likely represents the biologically significant unit of the complex in vivo, and we predict that two copies of the assembly bind the promoter and thus regulate transcription from the VapBC operon, either through deformation of the DNA or protein induced fit. Previous crystal structures of isolated VapBC complexes from N. gonorrhoeae, M. tuberculosis, and archaeal homologues showed hetero-tetrameric multimers, while the DNA-bound structure of N. gonorrhoeae FitAB revealed a hetero-octameric assembly similar to that of S. flexneri VapBC, which seems to be required for DNA binding.16–18 However, FitAB contains a helical DNA-binding domain, and the structure is much more open than VapBC, which is the first example of a TA system containing an AbrB/SpoVT-type DNA-binding domain. The DNA-binding motif in VapBC forms through homodimerization of the antitoxin, and it may therefore be one of the main determinants for whether TA complexes form tetramers or octamers. Future studies will hopefully reveal the structural basis for promoter recognition by S. flexneri VapBC and the mode of interaction with the toxin target tRNAfMet.
Materials and Methods
Protein expression and purification
The construct pKW812HB22 was used for expression of His6-VapB:VapC in E. coli C41 (DE3). Cells grew in 1× LB containing 100 μg/ml ampicillin and were induced with 1 mM IPTG at an OD600 of 0.5. Expression was carried out overnight at 25 °C with vigorous shaking (120 rpm) before harvesting cells (15 min, 12,000g) and resuspension in 50 mM Tris (pH 8.0), 500 mM NaCl, 5 mM MgCl2, 5 mM β-mercaptoethanol, 10 mM imidazole, and protease inhibitor tablets (Sigma). For cell disruption, high-pressure homogenization at 15,000 psi and sonication were combined, and the resulting cell lysate was cleared by centrifugation at 15,000 rpm for 45 min and was loaded onto a pre-packed 5-ml HiTrap Ni-NTA column (GE Healthcare) followed by extensive column washing (20 column volumes) in 50 mM Tris (pH 8.0), 500 mM NaCl, 5 mM MgCl2, 5 mM β-mercaptoethanol, and 35 mM imidazole. Finally, the nearly pure VapBC complex was eluted with 50 mM Tris (pH 8.0), 500 mM NaCl, 5 mM MgCl2, 5 mM β-mercaptoethanol, and 300 mM imidazole. For crystallization and cross-linking, the sample was concentrated to 7 mg/ml using a Vivaspin 6 spin filter with a 5-kDa molecular mass cutoff and was further purified by gel filtration in 25 mM Tris (pH 8.0), 500 mM NaCl, 5 mM MgCl2, and 5 mM β-mercaptoethanol on a Superdex 200 10/300 HR column (GE Healthcare). VapBC used for EMSA was purified from E. coli C41 (DE3) containing the plasmid pKW812HC,22 which has the His-tag on VapC to prevent interference with the DNA-binding domain (VapB:His6-VapC). The protein was purified once by Ni-NTA using phosphate buffers [50 mM NaH2PO4 (pH 8.0), 0.3 M NaCl, 10/25/500 mM imidazole, and 5 mM β-mercaptoethanol] and subsequently dialyzed overnight against storage buffer (1× phosphate-buffered saline, 20% glycerol, and 1 mM DTT) at 4 °C.
Chemical cross-linking
Prior to the experiment, the Tris buffer was exchanged for 100 mM sodium borate, pH 9.1, using a Vivaspin 6 spin filter with a 5-kDa molecular mass cutoff (GE Healthcare) to prevent reaction of the cross-linking agent with the free amines of the buffer molecules, and the protein was concentrated to 1 mg/ml. A reaction containing 50 μl of 1 mg/ml VapBC complex and 5 μl of 20 mg/ml DMSI was prepared, and11 μl aliquots was removed after 5, 10, 30, 60, and 120 min. The reaction was stopped with 4 μl of 2 M Tris, pH 8.0, and samples were analyzed by 15% Coomassie-stained SDS-PAGE.
Crystallization and structure determination
For crystallization, the protein sample from gel filtration was concentrated to 7 mg/ml in gel-filtration buffer. Large single VapBC crystals appeared after 3–4 days in 0.1 M Bis-Tris (2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol) (pH 5.5), 1 M ammonium sulfate, and 0.5% (v/v) polyethylene glycol 3350 at 19 °C using sitting-drop vapor diffusion in a 1:1 ratio of protein to crystallization buffer. Before data collection, crystals were cryo-protected by serial transfer into drops containing 10–20% glycerol and flash frozen in liquid nitrogen. Uranium derivative crystals were prepared by soaking the crystals for 3 h in mother liquor supplemented with 1.25 mM UO2(CH3OCOO−)2 (uranyl acetate). Data collection for native crystals was carried out at beamline ID23-1 at European Synchrotron Radiation Facility, Grenoble, with detectable diffraction to about 2.7 Å. The native data set has an unusually high redundancy (22-fold), giving rise to relatively high symmetry R-factors; however, the data are of very high quality, which are also witnessed by the redundancy-corrected R-factor Rmrdg,F (see Table 1). Uranium derivative crystals diffracted to 2.9 Å and data were collected at beamline i911-2 at MAX-Lab in Lund, Sweden. Indexing, integration, and scaling were carried out using XDS for both native and derivative data,27 and PHENIX was used to solve and refine the structure.28 Briefly, the uranium derivative data were used to solve the phase problem for VapBC by SIRAS. The initially obtained experimentally phased electron density map was submitted for automatic model building using phenix.autobuild, and the model was then iteratively updated manually and refined using Coot29,30 and phenix.refine, respectively.
Electrophoretic mobility shift assay
The control DNA fragment and DNA I containing both vapBC operator sites were amplified in a PCR reaction using primers 171SR14 (5′-GGGGCAGCTGGCGAAAGGGGGATGTGCTGC) and 171SR16 (5-GGGGCAGCTGAATTTCACACAGGAAACAGCTA) and SF-EMSA-f (5′-GGCCGGCCCAGCGTTCTC) and SF-EMSA-r (5′-TGCTGAGAAATACGGTGG), respectively. Prior to the PCR amplification, primers 171SR14 and SF-EMSA-f were 5′ end-labeled with [γ-32P]ATP using T4 polynucleotide kinase (New England Biolabs). 171SR14 and 171SR16 produce a 199-bp DNA fragment of pUC plasmid DNA, while SF-EMSA-f and SF-EMSA-r produce a 212-bp DNA fragment containing the entire vapBC promoter region. The DNA II and DNA III fragments were created by hybridizing SF-EMSA1-f (5′-ACAATAGATATACACAAGACATATCCACAT) and SF-EMSA1-r (5′-ATGTGGATATGTCTTGTGTATATCTATTGT) and SF-EMSA2-f (5′-ATAAACGTATATCCCTTTGACATATCCCGG) and SF-EMSA2-r (5′-CCGGGATATGTCAAAGGGATATACGTTTAT), respectively. Prior to hybridization, primers SF-EMSA1-f and SF-EMSA2-f were 5′ end-labeled as described above. Labeled DNA fragments (1 nM) were incubated with purified protein complex in binding buffer [20 mM Tris–HCl (pH 7.5), 100 mM KCl, 2 mM MgCl2, 1 mM DTT, 50 μg/ml bovine serum albumin, and 10% glycerol]. To avoid nonspecific DNA binding, we added sonicated salmon sperm DNA to a final concentration of 0.1 mg/ml. Reactions were incubated for 20 min at 37 °C, protein–DNA complexes were separated by native PAGE in 6% acrylamide gels with 0.5× Tris–borate–ethylenediaminetetraacetic acid, and the separated complexes were visualized by phosphor imaging.
Accession number
The structure of the S. flexneri VapBC complex has been deposited in the PDB with accession code 3TND.
Acknowledgements
We are thankful to the beamline staff at European Synchrotron Radiation Facility, Grenoble, and MAX-Lab, Lund, for help with data collection. This work was funded by the Danish National Science Foundation's Centre for mRNP Biogenesis and Metabolism and the Novo Nordisk Foundation and the Wellcome Trust.
Edited by R. Huber
Footnotes
Supplementary data to this article can be found online at doi:10.1016/j.jmb.2011.10.024
Appendix A.
Supplementary materials
References
- 1.Pandey D.P., Gerdes K. Toxin–antitoxin loci are highly abundant in free-living but lost from host-associated prokaryotes. Nucleic Acids Res. 2005;33:966–976. doi: 10.1093/nar/gki201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Makarova K.S., Wolf. Y.I, Koonin E.V. Comprehensive comparative-genomic analysis of type 2 toxin-antitoxin systems and related mobile stress response systems in prokaryotes. Biol. Direct. 2009;4:19. doi: 10.1186/1745-6150-4-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jørgensen M.G., Pandey D.P., Jaskolska M., Gerdes K. HicA of Escherichia coli defines a novel family of translation-independent mRNA interferases in bacteria and archaea. J. Bacteriol. 2009;191:1191–1199. doi: 10.1128/JB.01013-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yamaguchi Y., Inouye M. mRNA interferases, sequence-specific endoribonucleases from the toxin–antitoxin systems. Prog. Mol. Biol. Transl. Sci. 2009;85:467–500. doi: 10.1016/S0079-6603(08)00812-X. [DOI] [PubMed] [Google Scholar]
- 5.Gerdes K., Christensen S.K., Løbner-Olesen A. Prokaryotic toxin–antitoxin stress response loci. Nat. Rev. Microbiol. 2005;3:371–382. doi: 10.1038/nrmicro1147. [DOI] [PubMed] [Google Scholar]
- 6.Ahidjo B.A., Kuhnert D., McKenzie J.L., Machowski E.E., Gordhan B.G., Arcus V. VapC toxins from Mycobacterium tuberculosis are ribonucleases that differentially inhibit growth and are neutralized by cognate VapB antitoxins. PLoS One. 2011;6:e21738. doi: 10.1371/journal.pone.0021738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Christensen S.K., Pedersen K., Hansen F.G., Gerdes K. Toxin–antitoxin loci as stress-response-elements: ChpAK/MazF and ChpBK cleave translated RNAs and are counteracted by tmRNA. J. Mol. Biol. 2003;332:809–819. doi: 10.1016/s0022-2836(03)00922-7. [DOI] [PubMed] [Google Scholar]
- 8.Pedersen K., Zavialov A.V., Pavlov M.Y., Elf J., Gerdes K., Ehrenberg M. The bacterial toxin RelE displays codon-specific cleavage of mRNAs in the ribosomal A site. Cell. 2003;112:131–140. doi: 10.1016/s0092-8674(02)01248-5. [DOI] [PubMed] [Google Scholar]
- 9.Hurley J.M., Cruz J.W., Ouyang M., Woychik N.A. Bacterial toxin RelE mediates frequent codon-independent mRNA cleavage from the 5′ end of coding regions in vivo. J. Biol. Chem. 2011;286:14770–14778. doi: 10.1074/jbc.M110.108969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jensen R.B., Gerdes K. Programmed cell death in bacteria: proteic plasmid stabilization systems. Mol. Microbiol. 1995;17:205–210. doi: 10.1111/j.1365-2958.1995.mmi_17020205.x. [DOI] [PubMed] [Google Scholar]
- 11.Jiang Y., Pogliano J., Helinski D.R., Konieczny I. ParE toxin encoded by the broad-host-range plasmid RK2 is an inhibitor of Escherichia coli gyrase. Mol. Microbiol. 2002;44:971–979. doi: 10.1046/j.1365-2958.2002.02921.x. [DOI] [PubMed] [Google Scholar]
- 12.Liu M., Zhang Y., Inouye M., Woychik N.A. Bacterial addiction module toxin Doc inhibits translation elongation through its association with the 30S ribosomal subunit. Proc. Natl Acad. Sci. USA. 2008;105:5885–5890. doi: 10.1073/pnas.0711949105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Arcus V.L., Bäckbro K., Roos A., Daniel E.L., Baker E.N. Distant structural homology leads to the functional characterization of an archaeal PIN domain as an exonuclease. J. Biol. Chem. 2004;279:16471–16478. doi: 10.1074/jbc.M313833200. [DOI] [PubMed] [Google Scholar]
- 14.Arcus V.L., McKenzie J.L., Robson J., Cook G.M. The PIN-domain ribonucleases and the prokaryotic VapBC toxin–antitoxin array. Protein Eng. Des. Sel. 2011;24:33–40. doi: 10.1093/protein/gzq081. [DOI] [PubMed] [Google Scholar]
- 15.Maisonneuve E., Shakespeare L.J., Jørgensen M.G., Gerdes K. Bacterial persistence by RNA endonucleases. Proc. Natl Acad. Sci. USA. 2011;108:13206–13211. doi: 10.1073/pnas.1100186108. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 16.Bunker R.D., McKenzie J.L., Baker E.N., Arcus V.L. Crystal structure of PAE0151 from Pyrobaculum aerophilum, a PIN-domain (VapC) protein from a toxin–antitoxin operon. Proteins. 2008;72:510–518. doi: 10.1002/prot.22048. [DOI] [PubMed] [Google Scholar]
- 17.Miallau L., Faller M., Chiang J., Arbing M., Guo F., Cascio D., Eisenberg D. Structure and proposed activity of a member of the VapBC family of toxin–antitoxin systems. VapBC-5 from Mycobacterium tuberculosis. J. Biol. Chem. 2009;284:276–283. doi: 10.1074/jbc.M805061200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mattison K., Wilbur J.S., So M., Brennan R.G. Structure of FitAB from Neisseria gonorrhoeae bound to DNA reveals a tetramer of toxin–antitoxin heterodimers containing Pin domains and ribbon–helix–helix motifs. J. Biol. Chem. 2006;281:37942–37951. doi: 10.1074/jbc.M605198200. [DOI] [PubMed] [Google Scholar]
- 19.Wilbur J.S., Chivers P.T., Mattison K., Potter L., Brennan R.G., So M. Neisseria gonorrhoeae FitA interacts with FitB to bind DNA through its ribbon–helix–helix motif. Biochemistry. 2005;44:12515–12524. doi: 10.1021/bi0511080. [DOI] [PubMed] [Google Scholar]
- 20.Robson J., McKenzie J.L., Cursons R., Cook G.M., Arcus V.L. The vapBC operon from Mycobacterium smegmatis is an autoregulated toxin–antitoxin module that controls growth via inhibition of translation. J. Mol. Biol. 2009;390:353–367. doi: 10.1016/j.jmb.2009.05.006. [DOI] [PubMed] [Google Scholar]
- 21.Bodogai M., Ferenczi S., Bashtovyy D., Miclea P., Papp P., Dusha I. The ntrPR operon of Sinorhizobium meliloti is organized and functions as a toxin–antitoxin module. Mol. Plant-Microbe Interact. 2006;19:811–822. doi: 10.1094/MPMI-19-0811. [DOI] [PubMed] [Google Scholar]
- 22.Winther K.S., Gerdes K. Enteric virulence associated protein VapC inhibits translation by cleavage of initiator tRNA. Proc. Natl Acad. Sci. USA. 2011;108:7403–7407. doi: 10.1073/pnas.1019587108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hwang K.Y., Baek K., Kim H.Y., Cho Y. The crystal structure of flap endonuclease-1 from Methanococcus jannaschii. Nat. Struct. Biol. 1998;5:707–713. doi: 10.1038/1406. [DOI] [PubMed] [Google Scholar]
- 24.Coles M., Djuranovic S., Söding J., Frickey T., Koretke K., Truffault V. AbrB-like transcription factors assume a swapped hairpin fold that is evolutionarily related to double-psi β barrels. Structure. 2005;13:919–928. doi: 10.1016/j.str.2005.03.017. [DOI] [PubMed] [Google Scholar]
- 25.Kamada K., Hanaoka F., Burley S.K. Crystal structure of the MazE/MazF complex: molecular bases of antidote–toxin recognition. Mol. Cell. 2003;11:875–884. doi: 10.1016/s1097-2765(03)00097-2. [DOI] [PubMed] [Google Scholar]
- 26.Sullivan D.M., Bobay B.G., Kojetin D.J., Thompson R.J., Rance M., Strauch M.A., Cavanagh J. Insights into the nature of DNA binding of AbrB-like transcription factors. Structure. 2008;16:1702–1713. doi: 10.1016/j.str.2008.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kabsch W. XDS. Acta Crystallogr., Sect. D Biol. Crystallogr. 2010;66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Adams P.D., Afonine P.V., Bunkóczi G., Chen V.B., Davis I.W., Echols N. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr., Sect. D Biol. Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Emsley P., Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr., Sect. D Biol. Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 30.Emsley P., Lohkamp B., Scott W.G., Cowtan K. Features and development of Coot. Acta Crystallogr., Sect. D Biol. Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary materials