

Abstract
The active site of cytochrome c (Cyt c) consists of a heme covalently linked to a pentapeptide segment (Cys-X-X-Cys-His), which provides a link between the heme and the protein surface, where the redox partners of Cyt c bind. To elucidate the vibrational properties of heme c, nuclear resonance vibrational spectroscopy (NRVS) measurements were performed on 57Fe-labeled ferric Hydrogenobacter thermophilus cytochrome c552, including 13C8-heme–, 13C515N-Met–, and 13C15N-polypeptide (pp)–labeled samples, revealing heme-based vibrational modes in the 200- to 450-cm−1 spectral region. Simulations of the NRVS spectra of H. thermophilus cytochrome c552 allowed for a complete assignment of the Fe vibrational spectrum of the protein-bound heme, as well as the quantitative determination of the amount of mixing between local heme vibrations and pp modes from the Cys-X-X-Cys-His motif. These results provide the basis to propose that heme-pp vibrational dynamic couplings play a role in electron transfer (ET) by coupling vibrations of the heme directly to vibrations of the pp at the protein–protein interface. This could allow for the direct transduction of the thermal (vibrational) energy from the protein surface to the heme that is released on protein/protein complex formation, or it could modulate the heme vibrations in the protein/protein complex to minimize reorganization energy. Both mechanisms lower energy barriers for ET. Notably, the conformation of the distal Met side chain is fine-tuned in the protein to localize heme-pp mixed vibrations within the 250- to 400-cm−1 spectral region. These findings point to a particular orientation of the distal Met that maximizes ET.
Cytochromes c (Cyts c) are redox-active proteins involved in reactions crucial for diverse biological processes, particularly respiration, photosynthesis, apoptosis, detoxification of reactive oxygen species and gas sensing (1). The Cyt c active site is typified by a heme c prosthetic group, consisting of iron protoporphyrin IX covalently linked to the polypeptide (pp) backbone through two Cys residues in a Cys-X-X-Cys-His (CXXCH) arrangement. Here, “XX” may be any two amino acids [e.g., Met and Ala, as in Hydrogenobacter thermophilus cytochrome c552 (Ht Cyt c)], and His is the proximal heme ligand (Fig. 1). In the most frequently studied class (class I) of Cyts c, the side chain of a Met located near the C terminus coordinates to iron on the distal side of heme c, forming a six-coordinate, low-spin species. Our study focuses on ferric Ht Cyt c with the signature Cys12-Met13-Ala14-Cys15-His16 pentapeptide. The CXXCH segment is proposed to influence heme structure through covalent bonding to the porphyrin and through hydrogen bonding within the pentapeptide (2). The CXXCH loop has further been shown to influence the heme reduction potential (1). Notably, the CXXCH segment is near the protein-protein interaction interface for complexes of Cyts c with redox partners, as is seen in the yeast cytochrome c peroxidase (CCP)/Cyt c complex (3), where CCP binds in the area of the CXXCH loop (Fig. S1). This interaction mode suggests a role for the CXXCH and its environment as a link for direct “communication” of the heme active site with the protein surface. In this paper, we propose that this communication is achieved via strong vibrational dynamic couplings of heme and CXXCH peptide vibrations. Because the CXXCH motif is implicated in interactions with electron transfer (ET) partners, this coupling may then serve to trigger bimolecular ET.
Fig. 1.
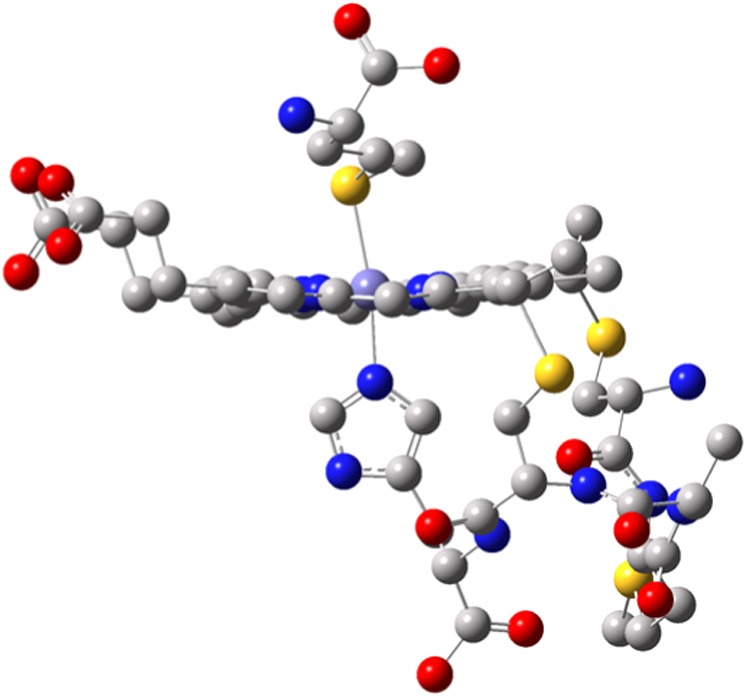
Active site of WT Fe(III) Cyt c from Hydrogenobacter thermophilus [Protein Data Bank (PDB) ID code: 1YNR] shows the heme, the Cys12-Met13-Ala14-Cys15-His16 pentapeptide loop on the proximal side of the heme, and Met61 on the distal side. The yellow, purple, red, blue, and gray spheres represent sulfur, heme iron, oxygen, nitrogen, and carbon, respectively.
The functions of proteins can generally be related to dynamic fluctuations of the protein structure that are thought to provide activation energy for function (4). A consequence of this concept is that modifications of the protein can influence the active site over a long distance. Our limited understanding of vibrational couplings between the heme active site and the protein matrix in Cyt c and their influence on function is a result of limited experimental data that probe this phenomenon. The vibrational properties of Cyt c have been previously studied (5–7), with particular focus on the heme cofactor, which is usually being treated as an isolated unit. More recent studies of the vibrational properties of the surrounding pp (8–10) highlight the importance of examining both the cofactor and peptide dynamics in Cyt c for elucidating their contributions to function.
Vibrational dynamics are challenging to investigate experimentally; therefore, computational studies have been a preferred technique to understand this phenomenon (11–13). Our work contributes to this greater effort by probing vibrational motions that involve the inner core of the heme and the surrounding peptide through a synchrotron-based technique, nuclear resonance vibrational spectroscopy (NRVS). This method measures the vibrations of a Mössbauer-active nucleus (here, 57Fe). The advantage of NRVS is that it detects all motions that involve the probe nucleus, which are coupled to the surrounding ligands (14) and, here also, vibrations of the pp. In addition, NRVS intensity scales with the amount of iron motion in a normal mode; hence, NRVS intensities can straightforwardly be simulated from normal coordinate analysis (15, 16).
This paper presents the complete vibrational assignment of the NRVS spectrum of a heme protein, ferric Ht Cyt c. This work includes the modes corresponding to the Fe-ligand vibrations, the assignments of which have been an ongoing matter of debate in the literature (17). Based on these assignments, the NRVS data obtained on 13C8-heme–labeled (labeled at 4 α and 4 β carbons; Fig. S2), 13C515N-Met–labeled, and 13C15N-pp–labeled ferric Ht Cyt c were analyzed (Fig. 2 and Fig. S3). These data allow us to probe the heme-protein vibrational couplings directly and to deduce implications for protein function. Finally, the effect of heme axial Met side-chain orientation on these coupled heme-pp vibrations is evaluated.
Fig. 2.
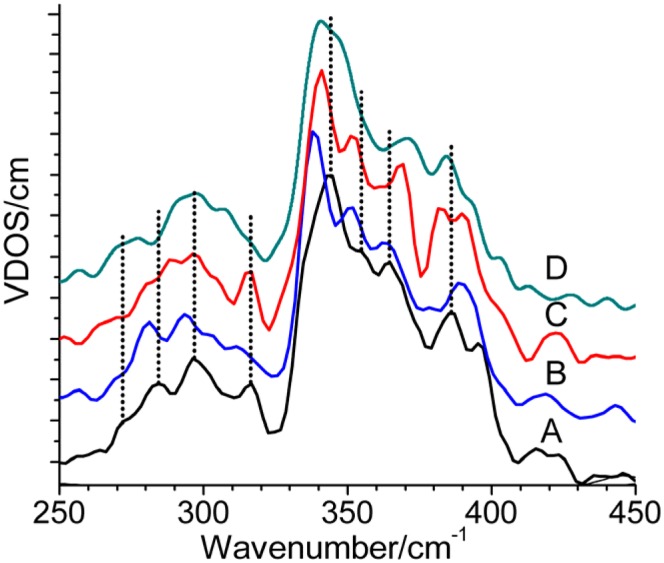
NRVS spectra of 57Fe(III) Ht Cyt c (A) and 13C8-heme–labeled (B), 13C15N-Met–labeled (C), and 13C15N-pp–labeled (D) protein in 50 mM Hepes buffer containing 10% (vol/vol) glycerol (pH 7.0, 16.5–18 mM). K2IrCl6 was added to a concentration of 25 mM to prevent sample reduction. VDOS, vibrational density of states.
Results and Discussion
Experimental Results.
The NRVS spectrum of 57Fe Ht ferric Cyt c is shown in black in Fig. 2. Peaks are observed in the 200- to 450-cm−1 region. The bands representing vibrational modes with the greatest displacement of iron, and hence the strongest intensities, are in the 325- to 400-cm−1 region, where seven distinct bands are resolved by simulation. At lower energy, a weaker three-peak spectral envelope in the 250- to 325-cm−1 region is resolved into five Gaussian bands (Fig. S3). Notably, these data resemble the NRVS spectrum of horse heart ferric Cyt c reported by Leu et al. (17). The sensitivity of the NRVS features to changes in the protein environment, particularly the CXXCH motif, is determined here through analysis of 13C515N-Met–, 13C8-heme–, and 13C15N-pp–labeled samples (Fig. 2). The vibrational assignments of these data are described in the next section.
Assignment of the NRVS Spectrum of Ferric Cyt c.
Density functional theory calculations.
To analyze the NRVS data of Ht Cyt c, we built a model of the active site that includes heme c, the covalently linked pentapeptide segment with the −R groups along the chain truncated at the α carbons, and formaldehyde, which takes the place of proline that forms a hydrogen bond to the proximal histidine (SI Materials and Methods). The optimized structure of our active site model of ferric Ht Cyt c (Fig. S4) reproduces the corresponding crystal structure [Protein Data Bank (PDB) ID code 1YNR] well. In particular, the Fe-SMet and average Fe-Npyr (pyr = pyrrole) bond distances and the ruffled nature of the heme show excellent agreement with the experiment (Table 1). Larger deviations in geometry, attributable to the nature of the applied model, are observed for the orientation of the Met distal ligand and the CXXCH pentapeptide side chains. Most importantly, a 23° difference in the NHis-Fe-SMet-Cγ dihedral angle is observed between the calculated and experimental structures. In addition, the propionate groups in the optimized geometry have a different conformation relative to the crystal structure. In the model, they interact via an H-bond, whereas in the crystal structure, they interact with the pp. This disparity is accounted for by manually adjusting the positions of the propionate groups according to the crystal structure in the quantum chemistry-centered normal coordinate analysis (QCC-NCA) simulation of the NRVS data. The propionate groups are modeled as protonated so as to take into account the fact that they are extensively hydrogen-bonded in the protein.
Table 1.
Selected bond distances (Å), angles (°), and force constants (mdyn/Å, mdyn⋅Å) for the ferric Ht Cyt c active site
| Fe(III) Cyt c | X-ray crystallography [Protein Data Bank (PDB) ID code 1YNR] | DFT structure BP86/LanL2DZ* | QCC-NCA force constants |
| Fe-NHis | 2.1 | 1.967 | 1.239 |
| Fe-SMet | 2.4 | 2.449 | 0.930 |
| Fe-NPyr (average) | 2.0 | 2.004 | 1.760 |
| NPyr-Fe-NPyr (average) | 90.0 | 90.0 | 0.751 |
| NHis-Fe-SMet-C | 127.0 | 150.0 | — |
The density functional theory (DFT)-predicted NRVS spectrum shows distinct frequency shifts and, more importantly, a dramatic redistribution of spectral intensity in the 230- to 350-cm−1 region compared with experimental data as shown in Fig. 3. The deviation is too large to simply use the DFT result to analyze/assign the experimental spectra by comparison, which has become a common practice in the literature (18, 19). However, the deviation between the DFT and experimental NRVS data can be resolved by QCC-NCA simulations, a procedure that allows for the stepwise and systematic optimization of the DFT-calculated force field to reproduce the vibrational energies, isotope shifts, and NRVS intensities of the experimental spectrum (20).
Fig. 3.

Experimental, DFT, and QCC-NCA NRVS spectra of ferric Ht Cyt c. VDOS, vibrational density of states.
QCC-NCA analysis and assignment of modes.
The Cyt c active-site model used here requires over 500 internal coordinates to define its force field properly. In the spirit of the QCC-NCA approach, large changes in the calculated force constants are avoided in the fitting procedure (Table S1). Fig. 3 shows the obtained QCC-NCA fit, which shows excellent agreement with the experiment. The only noticeable deviation is observed in the 250- to 325-cm−1 range. Here, the experimentally observed three-band pattern is well reproduced in the fit, but the vibrational energies are shifted to lower energy by ∼15 cm−1 compared with the experiment. This region of the vibrational spectrum is particularly sensitive to force constants of the NHis-Fe-SMet axial unit and the CXXCH residues, implicating a high degree of peptide character within this spectral range (vide infra). The energy shift in this region is therefore likely a result of the simplification of the distal Met and the pentapeptide segment in our model. The 325- to 400-cm−1 region is well reproduced in the fit; this region is mostly sensitive to modifications of the force constants of the first coordination sphere of iron, where the bond lengths and angles of the active site model adequately compare with the crystal structure.
Based on the excellent agreement between the experiment and the QCC-NCA simulation, the NRVS spectrum of Cyt c can be assigned, as shown in Table S2. The low-energy spectral region (145–325 cm−1) is dominated by pp modes, with contributions from heme and NHis-Fe-SMet axial unit modes. The assignments of the bands in this region are consistent with the data on the isotopically labeled proteins (Fig. 2). In our analysis, the long-debated ν(Fe-SMet) and ν(Fe-NHis) stretching modes are distributed over the entire 145- to 325-cm−1 region, where a majority of the bands have strong pp character. The weak features in the 160- to 280-cm−1 region that have ν(Fe-SMet) character show mostly Met moving perpendicular to the heme, consistent with the large mass effect of Met and the relatively weak Fe-SMet bond (Fe-SMet force constant = 0.93 mdyn/Å). The more intense bands between 280 cm−1 and 325 cm−1 that have ν(Fe-SMet) contributions display iron out-of-plane (oop) motions, indicating significant mixing with heme oop modes. Like ν(Fe-SMet), the ν(Fe-NHis) stretching mode where His is dominantly moving relative to heme occurs between 145 cm−1 and 205 cm−1 (less intense spectral features), whereas for the modes between 270 cm−1 and 315 cm−1 (more intense bands), iron shows predominant oop motion. This distribution of the Fe-SMet and Fe-NHis internal coordinates over many modes explains why distinct ν(Fe-SMet) and ν(Fe-NHis) modes have been challenging to detect. The occurrence of the ν(Fe-SMet) and ν(Fe-NHis) modes in ferric Ht Cyt c at quite low energy is consistent with a previous report on mitochondrial Cyt c, where bands at 176 cm−1 and 183 cm−1 were assigned to the ν(Fe-NHis) stretch coupled to ν(Fe-SMet) (21). Overall, the Fe(III)-His bond (force constant = 1.24 mdyn/Å) is found to be distinctly stronger than the Fe-SMet bond, in agreement with binding constant measurements on model complexes (22).
The most intense NRVS spectral features in the 325- to 400-cm−1 region correspond to modes involving iron moving in-plane (ip) [particularly the ν(Fe-NPyr) stretch at 344 cm−1, 355 cm−1, and 385 cm−1] and oop (e.g., pyrrole tilting modes at 366 cm−1 and 397 cm−1 and pyrrole swiveling modes at 375 cm−1 and 397 cm−1) (Table S2). The assignment of the most intense band at 344 cm−1 to a ν(Fe-NPyr) stretch is consistent with the assignment of a Raman peak observed at 347 cm−1 by Spiro and coworkers (5). The highly intense ν(Fe-NPyr) bands also have contributions from pp, heme, and NHis-Fe-SMet axial ligand internal coordinates but are mixed to a lesser extent compared with those bands observed in the lower energy region. Leu et al. (17) suggested that the ν(Fe-SMet) stretch in horse heart Cyt c is observed in the 340- to 370-cm−1 region, by comparison with ν(Fe-SCys) stretching frequencies in Cyt P450 (23) and chloroperoxidase (24). However, our data and analysis show that the heme-axial ligand vibrations are observed at much lower energy and, instead, that ip ν(Fe-NPyr) stretching modes dominate this energy region.
Importantly, the amount of pp character in the heme vibrations can directly be probed by pp isotopic labeling. The vibrational shifts to lower frequencies observed on heme and pp labeling in the 250- to 400-cm−1 region are consistent with the QCC-NCA result that these features show strong contributions of pp modes. An interesting exception is the band at 366 cm−1, which shifts to higher energy on pp labeling (Fig. 2 and Fig. S4). QCC-NCA analysis shows that this “inverse” shift is caused by pp-bending modes that shift into the 360- to 380-cm−1 spectral region on pp labeling and that gain much intensity by mixing with heme-based vibrations in this energy region. This redistribution of spectral intensity creates this unusual inverse isotope shift that is reproduced in our simulation. The higher energy region of the NRVS spectra (400–500 cm−1) shows weak intensity features that correspond either to mixed heme and pp vibrations (418 cm−1) or to “pure” heme modes (464 cm−1 and 485 cm−1) (Fig. S4). Assignments of these features are collected in Table S2.
In summary, NRVS data on pp- and 13C515N-Met–labeled ferric Ht Cyt c allow for the direct elucidation of heme-peptide vibrational couplings. Our results show strong mixing of heme and CXXCH peptide vibrations in the 145- to 400-cm−1 region, indicating that these two components cannot be treated separately. The long-debated oop ν(Fe-SMet) and ν(Fe-NHis) stretching vibrations are distributed throughout the 145- to 325-cm−1 region, which explains the previous difficulties in identifying these features. The 325- to 400-cm−1 region is dominated by ip heme vibrations, but these highly intense bands again have significant contributions from pp and heme oop internal coordinates. Nevertheless, the strongest couplings between heme and pp modes are observed in the 140- to 325-cm−1 region.
Effect of axial Met conformation.
The effect of the position of the distal Met side chain relative to the heme on the vibrational energies and iron vibrational amplitudes can be probed directly using the force field developed here. As shown in Fig. 4, this effect is surprisingly large, which means that the Fe-ligand modes are very sensitive to the heme-{axial Met} orientation. A rotation of Met leads to distinct shifts of vibrational energies, and particularly, dramatic spectral intensity redistributions in the 250- to 400-cm−1 energy range. The mode that is most sensitive to the Met orientation is the ν(Fe-NPyr) stretch, where contributions are observed at 410 cm−1 on a 180° Met rotation (Fig. 4). A redistribution of the ip and oop heme and pp modes is also observed, suggesting that the energies of these modes can be modulated to various extents via the Met orientation. The biological implications of this redistribution are discussed in the next section.
Fig. 4.

Simulated NRVS spectra of ferric Ht Cyt c as a function of the dihedral angle of the Fe-S-Met group. The final simulation of the experimental NRVS spectrum is shaded in gray (compare with Fig. 3). The vertical ticks correspond to the square of the iron amplitude (eFe2) of each mode and their contributions to the total simulated spectrum. (Upper Left) Vertical ticks for the final simulation are shown.
Aside from the orientation of the distal Met, the stereochemistry at sulfur, which is linked to the oxidation state of iron, also affects the NRVS spectrum of Cyt c (Fig. S5). The X-ray crystal structure of Ht Cyt c shows Met in the “R” configuration (25); hence, this is incorporated into our model. On the other hand, NMR analysis of Ht Cyt c demonstrates that the R form of Met is preferred in ferrous Cyt c (although both the R and “S” forms can be detected), whereas an ∼1:1 mixture of the R and S forms exists in the ferric form in solution (26). To test the significance of the Met stereochemistry, we changed Met to the S form in our QCC-NCA simulation. Interestingly, the resulting NRVS spectrum shows only slight intensity and energy shifts of ν(Fe-NPyr), ip and oop heme modes, and pp mode redistributions in the 250- to 400-cm−1 region. As shown in Fig. S5, these changes are minor compared with the effects of the rotation of the Met group relative to heme, corresponding to a change in the NHis-Fe-SMet-Cγ dihedral angle (vide supra). Finally, the overall features of the NRVS spectrum are not particularly sensitive to the orientation of the propionate groups relative to heme. The intensity of the 397-cm−1 band is slightly affected, but other changes are limited when comparing the simulated NRVS spectra with the crystallographic and geometry-optimized orientations of the propionates.
Biological Implications.
The role of Cyts c as electron carriers has long been known, although how structure and dynamics of ET protein complexes dictate rates of long-range ET is still actively being researched. In this study, we used Ht Cyt c as a subject for the complete assignment of iron vibrational modes because of its extreme stability and resistance to aggregation at the high concentrations required for NRVS. Furthermore, Ht Cyt c has structural homology to mitochondrial Cyts c, including similar positioning of the CXXCH residues and the solvent-exposed heme edge at pyrrole II relative to the protein fold (Fig. S6). In line with the known exponential distance dependence for long-range ET (27), the region of the protein around pyrrole II is the preferred region for the formation of functional complexes with ET partners (28). For example, in the crystal structure of the yeast iso-1-Cyt c/CCP complex, CCP is positioned near heme pyrrole II and the CXXCH of Cyt c, with the closest point-to-point distance of 3.5 Å occurring between Gln16 of the CXXCH motif and Ala193 on CCP (3). The proximity of the CXXCH residues of Cyt c to the protein surface of CCP points to a role of CXXCH in modulating ET rates (3). In addition, in eukaryotic Cyts c, a conserved basic residue (Lys13 or Arg13) located at the N-terminal side of the CXXCH motif has been shown to be a key site for coupling to redox partners (29).
Although our investigations in this paper focus on the ferric form of Cyt c, we expect that a similar degree of heme-pp vibrational couplings is also present in the ferrous form of the protein because the couplings are modulated by the heme-pp bonds. The key finding from our studies is that the strong vibrational coupling between the heme and the CXXCH vibrations provides a direct communication and/or energy transduction pathway between the heme and the protein surface. In other words, the heme may “sense” when a redox partner binds to Cyt c at the typical ET docking site at pyrrole II and the CXXCH motif. This coupling may affect Cyt c function in a number of different ways. It is known that vibrational (thermal) motions are crucial in protein function, particularly for ET. As described by Marcus theory (27), vibrational excitation (thermal energy) is key to providing the energy that is required to reach the transition state for ET. The free energy released on protein/protein complex formation may be converted into vibrational energy of local modes, which could bring the system toward the transition state for ET. In particular, lower frequency modes are thermally accessible and have been shown to play key roles as reaction coordinates in enzyme-facilitated processes (30). In this work, the QCC-NCA analysis of the NRVS data of Cyt c has revealed that most heme-based and Fe-{axial ligand} vibrations have major contributions from pp modes (Table S2). This strong coupling between the heme and pp vibrations is directly probed here via isotope-labeled Cyt c (Fig. S7).
One possible role of the heme-pp vibrational coupling is that on formation of a Cyt c/{Cyt c redox partner} complex, the released free energy could be transduced via the CXXCH motif vibrations (potentially involving the basic residue near the CXXCH) from the protein surface to the heme. As a result, vibrations that distort the heme along the ET reaction coordinate, for example, the noncentrosymmetrical (antisymmetrical) ν(Fe-NPyr) modes detected here via NRVS, may be activated to allow the complex to reach the transition state for ET (27). Because nuclei momenta and kinetic energies do not change during the ET step (27), the transfer of the electron could be faster than the dissipation of the vibrational excitation energy. One argument against this scenario is that protein/protein complex formation can be slow (diffusion-like), which could lead to continuous dissipation of the protein/protein complex formation energy along the protein/protein association pathway (31).
Alternatively, protein/protein complex formation could induce conformational changes of the CXXCH motif, which, via the observed vibrational couplings, could lead to a modulation of the vibrational Eigenstates of the heme. This could lead to a localization of the heme center in a local minimum that is more favorable for ET, thus minimizing the reorganization energy of the ET process. Indeed, high-frequency nuclear motions have been found to decrease inner-sphere reorganization energy significantly in a protein/protein complex (32). This effect would ensure that efficient ET occurs in the protein/protein complex, which is very short-lived in biological ET (33). This model is supported by the observation that each collision between cyt c and the cyt c oxidase CuA fragment is highly likely to result in efficient ET (34).
Finally, the energy of the NRVS spectral features is key, because lower energy modes are the most thermally accessible; therefore, these assist more efficiently in the ETs mediated by Cyt c. From a structure-function standpoint, the position of distal Met therefore plays an important role in optimizing the energy of the vibrational modes implicated in energy transduction. As shown in Fig. 4, rotating the distal Met away from the position observed in the crystal structure redistributes iron amplitude and shifts many modes to higher energy, which makes these modes harder to excite thermally and, hence, less efficient for ET. Based on these observations, the position of the distal Met seems optimized to promote ET in Cyt c.
Materials and Methods
Ht Cyt c was expressed in Escherichia coli using the pSHC552 plasmid (Ampr). To express 13C515N-Met Ht Cyt c, the protein was expressed in a Met auxotrophic strain of E. coli, B834. Competent B834 cells were purchased from Novagen. All other samples were expressed in BL21 (DE3) (Invitrogen). For incorporation of 57Fe, minimal growth medium was supplemented with 57FeSO4⋅1.2 H2O (20 mg/L). NRVS data were collected at beam line 3-ID-XOR of the Advanced Photon Source at Argonne National Laboratory. Details of the experimental procedures are provided in the SI Materials and Methods.
Supplementary Material
Acknowledgments
We thank Prof. Serena DeBeer (Max Planck Institute for Bioinorganic Chemistry) for help with the initial NRVS measurements, Violet Zhang (University of Rochester) for help with the sample preparation, and Prof. Alexei Stuchebrukhov (University of California, Davis) for helpful discussions. This work was supported by National Institutes of Health Grant R01-GM63170 (to K.L.B.) and by the Dow Corning Corp. (N.L.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1200345109/-/DCSupplemental.
References
- 1.Bowman SEJ, Bren KL. The chemistry and biochemistry of heme c: Functional bases for covalent attachment. Nat Prod Rep. 2008;25:1118–1130. doi: 10.1039/b717196j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ma JG, et al. Protein-induced changes in nonplanarity of the porphyrin in nickel cytochrome c probed by resonance Raman spectroscopy. Biochemistry. 1998;37:5118–5128. doi: 10.1021/bi972375b. [DOI] [PubMed] [Google Scholar]
- 3.Pelletier H, Kraut J. Crystal structure of a complex between electron transfer partners, cytochrome c peroxidase and cytochrome c. Science. 1992;258:1748–1755. doi: 10.1126/science.1334573. [DOI] [PubMed] [Google Scholar]
- 4.Hay S, Scrutton NS. Good vibrations in enzyme-catalysed reactions. Nat Chem. 2012;4:161–168. doi: 10.1038/nchem.1223. [DOI] [PubMed] [Google Scholar]
- 5.Hu S, Morris IK, Singh JP, Smith KM, Spiro TG. Complete assignment of cytochrome c resonance Raman spectra via enzymatic reconstitution with isotopically labeled hemes. J Am Chem Soc. 1993;115:12446–12458. [Google Scholar]
- 6.Desbois A. Resonance Raman spectroscopy of c-type cytochromes. Biochimie. 1994;76:693–707. doi: 10.1016/0300-9084(94)90145-7. [DOI] [PubMed] [Google Scholar]
- 7.Johannessen C, White PC, Abdali S. Resonance Raman optical activity and surface enhanced resonance Raman optical activity analysis of cytochrome c. J Phys Chem A. 2007;111:7771–7776. doi: 10.1021/jp0705267. [DOI] [PubMed] [Google Scholar]
- 8.Berezhna S, Wohlrab H, Champion PM. Resonance Raman investigations of cytochrome c conformational change upon interaction with the membranes of intact and Ca2+-exposed mitochondria. Biochemistry. 2003;42:6149–6158. doi: 10.1021/bi027387y. [DOI] [PubMed] [Google Scholar]
- 9.Yeo B-S, Madler S, Schmid T, Zhang W, Zenobi R. Tip-enhanced Raman spectroscopy can see more: The case of cytochrome c. J Phys Chem C. 2008;112:4867–4873. [Google Scholar]
- 10.Chin JK, Jimenez R, Romesberg FE. Direct observation of protein vibrations by selective incorporation of spectroscopically observable carbon-deuterium bonds in cytochrome c. J Am Chem Soc. 2001;123:2426–2427. doi: 10.1021/ja0033741. [DOI] [PubMed] [Google Scholar]
- 11.Freedman H, Martel P, Cruzeiro L. Mixed quantum-classical dynamics of an amide-I vibrational excitation in a protein α-helix. helix. Phys Rev B Condens Matter Mater Phys. 2010;82:174301–174308. [Google Scholar]
- 12.Reuveni S, Granek R, Klafter J. Anomalies in the vibrational dynamics of proteins are a consequence of fractal-like structure. Proc Natl Acad Sci USA. 2010;107:13696–13700. doi: 10.1073/pnas.1002018107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Song G, Jernigan RL. vGNM: A better model for understanding the dynamics of proteins in crystals. J Mol Biol. 2007;369:880–893. doi: 10.1016/j.jmb.2007.03.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Scheidt WR, Durbin SM, Sage JT. Nuclear resonance vibrational spectroscopy—NRVS. J Inorg Biochem. 2005;99:60–71. doi: 10.1016/j.jinorgbio.2004.11.004. [DOI] [PubMed] [Google Scholar]
- 15.Paulat F, et al. Vibrational assignments of six-coordinate ferrous heme nitrosyls: New insight from nuclear resonance vibrational spectroscopy. Inorg Chem. 2008;47:11449–11451. doi: 10.1021/ic801626w. [DOI] [PubMed] [Google Scholar]
- 16.Lehnert N, et al. Oriented single-crystal nuclear resonance vibrational spectroscopy of [Fe(TPP)(MI)(NO)]: Quantitative assessment of the trans effect of NO. Inorg Chem. 2010;49:7197–7215. doi: 10.1021/ic1010677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Leu BM, et al. Vibrational dynamics of iron in cytochrome C. J Phys Chem B. 2009;113:2193–2200. doi: 10.1021/jp806574t. [DOI] [PubMed] [Google Scholar]
- 18.Guo Y, et al. Characterization of the Fe site in iron-sulfur cluster-free hydrogenase (Hmd) and of a model compound via nuclear resonance vibrational spectroscopy (NRVS) Inorg Chem. 2008;47:3969–3977. doi: 10.1021/ic701251j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li J, et al. New perspectives on iron-ligand vibrations of oxyheme complexes. Chemistry. 2011;17:11178–11185. doi: 10.1002/chem.201101352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lehnert N. In: Computational Inorganic and Bioinorganic Chemistry. Solomon EI, King RB, Scott RA, editors. Chichester, UK: John Wiley & Sons; 2009. pp. 123–140. [Google Scholar]
- 21.Cartling B. Intermediate and stable redox states of cytochrome c studied by low temperature resonance Raman spectroscopy. Biophys J. 1983;43:191–205. doi: 10.1016/S0006-3495(83)84340-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tezcan FA, Winkler JR, Gray HB. Effects of ligation and folding on reduction potentials of heme proteins. J Am Chem Soc. 1998;120:13383–13388. [Google Scholar]
- 23.Champion PM, Stallard BR, Wagner GC, Gunsalus IC. Resonance Raman detection of an Fe-S bond in Cyt P450cam. J Am Chem Soc. 1982;104:5469–5472. [Google Scholar]
- 24.Bangcharoenpaurpong O, Champion PM, Hall KS, Hager LP. Resonance Raman studies of isotopically labeled chloroperoxidase. Biochemistry. 1986;25:2374–2378. doi: 10.1021/bi00357a011. [DOI] [PubMed] [Google Scholar]
- 25.Travaglini-Allocatelli C, et al. An obligatory intermediate in the folding pathway of cytochrome c552 from Hydrogenobacter thermophilus. J Biol Chem. 2005;280:25729–25734. doi: 10.1074/jbc.M502628200. [DOI] [PubMed] [Google Scholar]
- 26.Zhong L, et al. Heme axial methionine fluxionality in Hydrogenobacter thermophilus cytochrome c552. Proc Natl Acad Sci USA. 2004;101:8637–8642. doi: 10.1073/pnas.0402033101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Marcus RA, Sutin N. Electron transfers in chemistry and biology. Biochim Biophys Acta. 1985;811:265–322. [Google Scholar]
- 28.Bertini I, Cavallaro G, Rosato A. Principles and patterns in the interaction between mono-heme cytochrome c and its partners in electron transfer processes. Metallomics. 2011;3:354–362. doi: 10.1039/c0mt00108b. [DOI] [PubMed] [Google Scholar]
- 29.Niki K, et al. Coupling of Lysine-13 promotes electron tunneling through carboxylate-terminated alkanethiol self-assembled monolayers to Cytochrome c. J Phys Chem B. 2003;107:9947–9949. [Google Scholar]
- 30.Karunakaran V, et al. Investigations of low-frequency vibrational dynamics and ligand binding kinetics of cystathionine β-synthase. J Phys Chem B. 2010;114:3294–3306. doi: 10.1021/jp909700r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schreiber G. Kinetic studies of protein-protein interactions. Curr Opin Struct Biol. 2002;12:41–47. doi: 10.1016/s0959-440x(02)00287-7. [DOI] [PubMed] [Google Scholar]
- 32.Miyashita O, Okamura MY, Onuchic JN. Theoretical understanding of the interprotein electron transfer occurring between cytochrome c2 and the photosynthetic reaction center. J Phys Chem B. 2003;107:1230–1241. [Google Scholar]
- 33.Bashir Q, Scanu S, Ubbink M. Dynamics in electron transfer protein complexes. FEBS J. 2011;278:1391–1400. doi: 10.1111/j.1742-4658.2011.08062.x. [DOI] [PubMed] [Google Scholar]
- 34.Maneg O, Ludwig B, Malatesta F. Different interaction modes of two cytochrome-c oxidase soluble CuA fragments with their substrates. J Biol Chem. 2003;278:46734–46740. doi: 10.1074/jbc.M307594200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.