

Abstract
Apolipoproteins E3 and E4, proteins with a molecular mass of 34.15 kDa, differ by a single amino acid change. ApoE4 contains an arginine residue at position 112, whereas apoE3 has a cysteine at this position. ApoE4 is the major risk factor for late-onset Alzheimer’s disease, whereas apoE3, the common isoform, is neutral with respect to this disease. Here, using literature data from both hydrogen-deuterium exchange and site-directed mutations, we suggest structural differences between these two isoforms that are distant from the site of the arginine-to-cysteine change. These structural differences involve sequences from both the N- and C-terminal domains, sequentially far apart but structurally close. In addition, these regions are close to regions that bind lipid and to a region involved in association of apoE monomers to higher molecular weight forms. We discuss the possibility that these regions could be targeted preferentially to affect the function of apoE4 relative to apoE3.
Keywords: amyloid, charge relay, lipid binding, structure propagation
ApoE4 is the major causative factor for developing late-onset Alzheimer’s disease (LOAD) (1–5). This protein is one of three common isoforms of the apolipoproteins apoE2, apoE3, and apoE4, which are coded at a single gene locus. All apoE isoforms consist of 299 amino acids with the only differences being single amino acid changes. However, there are clearly functional differences because apoE3, the common isoform, is not associated with Alzheimer’s disease, whereas apoE2 appears to be protective (6, 7). Because these functional properties must relate to structural properties, it is essential to determine what the structural differences are between the apoE isoforms. In vitro, the issue has been examined by many investigators, but progress in this area has been thwarted by the fact that no structure of any full-length WT isoform has ever been reported. The reasons for this state of affairs are twofold: first, the protein aggregates at the concentrations needed for crystallographic determination, and second, the formation of oligomers of the WT 34-kDa apoE at micromolar concentrations makes NMR studies highly improbable. That situation has been somewhat alleviated by the recent publication of an NMR structure of a mutated apoE3 that remains monomeric at high concentrations (8). This structure is distinctly different from structures that have been proposed for the full-length protein (9), primarily with respect both to structure of the C-terminal domain and to its interaction with the N-terminal domain. Both biophysical and functional studies indicate that the structure determined by Chen et al. (8) is a true representation of the WT protein (10, 11).
In the discussion below, we have used the monomeric structure, plus data previously obtained for both the monomeric and WT proteins, to explore the structural differences between apoE3 and apoE4. We then suggest that these differences could lead to the development of therapeutic agents that would distinguish between these two isoforms. We conclude that such agents might delay or prevent the development of LOAD.
Results and Discussion
ApoE is a protein consisting of two domains (the N- and C-terminal domains) separated by a protease-sensitive hinge region. The differences between the apoE isoforms are arginine-to-cysteine changes in the N-terminal domain: ApoE4 contains arginines at positions 112 and 158, whereas apoE3 has a cysteine at position 112. As discussed later, the structure of the N-terminal domain (∼22 kDa) of apoE3 and apoE4 has been determined using both X-ray and NMR methods (12–14). The isolated C-terminal domain is known to aggregate (9), thereby preventing structure determination by standard methods. Shown in Fig. 1 is an average NMR structure recently obtained by Chen et al. (8) of a full-length monomeric mutant of apoE3 with the C-terminal domain (residues 238–299) shown in green. To obtain this structure, several amino acids in the C-terminal domain were modified so as to prevent association to higher molecular-weight forms (10). This structure differs significantly from the previous models of apoE3 (9). For example, it had been suggested, based on mutational data, that interaction between N- and C-terminal domains was a unique property of apoE4 attributable to a salt bridge formation between Arg61 and Glu255 (15). However, the solution NMR structure of apoE3 clearly indicates extensive interactions between the two domains in this isoform, showing, for example, that Arg61 forms a hydrogen bond with Thr194, whereas Glu255 forms a salt bridge with Lys95 (8). Indeed, Chen et al. (8) list at least 12 hydrophobic residues that are buried between C- and N-terminal domains. In addition, major portions of the C-terminal domain (residues 238–299), rather than being helical, appear disordered. This is important because the C-terminal domain is critical in defining the function of apoE. It contains not only residues involved in oligomerization, but the binding site for lipid and probably amyloid-β (Aβ). The C-terminal domain also covers an amphipathic helix (8) that contains residues important for binding to the cell surface receptor for LDL.
Fig. 1.
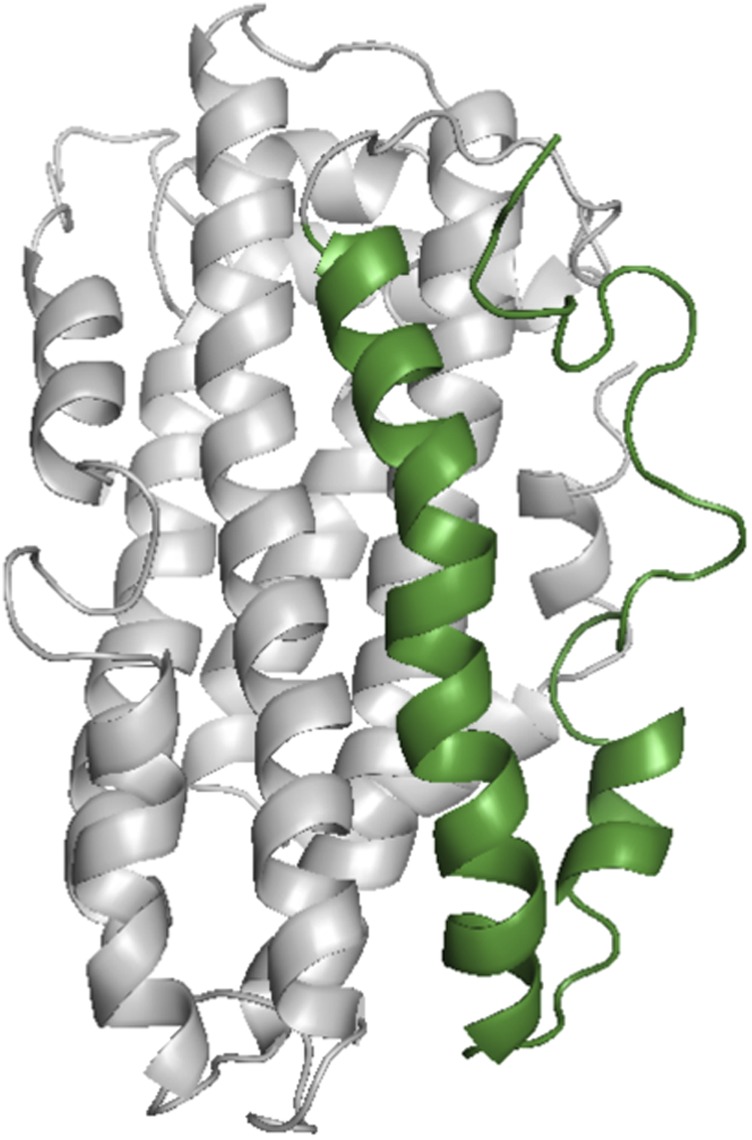
Average NMR structure of the monomeric form of apoE3 (Protein data Bank: 2L7B) as determined by Chen et al. (8). The C-terminal domain (residues 238–299) is shown in green. Note that the region from residue 277 to residue 299 appears structureless. Mutations used to make this monomeric form were Phe257Ala, Trp264Arg, Val269Ala, Leu279Gln, and Val287Glu.
To identify regions of the C-terminal domain involved in apoE oligomerization, we recently published data comparing deuterium uptake between a monomeric form of apoE and the WT oligomer for each apoE isoform (11). These experiments determined which residues were protected in the oligomer relative to the monomer, and therefore defined regions and residues important in the association of WT monomer to dimers and higher oligomeric forms. Although coverage of the protein was not complete, regions of the C-terminal domain involved in association of monomers to oligomers were reasonably, but not completely, defined. For example, in the C-terminal domain, the yield of the peptide representing residues 244–261 was too low to be detected (11, 16), but the inability to find this peptide does not affect the discussion of structural changes between apoE isoforms discussed below.
Association-Dissociation: Region of the C-Terminal Domain Involved.
As noted by Chen et al. (8), there are numerous exposed hydrophobic residues in the C-terminal domain. Using hydrogen/deuterium exchange (H/DX), coupled with electron transfer dissociation mass spectrometry, Huang et al. (11) found two major peptide regions (peptide regions 230–243 and 262–270) that differed in deuterium uptake between the monomer and oligomer. The H/DX results were similar for all three isoforms, a finding consistent with the observation that the rate constants that describe the monomer-dimer-tetramer association-dissociation only show slight differences between isoforms (17). As noted above, one region of the C-terminal domain (residues 244–261) is missing from these data (11, 16). Because residues on either side of this peptide are involved in the association-dissociation process, however, it seems reasonable to assume that region 244–261 may also be involved. In agreement with this conclusion is the observation that glutamate residues at positions 244 and 255 are much more reactive toward glycine ethyl ester in the monomeric mutant relative to the WT protein (16). We conclude, therefore, that residues within region 230–270 of the C-terminal domain are critical for oligomer formation. This region includes the major helix of the C-terminal domain (residues 238–266).
ApoE3/ApoE4 Structural Differences.
In the course of our studies, it also became clear that deuterium uptake of apoE monomers differed between the isoforms in regions that were not involved in association-dissociation. The data were shown in figure S6 of ref. 11. Four peptides stand out in this regard. Fig. 2 shows data for those peptides taken from the study of Huang et al. (11). Surprising are the data for peptides 5–14, 15–30, and 271–279, which are structurally distant from the site of the arginine-to-cysteine change at position 112. Peptide 5–14, a region of peptide 15–30 (residues 15–21), and peptide 271–279 are sequentially distant but next to one another structurally. Based on the program GETAREA (Provided by the Sealy Center for Structural Biology at the University of Texas Medical Branch) (18), there are at least 11 highly solvent exposed side chains from these regions. These peptides are shown in Fig. 3 (residues 5–21, shown in orange, and residues 271–279, shown in red). Shown in yellow is the cysteine at position 112. The data of Huang et al. (11) also show that peptide 5–14 is more protected and that peptide 271–279 is less protected in apoE4 relative to apoE3. We thus conclude that the amino acid change at position 112 must be propagated through the structure to the regions shown in Fig. 3. A proposal for how the arginine-to-cysteine change affects these distant regions is discussed later. The regions shown in Fig. 3 are somewhat different from those suggested by Barbier et al. (19), who, on the basis of proteolytic digestion experiments, concluded that residues 230–260 were protected in apoE4 relative to apoE3 or apoE2. In either case, however, structural differences were distant from the cysteine/arginine change.
Fig. 2.

Peptides that differ in H/DX between apoE2 (blue), apoE3 (pink), and apoE4 (green). Adapted with permission from ref. 11. Copyright (2011) American Chemical Society. All other peptides examined by HD/X show essentially no differences between apoE isoforms.
Fig. 3.
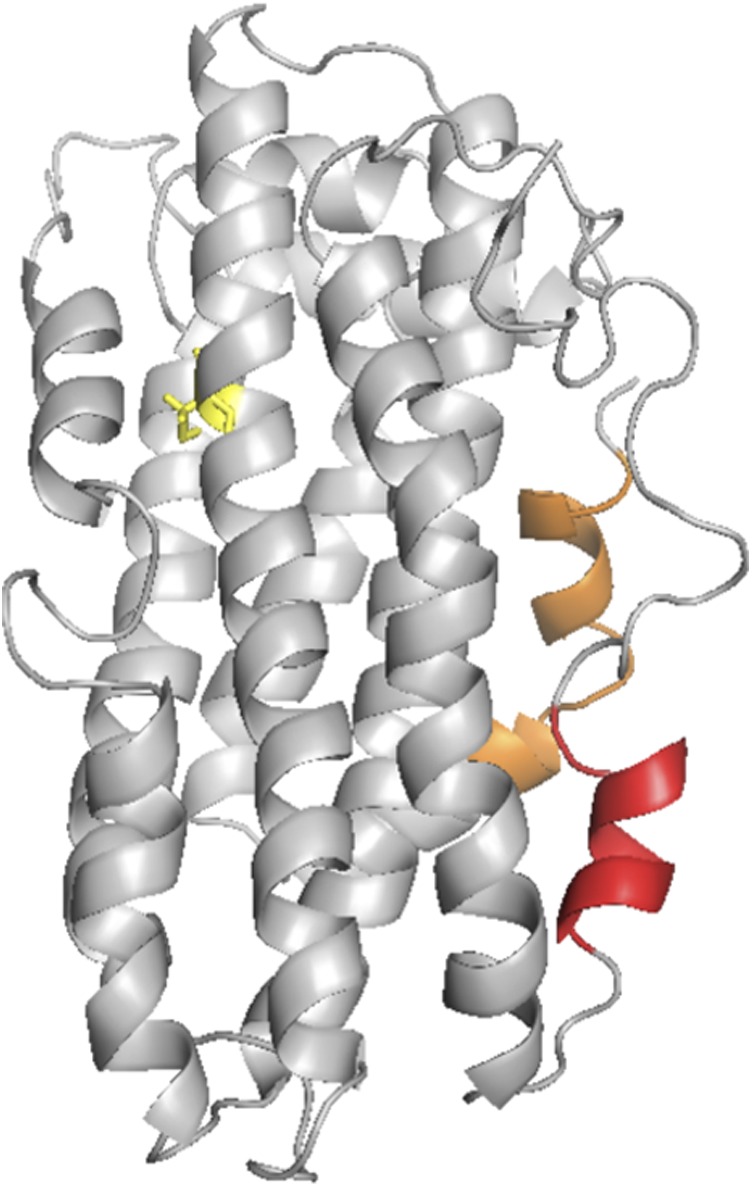
Regions of apoE (residues 5–21 shown in orange and residues 271–279 shown in red) taken from Fig. 2 that show different H/DX behavior between apoE3 and ApoE4. Cysteine 112 is shown in yellow.
Clearly, there should be structural changes at the site of the arginine-cysteine change, but data for peptide 105–115 could also not be determined in the H/DX experiments. However, deuterium uptake differs for peptide 116–123 (Fig. 2), and Met108 shows considerably greater free radical labeling in apoE4 relative to apoE3, supporting the idea of a structural difference in this region of the protein (16).
Overlap Between Regions Involving Structural Differences, Association-Dissociation, and Lipid Binding.
Two reports, using C-terminal truncated versions of apoE, conclude that the residues important for the initiation of lipid binding to apoE involved residues in the region 261–272 (20, 21). These residues and those involved in the structural differences between apoE3 and apoE4 are close. In addition, regions involved in the association to oligomers overlap those of lipid binding. We have discussed elsewhere our results demonstrating that the apoE oligomer, specifically the dimer, must dissociate to monomer in order for lipid to bind to this region (22). Table 1 summarizes those regions discussed here.
Table 1.
Summary of some structural and functional regions of apoE
| Peptide region | Comments |
| 5–14 | In contact with a portion of the C-terminal domain; structurally different between apoE3 and apoE4 |
| 15–30 | Structurally different between apoE3 and apoE4 |
| 110–123 | Contains site of amino acid differences between apoE3 and apoE4 at position 112; some structural differences in peptide 116–123 between apoE3 and apoE4 |
| 131–164 | Helix 4; contains the LDL receptor binding site; highly charged from residues 130–154; Arg158 is a cysteine in apoE2 |
| 238–299 | C-terminal domain |
| 230–243 | Involved in oligomer formation; Aβ may also bind in this region |
| 261–272 | Residues involved in initiation of lipid binding |
| 263–270 | Includes residues involved in oligomerization: 263, 264, 265, 268, and 270 |
| 271–279 | Structurally different between apoE3 and apoE4 |
Fig. 4 is a surface representation of residues involved in the structural differences between apoE3 and apoE4 (orange and red), those involved in the association-dissociation process (blue), and those involved in lipid binding (residues 261–272, shown in green mesh). Only solvent-exposed residues are shown. Of considerable interest is that the regions involving the structural differences between apoE3 and apoE4, those involved in lipid binding, and those involved in monomer association form a continuous surface on the protein consisting of regions of both the C- and N-terminal domains. There is some overlap in specific residues between those involved in the association-dissociation and those that are structurally different, but the overlap is small. The structural differences between apoE3 and apoE4 do not sufficiently overlap with lipid binding regions, in agreement with only small differences in lipid binding between isoforms (22). On the other hand, as noted above, apoE must dissociate before binding lipid.
Fig. 4.
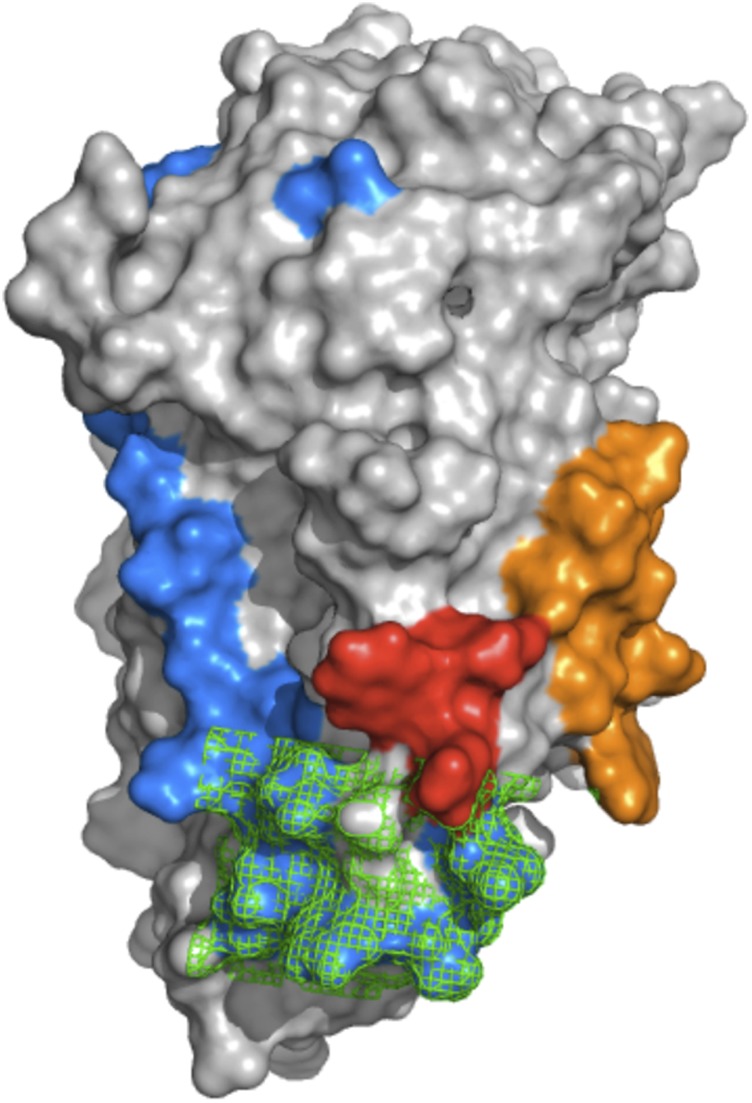
Surface representation of apoE3. Shown in orange and red are solvent-exposed residues, using GETAREA (18), for peptides 5–21 and 271–279, respectively. Residues in blue are those involved the association-dissociation process. Shown in green (as a mesh) are those residues involved in lipid binding. The overlap between lipid binding residues and those involved in the association to higher oligomers is consistent with the observation that apoE must dissociate before binding lipid.
Focus on the N-Terminal Region.
Amino acid residues in this region (residues 5–30) are not involved in apoE oligomerization (11, 16). One highly polar 11-residue stretch in these N-terminal peptides (residues 10–20) contains 4 glutamic and 3 glutamine residues (EPEPELRQQTEWQ). The H/DX data suggest less dynamic motion (less exchange and greater stability) in residues 5–14 but more in residues 15–30 (less stability) in apoE4 relative to apoE3.
Focus on Peptide 271–279.
This peptide sequence (MQRQWAGQ) is considerably more hydrophobic than that from the N-terminal region. As mentioned above, there are two major peptide regions in the C-terminal domain (peptides 230–243 and 262–270) that differ in deuterium uptake between the monomer and oligomer and are involved in the association to oligomer (11). Thus, there may be a small degree of overlap between regions responsible for oligomerization and those that differ between apoE3 and apoE4. Specific residues near this region involved in oligomerization are Ser263, Trp(or Arg)264, Phe265, Leu268, and Glu270 (11).
Structural Changes Around Residue 112.
Dong et al. (23) have compared the X-ray structures of the N-terminal domain of apoE3 and apoE4. They conclude that in apoE4, Arg112 forms a salt bridge with Glu109 lacking in apoE3 and that, consequently, the side chain of Arg61 moves away from the surface of the helix bundle. The structure shown in Fig. 3 is that of apoE3 with cysteine at this position. Although the larger and positively charged arginine in apoE4 must perturb the structure, the changes appear small and not related to the association-dissociation process because the kinetic and equilibrium constants for dissociation are very similar for apoE3 and apoE4 (17). As mentioned above, Met108 is considerably more reactive toward free radical labeling in apoE4 relative to apoE3, supporting the idea of a structural difference in this region of the protein (16).
Monomeric Mutant Relative to the WT ApoE3.
To make monomeric apoE3, Chen et al. (8) introduced five mutations in the C-terminal domain. It is crucial to determine whether these mutations (F257A, W264R, V269A, L279Q, and V287E) affect the structure of the monomeric mutant relative to the WT. There are several indications that the monomeric mutant is very nearly identical to the WT protein. First, the H/DX experiments show essentially identical exchange for WT as for the monomeric mutants, except for those regions discussed here and those involved in the association-dissociation process (11). It should be noted, however, that there was not complete coverage of the whole protein (65%). Second, Zhang et al. (10) carefully examined a number of properties of the monomeric mutant relative to the WT protein. These included CD spectroscopy, denaturation curves, lipid binding activity, and LDL receptor binding activity. These authors also showed that the N-terminal domain of the full-length protein adopts a quite similar, but not identical, structure as the isolated N-terminal domain (8). Finally, the 19F-NMR spectra of WT and monomeric apoE3 labeled with 19F-tryptophan show similar chemical shifts (24).
How the Cysteine/Arginine Change at Position 112 Is Propagated Through the Structure: Hypothesis.
As discussed earlier, the recently published solution NMR structure by Chen et al. (8) differs greatly from the suggested domain-domain interactions in an earlier proposed model of apoE3 (9). Thus, new insights based on current knowledge of the apoE structure are needed to explain the structural differences between apoE3 and apoE4. We speculate that the structural differences observed as a consequence of the cysteine/arginine change at position 112 may involve the highly charged helix 4 of the N-terminal domain. Fig. 5 shows that helix 4 is rich in charged residues (aspartate residues are shown in red, and arginine and lysine residues are shown in blue). Peptides 5–21 and 271–279 are spatially close to helix 4, and, as shown in Fig. 5, those residue pairs that lie within a distance of 6 Å between these peptides and helix 4 are connected by pink lines. We then hypothesize a possible pathway leading to structural differences between apoE3 and apoE4. Here, the relevant residues may be Arg114 and His140. We propose that the arginine/cysteine change at position 112 affects the movement of Arg114, which, in turn, perturbs the ionization of His140, which affects the charge distribution of helix 4, finally resulting in the structural difference between apoE3 and apoE4. We note here that there are only two histidine residues in apoE, with the other being His299 at the C-terminal end of the protein. Mutation of Arg114 or His140 to alanine in apoE4 may affect the behavior of those distant structural regions that differ between apoE3 and apoE4.
Fig. 5.

Proposed mechanism for propagation of the arginine-to-cysteine change at position 112 (in yellow) to the structural differences between apoE3 and apoE4 via Arg114 (blue) and His140 (green). In helix 4, arginines and lysines are shown in blue and aspartate is shown in red. Peptide 271–279 is shown in red, and peptide 5–21 is shown in orange; these are the peptides that are structurally different between apoE3 and apoE4. The pink lines connect pairs of residues that lie within 6 Å between helix 4 and the peptides.
What About Aβ?
The major protein component of amyloid plaques is Aβ1–42, and the relationship of Aβ to Alzheimer’s disease has been the subject of intense study by numerous investigators. Current thinking suggests that Aβ oligomers, rather than amyloid fibrils, are the toxic species in Alzheimer’s disease. Many in vitro studies have shown that apoE binds oligomeric Aβ, but the mechanism of binding, the Aβ binding site, and the oligomeric form of apoE that binds are still subject to investigation. As long ago as 1993, Strittmatter et al. (25) suggested that the isoforms apoE3 and apoE4 differed in the rate of Aβ binding. There is also evidence that apoE delays Aβ aggregation (26, 27), and recent data appear to confirm isoform-dependent differences of either the lipid-free (28) or lipid-bound (29) apoE. Liu et al. (30) suggest that the region 244–272 is involved in Aβ binding. It seems likely that apoE binds to some intermediate in Aβ1–40 aggregation rather than to monomeric Aβ. Careful examination of the full time course of Aβ aggregation using, for example, Thioflavin T fluorescence coupled with site-directed mutagenesis would clarify apoE isoform differences.
What About Lipidated ApoE?
In vivo, apoE is almost always associated with lipids and cholesterol, and the actual amount of lipid-free apoE is expected to be small. On lipidation, the protein undergoes large structural changes (31, 32), and although there is no high-resolution structure of lipid-bound apoE, the structural differences between the apoE isoforms lead to their differential association with the lipoprotein particle classes in the plasma (21). The more important question is whether the structural differences observed in the lipid-free apoE, as described here, are retained in the lipidated apoE. At this time, we cannot answer this question. The model of lipid-bound apoE proposed by Hatters et al. (33), however, does suggest that these peptides, which are close in the lipid-free apoE, are also close in the lipidated form. We propose that the structural differences observed in the lipid-free protein persist in the lipid-bound protein. Although obtaining direct structural information may be difficult, differences in association of 1, 2-dimyristoyl-sn-glycero-3-phosphocholine (DMPC)-apoE3 or DMPC-apoE4 with ligands, such as Aβ, might indicate whether the lipidated apoE isoforms differ structurally. There are indications in the literature that such differences exist (29), but more careful experiments are needed to settle this issue.
Can Therapeutic Agents Take Advantage of the Structural Differences Between ApoE3 and ApoE4?
As noted earlier, the major risk factor for LOAD (after 60 y of age) is APOE genotype. The increased relative risk (as measured by the odds ratio) for individuals homozygous for apoE4 is over 12 and over 3 for heterozygotes of the E4/E3 genotype compared with the E3/E3 genotype (34, 35). A therapeutic agent that might delay or prevent the onset of Alzheimer’s disease could be one that would alter the functional characteristics of apoE4, such that they would become similar to those of apoE3. Such an agent would preferentially affect ligand binding, and therefore the function properties of the apoE4 isoform. It is of interest that using high-throughput screening, several small molecules that alter the detrimental effects of apoE4 in neuronal cell culture have been found (36, 37). The authors conclude that these compounds function by abolishing domain-domain interactions preferentially in apoE4 compared with apoE3.
In this paper, we have pointed out structural differences between apoE3 and apoE4 that are distant from the site of the arginine-to-cysteine change. We note that ligand binding sites are located in the C-terminal domain, in regions close to the structural differences we have observed between apoE3 and apoE4. Assuming that the functional differences between apoE isoforms are a consequence of the structural differences, the data in Fig. 2 may provide insight into those regions of monomeric apoE that could be targeted. The ability to alter ligand specificity, rather than simply a binding constant, by binding a small molecule distant from the ligand binding site presents a difficult problem. However, that is what would be required to alter the properties of apoE4 to mirror more closely those of apoE3. On the other hand, it may not be difficult to develop methods that could be used to determine the effect of small molecule binding. The most obvious characteristic would be to alter Aβ binding. As suggested by data in the literature (26–29), the different isoforms appear to affect Aβ aggregation quite differently in vitro even though Aβ binding is currently poorly understood. Additionally, the specificity of lipidated apoE for lipoprotein particles is quite different: ApoE3 binds preferentially to HDL, whereas apoE4 binds preferentially to very low-density lipoprotein (VLDL). Nguyen et al. (21) suggest that the molecular basis for apoE3 and apoE4 partitioning differently between VLDL and HDL rests in region 261–272, a region involved in lipid binding. As pointed out from the H/DX data, at least three peptides differ between apoE3 and apoE4. Residues in peptides 15–30, 116–123, and 271–279 show greater exchange in apoE4 relative to apoE3, suggesting greater dynamic motion. Thus, stabilizing these regions may alter the properties of apoE4 to those similar to apoE3. Site-directed mutagenesis within these peptides or small molecule screening should also be able to identify residues that change the behavior of apoE4 to that similar to apoE3. We are currently carrying out such experiments.
Acknowledgments
The H/DX experiments were performed at the Washington University National Institutes of Health Mass Spectrometry Resource supported by National Institute of General Medical Sciences (8 P41 GM103422-35).
Footnotes
The authors declare no conflict of interest.
References
- 1.Frank A, Díez-Tejedor E, Bullido MJ, Valdivieso F, Barreiro P. APOE genotype in cerebrovascular disease and vascular dementia. J Neurol Sci. 2002;203-204:173–176. doi: 10.1016/s0022-510x(02)00286-1. [DOI] [PubMed] [Google Scholar]
- 2.Corder EH, et al. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science. 1993;261:921–923. doi: 10.1126/science.8346443. [DOI] [PubMed] [Google Scholar]
- 3.Saunders AM, et al. Association of apolipoprotein E allele epsilon 4 with late-onset familial and sporadic Alzheimer’s disease. Neurology. 1993;43:1467–1472. doi: 10.1212/wnl.43.8.1467. [DOI] [PubMed] [Google Scholar]
- 4.Kim J, Basak JM, Holtzman DM. The role of apolipoprotein E in Alzheimer’s disease. Neuron. 2009;63:287–303. doi: 10.1016/j.neuron.2009.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Verghese PB, Castellano JM, Holtzman DM. Apolipoprotein E in Alzheimer’s disease and other neurological disorders. Lancet Neurol. 2011;10:241–252. doi: 10.1016/S1474-4422(10)70325-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rebeck GW, Kindy M, LaDu MJ. Apolipoprotein E and Alzheimer’s disease: The protective effects of ApoE2 and E3. J Alzheimers Dis. 2002;4:145–154. doi: 10.3233/jad-2002-4304. [DOI] [PubMed] [Google Scholar]
- 7.Mahley RW, Weisgraber KH, Huang Y. Apolipoprotein E: Structure determines function, from atherosclerosis to Alzheimer’s disease to AIDS. J Lipid Res. 2009;50(Suppl):S183–S188. doi: 10.1194/jlr.R800069-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen J, Li Q, Wang J. Topology of human apolipoprotein E3 uniquely regulates its diverse biological functions. Proc Natl Acad Sci USA. 2011;108:14813–14818. doi: 10.1073/pnas.1106420108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hatters DM, Peters-Libeu CA, Weisgraber KH. Apolipoprotein E structure: Insights into function. Trends Biochem Sci. 2006;31:445–454. doi: 10.1016/j.tibs.2006.06.008. [DOI] [PubMed] [Google Scholar]
- 10.Zhang Y, et al. A monomeric, biologically active, full-length human apolipoprotein E. Biochemistry. 2007;46:10722–10732. doi: 10.1021/bi700672v. [DOI] [PubMed] [Google Scholar]
- 11.Huang RYC, Garai K, Frieden C, Gross ML. Hydrogen/deuterium exchange and electron-transfer dissociation mass spectrometry determine the interface and dynamics of apolipoprotein E oligomerization. Biochemistry. 2011;50:9273–9282. doi: 10.1021/bi2010027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wilson C, Wardell MR, Weisgraber KH, Mahley RW, Agard DA. Three-dimensional structure of the LDL receptor-binding domain of human apolipoprotein E. Science. 1991;252:1817–1822. doi: 10.1126/science.2063194. [DOI] [PubMed] [Google Scholar]
- 13.Sivashanmugam A, Wang J. A unified scheme for initiation and conformational adaptation of human apolipoprotein E N-terminal domain upon lipoprotein binding and for receptor binding activity. J Biol Chem. 2009;284:14657–14666. doi: 10.1074/jbc.M901012200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dong J, et al. Interaction of the N-terminal domain of apolipoprotein E4 with heparin. Biochemistry. 2001;40:2826–2834. doi: 10.1021/bi002417n. [DOI] [PubMed] [Google Scholar]
- 15.Dong LM, Weisgraber KH. Human apolipoprotein E4 domain interaction. Arginine 61 and glutamic acid 255 interact to direct the preference for very low density lipoproteins. J Biol Chem. 1996;271:19053–19057. doi: 10.1074/jbc.271.32.19053. [DOI] [PubMed] [Google Scholar]
- 16.Gau B, Garai K, Frieden C, Gross ML. Mass spectrometry-based protein footprinting characterizes the structures of oligomeric apolipoprotein E2, E3, and E4. Biochemistry. 2011;50:8117–8126. doi: 10.1021/bi200911c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Garai K, Frieden C. The association dissociation behavior of the ApoE proteins: Kinetic and equilibrium studies. Biochemistry. 2010;49:9533–9541. doi: 10.1021/bi101407m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fraczkiewicz R, Braun W. Exact and efficient analytical calculation of the accessible surface areas and their gradients for macromolecules. J Comput Chem. 1998;19:319–333. [Google Scholar]
- 19.Barbier A, et al. The structure of human apolipoprotein E2, E3 and E4 in solution 1. Tertiary and quaternary structure. Biophys Chem. 2006;119:158–169. doi: 10.1016/j.bpc.2005.07.010. [DOI] [PubMed] [Google Scholar]
- 20.Tanaka M, et al. Effect of carboxyl-terminal truncation on structure and lipid interaction of human apolipoprotein E4. Biochemistry. 2006;45:4240–4247. doi: 10.1021/bi060023b. [DOI] [PubMed] [Google Scholar]
- 21.Nguyen D, et al. Molecular basis for the differences in lipid and lipoprotein binding properties of human apolipoproteins E3 and E4. Biochemistry. 2010;49:10881–10889. doi: 10.1021/bi1017655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Garai K, Baban B, Frieden C. Dissociation of apolipoprotein E oligomers to monomer is required for high-affinity binding to phospholipid vesicles. Biochemistry. 2011;50:2550–2558. doi: 10.1021/bi1020106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dong LM, et al. Human apolipoprotein E. Role of arginine 61 in mediating the lipoprotein preferences of the E3 and E4 isoforms. J Biol Chem. 1994;269:22358–22365. [PubMed] [Google Scholar]
- 24.Garai K, Mustafi SM, Baban B, Frieden C. Structural differences between apolipoprotein E3 and E4 as measured by (19)F NMR. Protein Sci. 2010;19:66–74. doi: 10.1002/pro.283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Strittmatter WJ, et al. Binding of human apolipoprotein E to synthetic amyloid beta peptide: Isoform-specific effects and implications for late-onset Alzheimer disease. Proc Natl Acad Sci USA. 1993;90:8098–8102. doi: 10.1073/pnas.90.17.8098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Webster S, Rogers J. Relative efficacies of amyloid beta peptide (A beta) binding proteins in A beta aggregation. J Neurosci Res. 1996;46:58–66. doi: 10.1002/(SICI)1097-4547(19961001)46:1<58::AID-JNR8>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 27.Wood SJ, Chan W, Wetzel R. An ApoE-Abeta inhibition complex in Abeta fibril extension. Chem Biol. 1996;3:949–956. doi: 10.1016/s1074-5521(96)90183-0. [DOI] [PubMed] [Google Scholar]
- 28.Cerf E, Gustot A, Goormaghtigh E, Ruysschaert J-M, Raussens V. High ability of apolipoprotein E4 to stabilize amyloid-β peptide oligomers, the pathological entities responsible for Alzheimer’s disease. FASEB J. 2011;25:1585–1595. doi: 10.1096/fj.10-175976. [DOI] [PubMed] [Google Scholar]
- 29.Stratman NC, et al. Isoform-specific interactions of human apolipoprotein E to an intermediate conformation of human Alzheimer amyloid-beta peptide. Chem Phys Lipids. 2005;137:52–61. doi: 10.1016/j.chemphyslip.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 30.Liu Q, et al. Mapping ApoE/Aβ binding regions to guide inhibitor discovery. Mol Biosyst. 2011;7:1693–1700. doi: 10.1039/c1mb05019b. [DOI] [PubMed] [Google Scholar]
- 31.Peters-Libeu CA, Newhouse Y, Hatters DM, Weisgraber KH. Model of biologically active apolipoprotein E bound to dipalmitoylphosphatidylcholine. J Biol Chem. 2006;281:1073–1079. doi: 10.1074/jbc.M510851200. [DOI] [PubMed] [Google Scholar]
- 32.Peters-Libeu CA, Newhouse Y, Hall SC, Witkowska HE, Weisgraber KH. Apolipoprotein E*dipalmitoylphosphatidylcholine particles are ellipsoidal in solution. J Lipid Res. 2007;48:1035–1044. doi: 10.1194/jlr.M600545-JLR200. [DOI] [PubMed] [Google Scholar]
- 33.Hatters DM, Voss JC, Budamagunta MS, Newhouse YN, Weisgraber KH. Insight on the molecular envelope of lipid-bound apolipoprotein E from electron paramagnetic resonance spectroscopy. J Mol Biol. 2009;386:261–271. doi: 10.1016/j.jmb.2008.12.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bertram L, McQueen MB, Mullin K, Blacker D, Tanzi RE. Systematic meta-analyses of Alzheimer disease genetic association studies: The AlzGene database. Nat Genet. 2007;39:17–23. doi: 10.1038/ng1934. [DOI] [PubMed] [Google Scholar]
- 35.Bertram L. Alzheimer’s disease genetics current status and future perspectives. Int Rev Neurobiol. 2009;84:167–184. doi: 10.1016/S0074-7742(09)00409-7. [DOI] [PubMed] [Google Scholar]
- 36.Brodbeck J, et al. Structure-dependent impairment of intracellular apolipoprotein E4 trafficking and its detrimental effects are rescued by small-molecule structure correctors. J Biol Chem. 2011;286:17217–17226. doi: 10.1074/jbc.M110.217380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen HK, et al. Small molecule structure correctors abolish detrimental effects of apolipoprotein E4 in cultured neurons. J Biol Chem. 2012;287:5253–5266. doi: 10.1074/jbc.M111.276162. [DOI] [PMC free article] [PubMed] [Google Scholar]