

Abstract
The diversity of metabolites found in plants is by far greater than in most other organisms. Metabolic profiling techniques, which measure many of these compounds simultaneously, enabled investigating the regulation of metabolic networks and proved to be useful for predicting important agronomic traits. However, little is known about the genetic basis of metabolites in crops such as maize. Here, a set of 289 diverse maize inbred lines was genotyped with 56,110 SNPs and assayed for 118 biochemical compounds in the leaves of young plants, as well as for agronomic traits of mature plants in field trials. Metabolite concentrations had on average a repeatability of 0.73 and showed a correlation pattern that largely reflected their functional grouping. Genome-wide association mapping with correction for population structure and cryptic relatedness identified for 26 distinct metabolites strong associations with SNPs, explaining up to 32.0% of the observed genetic variance. On nine chromosomes, we detected 15 distinct SNP–metabolite associations, each of which explained more then 15% of the genetic variance. For lignin precursors, including p-coumaric acid and caffeic acid, we found strong associations (P values 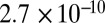 to
to 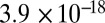 ) with a region on chromosome 9 harboring cinnamoyl-CoA reductase, a key enzyme in monolignol synthesis and a target for improving the quality of lignocellulosic biomass by genetic engineering approaches. Moreover, lignin precursors correlated significantly with lignin content, plant height, and dry matter yield, suggesting that metabolites represent promising connecting links for narrowing the genotype–phenotype gap of complex agronomic traits.
) with a region on chromosome 9 harboring cinnamoyl-CoA reductase, a key enzyme in monolignol synthesis and a target for improving the quality of lignocellulosic biomass by genetic engineering approaches. Moreover, lignin precursors correlated significantly with lignin content, plant height, and dry matter yield, suggesting that metabolites represent promising connecting links for narrowing the genotype–phenotype gap of complex agronomic traits.
Keywords: genetic association, metabolomics, Zea mays
Plants produce a huge array of biochemical compounds estimated to exceed 200,000 in the plant kingdom (1). Recent progress in analytical capabilities together with advanced data processing techniques enabled the quantitative measurement of hundreds of compounds from a wide range of chemical classes within a single sample of plant material (2). These advances have made it possible to deeply investigate the regulation of metabolic networks and to study their influence on complex traits (3).
Empirical evidence suggested that an array of metabolites can be linked to biomass accumulation in Arabidopsis thaliana (4, 5), illustrating their central role for traits connected to growth and development. Metabolomics approaches are also increasingly applied in crop breeding (6). Metabolic profiling could be successfully adopted to predict yield of potato tubers (7) or to distinguish sunflower genotypes with contrasting response to pathogen infections (8). Recently, we showed that metabolic profiles of diverse maize inbred lines allow prediction of their testcross performance in multilocation field trials (9).
Despite these successes, the genetic basis of the metabolic profile in important crops such as maize remains largely unclear. Although certain metabolic products, such as carotenoids in kernels (10), anthocyanins in leaves (11), or maysin (12), have been genetically well characterized, a global picture of the genetic basis of the leaf metabolome is missing. First approaches for studying the genetic basis of concentrations of many distinct metabolites in Arabidopsis used populations such as recombinant inbred lines (RIL) that carry genetic mosaics of two contrasting parental genotypes to map metabolic quantitative trait loci (mQTL) (13). Such linkage mapping approaches revealed a large number of mQTL, but most of them did not explain a substantial amount of genetic variance (14). However, linking genetic variability in metabolite concentrations to genetic variants is of high interest for several reasons.
First, mapping mQTL and ultimately the underlying causal genes can help in annotating the biological function of a metabolite, which may lead to the discovery of new biosynthetic pathways (3). Second, novel enzymatic and regulatory genes controlling metabolic pathways may be identified. Third, mQTL mapping may add functional links to bridge the genotype–phenotype gap of complex traits. In agricultural species, many agronomically important traits are controlled by a large number of genes with small effects (15). Consequently, QTL-based marker-assisted selection is increasingly replaced by whole-genome prediction approaches using thousands of single nucleotide polymorphisms (SNPs) in a black-box prediction model (9, 16). Though this approach is anticipated to be highly successful, it does not provide biological or mechanistic insights into how genetic information is translated into the genetic variability of complex traits. Bridging this apparent genotype–phenotype gap remains a big challenge. A promising approach might therefore be to investigate the genetic basis of intermediate phenotypes with lower genetic complexity, such as yield components or metabolites, and link these results back with the complex trait of interest (17).
Although linkage mapping has a high power for detecting QTL specific to the parental lines of the mapping population, its mapping resolution is very limited due to the few recombination events and, hence, long linkage blocks (18). With the advances in high-throughput genotyping technologies, genome-wide association (GWA) became available as a powerful alternative for dissecting quantitative traits in plants (19). GWA mapping relies on natural linkage disequilibrium (LD) generated by ancestral recombination events in diverse populations. Depending on the level of LD in the population investigated, the mapping resolution can be up to the single nucleotide level. Although plant populations are often prone to inherent population structuring and cryptic relatedness, which can lead to spurious associations in GWA scans (20), powerful techniques are available for decoupling genetic associations with confounding factors (21), and encouraging results have been reported for Arabidopsis (22) and crops such as rapeseed, potato, sugar beet, and maize (23).
Here, we studied the association of genetic variants and concentrations of 118 metabolites in leaves of 289 diverse maize inbred lines from worldwide sources. The population has been previously described in detail (9) and shows a level of LD high enough (r2 = 0.1 at  500 kb) to detect strong associations with 56,110 SNPs. Specifically, we asked the following questions: (i) Which portion of the measurable concentrations of metabolites are controlled by only a few genetic loci so that we can find significant associations, and what portion of the genetic variance do they explain? (ii) Can candidate genes be assigned to the detected associations, and are they plausible regarding the function of the metabolite in the plant? (iii) To what extent are metabolites with significant associations correlated with agronomic traits, and does this allow building bridges across candidate genes, metabolites, and agronomic traits?
500 kb) to detect strong associations with 56,110 SNPs. Specifically, we asked the following questions: (i) Which portion of the measurable concentrations of metabolites are controlled by only a few genetic loci so that we can find significant associations, and what portion of the genetic variance do they explain? (ii) Can candidate genes be assigned to the detected associations, and are they plausible regarding the function of the metabolite in the plant? (iii) To what extent are metabolites with significant associations correlated with agronomic traits, and does this allow building bridges across candidate genes, metabolites, and agronomic traits?
Results
Metabolic Profiling.
We determined the concentrations of 118 metabolites present in the third fully developed leaves using gas chromatography separation coupled with mass spectrometry (GC-MS). The set of metabolites was comprised of 21 amino acids, 13 organic acids, 7 phenylpropanoids, and 20 other metabolites with known, and 57 with unknown, chemical structure. Repeatabilities showed a broad range with a mean of 0.73 (Dataset S1). Additive estimates of heritabilities correlated strongly (r = 0.88) with the repeatabilities.
The correlation pattern revealed hotspots of highly correlated compounds in the metabolic profile that largely corresponded to a functional classification of the metabolites (Fig. 1 and Dataset S2). As examples, the concentration levels of 14 proteinogenic and three nonproteinogenic amino acids, as well as seven organic acids, clustered together.
Fig. 1.

Correlation pattern among measured metabolites. Pairwise Pearson correlations (r) are shown with P < 0.01. Metabolites are ordered using complete linkage hierarchical cluster analysis on pairwise dissimilarity calculated as 1 − r. Hotspots in the correlation pattern are surrounded by a black box. In the dendrogram on the left, the hotspots encompassing predominately amino acids and organic acids are colored in red and blue, respectively. Repeatabilities 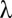 of metabolite concentrations, with additive estimates of heritabilities shown as blue crosses, are shown at the top.
of metabolite concentrations, with additive estimates of heritabilities shown as blue crosses, are shown at the top.
GWA Mapping.
Compared with a one-way ANOVA model, genomic inflation factors ( ), which measure the extent of systematic bias leading to false positive signals, could be drastically reduced by decoupling associations from population structure and cryptic relatedness, which are regarded as two different confounding factors (20, 21). Although it has been argued that correction for cryptic relatedness (
), which measure the extent of systematic bias leading to false positive signals, could be drastically reduced by decoupling associations from population structure and cryptic relatedness, which are regarded as two different confounding factors (20, 21). Although it has been argued that correction for cryptic relatedness (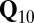 model) would be sufficient, we consistently observed lower genomic inflation factors when we corrected additionally for the main directions of population structure by regressing on the first 10 principal components on SNP data (
model) would be sufficient, we consistently observed lower genomic inflation factors when we corrected additionally for the main directions of population structure by regressing on the first 10 principal components on SNP data (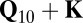 ), especially for metabolites like lysine, which show a strong confounding (Fig. 2). All further results refer to this
), especially for metabolites like lysine, which show a strong confounding (Fig. 2). All further results refer to this 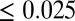 model.
model.
Fig. 2.

Comparison of QQ plots for different GWA models for two metabolites with contrasting extent of confounding with population structure. Observed vs. expected P values are shown for (A) p-coumaric acid and (B) lysine using six different models: a one-way ANOVA model and five models with different corrections of confounding factors (Materials and Methods). The lowest values for 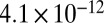 were consistently obtained for all metabolites using the
were consistently obtained for all metabolites using the 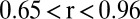 model for which
model for which 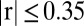 for p-coumaric acid and
for p-coumaric acid and  for lysine.
for lysine.
Several approaches have been suggested to derive an appropriate significance threshold for SNP–trait associations. Whereas the Bonferroni correction leads to a too-conservative overcorrection, empirical resampling approaches have been criticized for breaking both SNP–trait as well as SNP–population structure relationships (24). Here, we controlled the false discovery rate (FDR) (25), as has been previously suggested for GWA mapping (26).
Significant SNPs with FDR  were found for 26 metabolites, of which 17 had an unknown chemical structure (Fig. 3, Fig. S1, and Table S1). Metabolites with significant associations showed repeatability above 0.64 and were approximately normally distributed except for dopamine and three unknown metabolites, which showed a stronger bimodal distribution (Fig. S2). Significant associations were found on all chromosomes with SNPs, explaining up to 32.0% of the observed genetic variance (median 16.2%). On nine chromosomes, we found 15 distinct SNP–metabolite association signals, each of which explained more than 15% of the genetic variance (median 22.1%).
were found for 26 metabolites, of which 17 had an unknown chemical structure (Fig. 3, Fig. S1, and Table S1). Metabolites with significant associations showed repeatability above 0.64 and were approximately normally distributed except for dopamine and three unknown metabolites, which showed a stronger bimodal distribution (Fig. S2). Significant associations were found on all chromosomes with SNPs, explaining up to 32.0% of the observed genetic variance (median 16.2%). On nine chromosomes, we found 15 distinct SNP–metabolite association signals, each of which explained more than 15% of the genetic variance (median 22.1%).
Fig. 3.

Manhattan plots for metabolites with known chemical structure and significant association signals. P values are shown on a 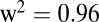 scale and colored in red if significant with FDR ≤ 0.025. For the Manhattan plots of the metabolites with unknown chemical structure, see Fig. S1.
scale and colored in red if significant with FDR ≤ 0.025. For the Manhattan plots of the metabolites with unknown chemical structure, see Fig. S1.
The two amino acids lysine and tyrosine showed their strongest signals for association at a location on chromosome 5 that was separated by 28.3 kb from a putative cationic amino acid transporter. Although amino acids clustered together in the metabolic profile (Fig. 2), this signal was specific to lysine and tyrosine; it was not found for either any other amino acids or the three main principal components calculated from all amino acids.
γ-Aminobutyric acid (GABA) showed a significant association with an SNP located in a gene on chromosome 1 encoding a phytochrome photoreceptor system. The sugar alcohol ribitol and four unknown metabolites showed significant associations with SNPs located on chromosome 10 within a 91-kb region containing two sugar/inositol transporter genes as the only genes in this genomic region.
We found three unknown metabolites to show significant associations with an SNP 1.5 kb apart of a cytochrome P450 protein on chromosome 3. Other candidate genes for unknown metabolites included galactinol-sucrose galactosyltransferase, 40S and 60S ribosomal proteins, cellulose synthase-like protein, cysteine synthase, transcription factors, GDSL esterase/lipase, and ubiquitin-associated protein.
The catecholamine dopamine, the phenylpropanoids p-coumaric acid, and caffeic acid, as well as two metabolites with unknown chemical structure (1016200-307 and 1044100-307), consistently showed their two strongest significant association signals in a 762-kb region on chromosome 9. To increase the mapping resolution in GWA with a limited amount of SNPs, it has been suggested to impute the allelic states of ungenotyped SNPs based on data from a reference population that has been genotyped at a much higher density (27). We therefore imputed SNPs surrounding the chromosomal region on chromosome 9 using the first-generation HapMap data (1.6 million SNPs) available for 27 maize inbred lines (28). Imputation revealed several closely located SNPs in strong LD and lower P values compared with the two surrounding genotyped SNPs located in a cellulose synthase A (CESA) and a bZIP transcription factor (BZIP) (Fig. 4). For three of the five metabolites (caffeic acid, 1016200-307, and 1044100-307), the strongest signal was consistently observed for a SNP 19.9 kb away from a putative cinnamoyl-CoA reductase (CCR), an oxidoreductase important in the monolignol biosynthesis (Fig. 5). This SNP showed also the second lowest P value of 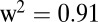 for p-coumaric acid. Imputation did not lead to a higher resolution of the other weaker association signals.
for p-coumaric acid. Imputation did not lead to a higher resolution of the other weaker association signals.
Fig. 4.

Regional association plot of the region on chromosome 9 for caffeic acid. Imputation revealed several closely located SNPs in strong LD (r2) with the genotyped SNP at position 96,124,914 (black). The strongest association signal was obtained for an SNP 19.9 kb apart from the candidate gene CCR.
Fig. 5.

Biochemical connection of p-coumaric acid, caffeic acid, and dopamine with the candidate genes CCR and CESA. Shown are the intermediates and enzymes (blue) of the relevant sections of the simplified phenylpropanoid and catecholamine pathways. Dashed arrows indicate complex transition steps, which can be found in detail in Humphreys and Chapple (38). The cellulose synthase complex is a hexametric rosette complex whose subunits consist of three different cellulose synthase A (CESA) proteins. The double-headed arrow between cellulose and lignin illustrates that synthesis of lignin is negatively coregulated with synthesis of cellulose (39). 4CL, 4-coumarate:CoA ligase; AAH, amino acid hydroxylase; C3H, p-coumarate 3-hydroxylase; C4H, cinnamate 4-hdroxylase; CAD, cinnamyl alcohol dehydrogenase; CCoAOMT, caffeoyl-CoA O-methyltransferase; CCR, cinnamoyl-CoA reductase; DDC, dopa decarboxylase; MH, monophenol hydroxylase; PAL, phenylalanine ammonia lyase; TAL, tyrosine ammonia lyase; TH, tyrosine hydroxylase; TDC, tyrosine decarboxylase. Adapted and modified from refs. 37, 38, and 40.
p-Coumaric acid showed strong negative correlations with dopamine, caffeic acid, 1016200-307, and 1044100-307, which were positively correlated with each other ( ; Table 1). These five metabolites with significant signals at the same position on chromosome 9 showed weak but highly significant correlations with the agronomic traits lignin content, plant height, and whole-plant dry matter yield determined in mature plants grown in the same environment. Correlations with early biomass determined at the time of metabolite measurements were highly significant for four of the five metabolites, but lower compared with the other agronomic traits determined at the end of the vegetation period.
; Table 1). These five metabolites with significant signals at the same position on chromosome 9 showed weak but highly significant correlations with the agronomic traits lignin content, plant height, and whole-plant dry matter yield determined in mature plants grown in the same environment. Correlations with early biomass determined at the time of metabolite measurements were highly significant for four of the five metabolites, but lower compared with the other agronomic traits determined at the end of the vegetation period.
Table 1.
Correlations between lignin content, plant height, early biomass, dry matter yield, and the five metabolites that show significant associations with the same genomic region on chromosome 9
| Caffeic acid | Dopamine | p-CA* | 1016200-307† | 1044100-307† | Lignin | PH | EB | DMY | |
| Caffeic acid | — | 0.65 | −0.45 | 0.79 | 0.72 | −0.18 | −0.21 | −0.18 | −0.28 |
| Dopamine | <1 E-15 | — | −0.70 | 0.75 | 0.69 | −0.16 | −0.23 | −0.13 | −0.23 |
| p-CA* | 4.4 E-15 | <1 E-15 | — | −0.72 | −0.72 | 0.15 | 0.16 | 0.07 | 0.12 |
| 1016200-307† | <1 E-15 | <1 E-15 | <1 E-15 | — | 0.96 | −0.20 | −0.20 | −0.26 | −0.33 |
| 1044100-307† | <1 E-15 | <1 E-15 | <1 E-15 | <1 E-15 | — | −0.19 | −0.21 | −0.25 | −0.35 |
Lignin (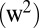 ) ) |
3.3 E-3 | 6.8 E-3 | 1.1 E-2 | 1.0 E-3 | 1.8 E-3 | — | 0.30 | −0.04 | 0.07 |
PH (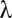 ) ) |
4.4 E-4 | 1.6 E-4 | 8.4 E-3 | 9.4 E-4 | 5.5 E-4 | 6.4 E-7 | — | 0.00 | 0.50 |
EB ( ) ) |
3.2 E-3 | 3.6 E-2 | 2.8 E-1 | 3.4 E-5 | 4.9 E-5 | 5.3 E-1 | 9.7 E-1 | — | 0.45 |
DMY (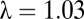 ) ) |
2.7 E-6 | 1.1 E-4 | 5.0 E-2 | 2.8 E-8 | 4.4 E-9 | 2.4 E-1 | <1 E-15 | 3.1 E-15 | — |
Pairwise Pearson correlations are shown above the diagonal, and associated P values are shown below the diagonal. DMY, dry matter yield; EB, early biomass; PH, plant height.
*p-Coumaric acid.
†Metabolite with unknown chemical structure.
Discussion
In this study, we showed that GWA mapping is a powerful tool for linking metabolic composition of leaves from field-grown maize inbred lines with genetic variants at a high resolution. Compared with previous mQTL linkage mapping experiments reporting hundreds to several thousands mQTL for the plant metabolome, our results differ concerning both the number of associations and their explained genetic variance. Lisec et al. (13) reported 157 mQTL that account for a median of 4.3% of the phenotypic variation for 181 metabolites with an average repeatability of 0.4. Schauer et al. (29) detected 104 mQTL for 22 distinct amino acids in tomato. Keurentjes et al. (30) found on average 2 mQTL for each of the 1,592 mass signals obtained from a LC-MS analysis of 14 Arabidopsis accessions, resulting in 4,213 significant mQTL. It was, however, unclear whether the distinct mass signals correspond to chemically distinct compounds or fragments of the same compounds, and whether these massive amounts of mQTL account for a nontrivial amount of genetic variability of distinct plant metabolites.
In comparison, we found significant associations for only 26 of the 118 metabolites with 15 distinct SNP–metabolite associations, which account for up to 32.0% of genetic variance with a median of 22.1%. Possible reasons for the, on average, larger proportion of explained genetic variance for the set of metabolites measured in our study include (i) the greater genetic variability compared with the genetically narrow populations used in linkage mapping experiments, and (ii) the higher repeatabilities and, hence, higher precision in measuring the metabolite concentrations in the field.
Although we found significant GWA signals only for metabolites with a repeatability above 0.64, the correlation between total explained genetic variance and repeatability was not significant for the 26 metabolites for which significant association signals were found (Fig. S3). The metabolites for which we obtained high repeatabilities but no significant associations are therefore either under the control of very complex genetic architecture or the association signals could not be detected because of limited sample size or insufficient LD with potential causal variants. To investigate the latter, we applied a SNP hiding test (28, 31). We found that for 48.1% of the SNPs, there exists at least one SNP in strong LD (r2 > 0.8) within a surrounding window of 200 SNPs, indicating that a much larger number of SNPs would be necessary to ensure that every potential causal variant is in strong LD with at least one SNP.
Other reasons for the moderate number of associations compared with mQTL linkage mapping experiments include the more stringent significant threshold as well as possible confounding influence of population structure, as has been observed in a GWA study of glucosinolate metabolites in Arabidopsis (32). Despite these limitations, our results demonstrate that levels of at least some of the metabolites found in leaves of young maize plants can be under a relatively simple genetic control. Therefore, the maize leaf metabolome seems to be highly diverse not only in terms of biochemical composition but also in terms of genetic architecture. Interestingly, similar simple genetic architectures have been recently observed in GWA studies of metabolic traits in human blood (33) and urine (34), illustrating that major mQTL are not uncommon in nature.
Though chromosomal hotspots are frequently found for mQTL (13, 30, 35), we could not determine any agglomeration of clustered associations. Besides the small number of total associations, a simple explanation for the reported hotspots of mQTL might be the occurrence of biochemically connected or otherwise highly correlated metabolites (including those with unknown chemical structure), pointing to the same genomic position, as was observed in this study for p-coumaric acid, caffeic acid, dopamine, and two highly correlated metabolites with unknown chemical structure.
We found that these five metabolites show a significant association in the same region on chromosome 9, indicating that they have similar genetic control. Results of SNP imputation indicate that the causal mutation in this region was likely not hit by either one of the two genotyped SNPs located in the genes CESA and BZIP. In the near future, genotyping by sequencing (36) will make it possible to build a much larger reference set for imputation and to assess in more detail the accuracy for imputing millions of SNPs in maize. Nevertheless, the result that for three metabolites, the lowest P values were consistently obtained for an imputed SNP close to CCR lifted this gene to the most promising candidate gene in this genomic region, which is supported by the biochemistry of lignin synthesis. The phenylpropanoids caffeic acid and p-coumaric acid are both intermediates in monolignol biosynthesis and, hence, precursors of lignin. After activation by ATP-dependent addition to CoA, both are direct substrates of CCR (37) (Fig. 5). Although the catecholamine dopamine is not directly involved in lignin synthesis, its negative correlation with p-coumaric acid (r = −0.70; Table 1) suggests that a conversion of phenylalanine to monolignols is negatively coregulated with a conversion of phenylalanine to catecholamines, leading to an indirect association with the same genomic region. The two unknown metabolites 1016200-307 and 1044100-307 are strongly positively correlated with caffeic acid, suggesting that both are likely intermediates in the phenylpropanoid pathway.
These five coregulated compounds correlated significantly with lignin content of the mature plants as well as with the higher integrated phenotypic traits, plant height and dry matter yield, measured at the end of the vegetation period (Table 1). These significant correlations suggest that the identified region on chromosome 9 not only changes concentrations of multiple metabolites involved in cell wall liginification, but is also an important control point for plant growth. This association supports the idea that the quality of lignocellulosic biomass can be improved for optimal conversion to biofuels through genetic engineering of key regulators in the monolignol synthesis pathway, as suggested by numerous studies (41, 42).
However, the different signs of these correlations illustrate that the relationship between pathway intermediates and lignin content as the final product in the mature plants is not simple and may require consideration of feedback loops. Moreover, the strong correlations of CCR substrates with dopamine, which is known to be stress induced (40), suggests that a change in carbon flux in monolignol synthesis impacts biochemical composition of other secondary metabolites related to, e.g., stress resistance. In fact, the results from several studies showed that perturbing individual steps of the lignin synthesis pathway affects the expression of other genes not only involved in lignin synthesis (43). Although encouraging results of modifying lignin content and composition through down-regulation of CCR have been achieved in poplar (44) and tobacco (45), deeper investigations into the regulatory mechanisms of monolignol biosynthesis seems to be crucial for a successful genetic engineering of lignin synthesis without detrimental side effects on biotic or abiotic stress resistance (46).
Because the generated metabolic profile is a snapshot at a certain moment in time during early development, it would be also of interest to quantitatively measure metabolic fluxes to capture the dynamic component of plant metabolism. Successive measurements of isotope-labeled metabolites have been successfully applied for measuring phenylpropanoids derived from p-coumaric acid in potato (47), and a similar approach could shed more light on how lignin synthesis is regulated at the metabolic level in maize.
The established associations with agronomic traits rely on phenotypic correlations of 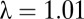 , making it difficult to assess quantitatively the direct impact of these genetic variants on the agronomic traits in the field. As expected with a population of our size, the two top significant SNPs on chromosome 9 were not significant in GWA scans of the agronomic traits using a
, making it difficult to assess quantitatively the direct impact of these genetic variants on the agronomic traits in the field. As expected with a population of our size, the two top significant SNPs on chromosome 9 were not significant in GWA scans of the agronomic traits using a  model, and explained less than 1.7% of their genetic variances. Whole-genome prediction, which simultaneously estimates genetic effects over the whole genome instead of focusing on single genomic regions only, remains therefore the method of choice for predicting complex agronomic traits (9).
model, and explained less than 1.7% of their genetic variances. Whole-genome prediction, which simultaneously estimates genetic effects over the whole genome instead of focusing on single genomic regions only, remains therefore the method of choice for predicting complex agronomic traits (9).
Given the fact that GWA mapping in elite maize inbred lines is (i) limited in its resolution due to the high level of LD in elite breeding germplasm of maize (9) and (ii) provides only statistical (i.e., indirect) evidence for the association of the genomic region with the investigated metabolites, biological validations of the detected associations remain to be conducted. Possible approaches include RNAi, antisense methods, or the production of knock-out mutants for inducing loss-of-function point mutations in the candidate genes.
In conclusion, we identified strong genetic associations for concentrations of metabolites, especially multiple lignin precursors, to characterize candidate genetic building blocks for lignin content and other agronomic traits. The molecular mechanisms underpinning these associations represent promising targets for genetic engineering approaches. Moreover, our results suggest that studying genetically less complex connecting links between genotype and phenotype, such as metabolites, may be a reasonable alternative for GWA mapping of highly complex traits in plants.
Materials and Methods
Genetic Material and Field Trials.
The population consisted of the 285 diverse inbred lines described previously (9), with additional four European Flint lines that served as the check genotypes in the field trials. In the trials of each of the three maturity groups, 100 genotypes, including five common check genotypes, were randomized as a 20 × 5 α-lattice design with two replications and planted in two-row plots. Plots were thinned to a final plant density of 100,000 plants per hectare. Early biomass was determined by measuring fresh weight of eight plants per field plot 32 d after sowing. Plant height (m) and dry matter yield of whole-plant biomass (t/ha) were measured for each field plot at the end of the vegetation period. Lignin (%) was measured in the harvested plant material using calibrated near-infrared spectroscopy as described previously (48).
Metabolic Profiling.
Leaf samples of the inbred lines were collected 33 d after sowing. Samples of ∼5 cm were cut from the middle part of the fully developed third leaf of 10 plants per plot, bulked, and immediately frozen using dry ice. The five plots of every incomplete block were sampled within a period of 15 s to minimize within-block error due to metabolic changes over time. All 600 plots were sampled within 69 min. The 50 samples from 10 randomly chosen blocks of one field replication of one maturity group were subsequently processed together as one batch. With this blocking structure, we could account for systematic shifts among batches while keeping the field randomization intact. Analysis of volatizable metabolites was conducted using an established GC-MS method (2) with the assistance of recently developed software (49).
Genotyping.
Genotyping was performed using the Illumina SNP chip MaizeSNP50 (Illumina Inc.) containing 56,110 unique SNPs. A quality preprocessing was done by applying the following criteria: (i) call rate above 0.95, (ii) unique allele assignment for the 22 replicated checks of genotype B73, (iii) minor allele frequency greater than 2.5%, and (iv) no more than three heterozygous genotypes. A total of 37,227 SNPs met theses criteria. Five genotypes with a residual heterozygosity above 5% were excluded. The chromosomal positions of the SNPs refer to the B73 reference genome (B73 RefGen_v1). Candidate genes were taken from the B73 filtered gene set (release 4a.53).
Statistical Analysis of Phenotypic and Metabolic Data.
Linear mixed models were used for obtaining least squares means for the phenotypic traits and metabolites. The model for the phenotypic traits was 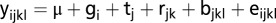 , where
, where 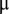 is the grand mean,
is the grand mean,  the fixed effect of the
the fixed effect of the  genotype,
genotype, 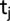 the fixed effect of the
the fixed effect of the 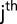 maturity group trial captured with the common check genotypes,
maturity group trial captured with the common check genotypes, 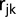 the fixed effect of the
the fixed effect of the 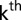 field replication within the
field replication within the  maturity group trial,
maturity group trial, 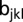 the random effect of the
the random effect of the  incomplete block within the
incomplete block within the 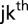 field replication, and the residual error
field replication, and the residual error  . For the metabolic traits, preprocessing was the same as described previously (9). A fixed effect
. For the metabolic traits, preprocessing was the same as described previously (9). A fixed effect  for the
for the 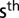 batch was included. To achieve homoscedasticity of the residuals of the metabolites, the flexible Box–Cox power transformation was applied. For each metabolite, the optimum transformation value was determined as described by Piepho (50) using a grid search between 0 and 1 with 100 steps. Repeatabilities
batch was included. To achieve homoscedasticity of the residuals of the metabolites, the flexible Box–Cox power transformation was applied. For each metabolite, the optimum transformation value was determined as described by Piepho (50) using a grid search between 0 and 1 with 100 steps. Repeatabilities  were calculated as
were calculated as 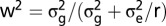 , where r is the number of field replications. Genotypic variance
, where r is the number of field replications. Genotypic variance 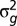 was estimated by restricted maximum likelihood (REML) assuming that
was estimated by restricted maximum likelihood (REML) assuming that 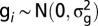 . REML-based additive estimates of heritabilities were calculated using the function polygenic_hglm of GenABEL (51) assuming random genotype effects with kinship matrix
. REML-based additive estimates of heritabilities were calculated using the function polygenic_hglm of GenABEL (51) assuming random genotype effects with kinship matrix  of proportion of shared SNP alleles as variance-covariance matrix.
of proportion of shared SNP alleles as variance-covariance matrix.
GWA Mapping.
Single-marker analysis was initially carried out using a one-way ANOVA model without considering confounding factors. Phenotypes were regressed on the number of copies of SNP alleles. Quantile–quantile (QQ) plots of the expected vs. observed P values were inspected for an inflation indicating false positive signals of association. Genome-wide inflation factors (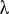 ) were calculated as the regression coefficient in the QQ plot with a zero intercept. Because of the high inflation factors, we next applied a
) were calculated as the regression coefficient in the QQ plot with a zero intercept. Because of the high inflation factors, we next applied a 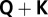 mixed linear model approach with correction for (i) main directions of population structure by regressing on the first three (
mixed linear model approach with correction for (i) main directions of population structure by regressing on the first three (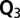 ) or 10 (
) or 10 (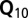 ) principal components on SNP data, and (ii) cryptic relatedness using the kinship matrix
) principal components on SNP data, and (ii) cryptic relatedness using the kinship matrix 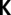 as variance-covariance matrix for random genotype effects (52). GWA models were fitted using the maximum likelihood implementation in the function polygenic of GenABEL (51). P values were obtained with the 1 degree of freedom score test implemented in the function mmscore of GenABEL (53). P values were transformed to q-values and regarded significant if ≤0.025 to control for a FDR (25) of 2.5%. The proportion of genetic variance explained by a certain SNP was calculated as
as variance-covariance matrix for random genotype effects (52). GWA models were fitted using the maximum likelihood implementation in the function polygenic of GenABEL (51). P values were obtained with the 1 degree of freedom score test implemented in the function mmscore of GenABEL (53). P values were transformed to q-values and regarded significant if ≤0.025 to control for a FDR (25) of 2.5%. The proportion of genetic variance explained by a certain SNP was calculated as 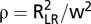 using the likelihood-ratio statistic
using the likelihood-ratio statistic  with
with  , where
, where 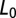 is the maximum likelihood of the baseline
is the maximum likelihood of the baseline 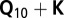 model without considering the SNP, and
model without considering the SNP, and 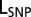 is the maximum likelihood of the full
is the maximum likelihood of the full 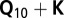 model including the SNP as cofactor, and n is the number of genotypes (54). For the regional association scan on chromosome 9, imputation was performed using BEAGLE 3.3 (55). BEAGLE parameters were set to nSamples = 50 and nIterations = 20.
model including the SNP as cofactor, and n is the number of genotypes (54). For the regional association scan on chromosome 9, imputation was performed using BEAGLE 3.3 (55). BEAGLE parameters were set to nSamples = 50 and nIterations = 20.
Supplementary Material
Acknowledgments
We thank the staff of the experimental research stations of the University of Hohenheim for assistance in conducting the field experiments. This research was supported by the Max Planck Society and the German Federal Ministry of Education and Research (BMBF) within the project GABI-Energy (FKZ: 0315045).
Footnotes
The authors declare no conflict of interest
This article is a PNAS Direct Submission. E.S.B. is a guest editor invited by the Editorial Board.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1120813109/-/DCSupplemental.
References
- 1.Dixon RA, Strack D. Phytochemistry meets genome analysis, and beyond. Phytochemistry. 2003;62:815–816. doi: 10.1016/s0031-9422(02)00712-4. [DOI] [PubMed] [Google Scholar]
- 2.Lisec J, Schauer N, Kopka J, Willmitzer L, Fernie AR. Gas chromatography mass spectrometry-based metabolite profiling in plants. Nat Protoc. 2006;1:387–396. doi: 10.1038/nprot.2006.59. [DOI] [PubMed] [Google Scholar]
- 3.Saito K, Matsuda F. Metabolomics for functional genomics, systems biology, and biotechnology. Annu Rev Plant Biol. 2010;61:463–489. doi: 10.1146/annurev.arplant.043008.092035. [DOI] [PubMed] [Google Scholar]
- 4.Meyer RC, et al. The metabolic signature related to high plant growth rate in Arabidopsis thaliana. Proc Natl Acad Sci USA. 2007;104:4759–4764. doi: 10.1073/pnas.0609709104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sulpice R, et al. Starch as a major integrator in the regulation of plant growth. Proc Natl Acad Sci USA. 2009;106:10348–10353. doi: 10.1073/pnas.0903478106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fernie AR, Schauer N. Metabolomics-assisted breeding: A viable option for crop improvement? Trends Genet. 2009;25:39–48. doi: 10.1016/j.tig.2008.10.010. [DOI] [PubMed] [Google Scholar]
- 7.Steinfath M, et al. Discovering plant metabolic biomarkers for phenotype prediction using an untargeted approach. Plant Biotechnol J. 2010;8:900–911. doi: 10.1111/j.1467-7652.2010.00516.x. [DOI] [PubMed] [Google Scholar]
- 8.Peluffo L, et al. Metabolic profiles of sunflower genotypes with contrasting response to Sclerotinia sclerotiorum infection. Phytochemistry. 2010;71:70–80. doi: 10.1016/j.phytochem.2009.09.018. [DOI] [PubMed] [Google Scholar]
- 9.Riedelsheimer C, et al. Genomic and metabolic prediction of complex heterotic traits in hybrid maize. Nat Genet. 2012;44:217–220. doi: 10.1038/ng.1033. [DOI] [PubMed] [Google Scholar]
- 10.Wong JC, Lambert RJ, Wurtzel ET, Rocheford TR. QTL and candidate genes phytoene synthase and zeta-carotene desaturase associated with the accumulation of carotenoids in maize. Theor Appl Genet. 2004;108:349–359. doi: 10.1007/s00122-003-1436-4. [DOI] [PubMed] [Google Scholar]
- 11.Christie PJ, Alfenito MR, Walbot V. Impact of low-temperature stress on general phenylpropanoid and anthocyanin pathways: Enhancement of transcript abundance and anthocyanin pigmentation in maize seedlings. Planta. 1994;194:541–549. [Google Scholar]
- 12.Byrne PF, et al. Quantitative trait loci and metabolic pathways: Genetic control of the concentration of maysin, a corn earworm resistance factor, in maize silks. Proc Natl Acad Sci USA. 1996;93:8820–8825. doi: 10.1073/pnas.93.17.8820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lisec J, et al. Identification of metabolic and biomass QTL in Arabidopsis thaliana in a parallel analysis of RIL and IL populations. Plant J. 2008;53:960–972. doi: 10.1111/j.1365-313X.2007.03383.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brotman Y, et al. Identification of enzymatic and regulatory genes of plant metabolism through QTL analysis in Arabidopsis. J Plant Physiol. 2011;168:1387–1394. doi: 10.1016/j.jplph.2011.03.008. [DOI] [PubMed] [Google Scholar]
- 15.Bernardo R. Molecular markers and selection for complex traits in plants: Learning from the last 20 years. Crop Sci. 2008;48:1649–1664. [Google Scholar]
- 16.Heffner E, Sorrells M, Jannink JL. Genomic selection for crop improvement. Crop Sci. 2009;49:1–12. [Google Scholar]
- 17.Keurentjes JJB. Genetical metabolomics: Closing in on phenotypes. Curr Opin Plant Biol. 2009;12:223–230. doi: 10.1016/j.pbi.2008.12.003. [DOI] [PubMed] [Google Scholar]
- 18.Mauricio R. Mapping quantitative trait loci in plants: Uses and caveats for evolutionary biology. Nat Rev Genet. 2001;2:370–381. doi: 10.1038/35072085. [DOI] [PubMed] [Google Scholar]
- 19.Stich B, Melchinger A. An introduction to association mapping in plants. CAB Reviews. 2010;5:1–9. [Google Scholar]
- 20.Astle W, Balding DJ. Population structure and cryptic relatedness in genetic association studies. Stat Sci. 2009;24:451–471. [Google Scholar]
- 21.Sillanpää MJ. Overview of techniques to account for confounding due to population stratification and cryptic relatedness in genomic data association analyses. Heredity (Edinb) 2011;106:511–519. doi: 10.1038/hdy.2010.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Atwell S, et al. Genome-wide association study of 107 phenotypes in Arabidopsis thaliana inbred lines. Nature. 2010;465:627–631. doi: 10.1038/nature08800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stich B, Melchinger AE. Comparison of mixed-model approaches for association mapping in rapeseed, potato, sugar beet, maize, and Arabidopsis. BMC Genomics. 2009;10:94. doi: 10.1186/1471-2164-10-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Aulchenko YS, de Koning DJ, Haley C. Genomewide rapid association using mixed model and regression: A fast and simple method for genomewide pedigree-based quantitative trait loci association analysis. Genetics. 2007;177:577–585. doi: 10.1534/genetics.107.075614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Benjamini Y, Hochberg Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J R Stat Soc, B. 1995;57:289–300. [Google Scholar]
- 26.Storey JD, Tibshirani R. Statistical significance for genomewide studies. Proc Natl Acad Sci USA. 2003;100:9440–9445. doi: 10.1073/pnas.1530509100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Marchini J, Howie B. Genotype imputation for genome-wide association studies. Nat Rev Genet. 2010;11:499–511. doi: 10.1038/nrg2796. [DOI] [PubMed] [Google Scholar]
- 28.Gore MA, et al. A first-generation haplotype map of maize. Science. 2009;326:1115–1117. doi: 10.1126/science.1177837. [DOI] [PubMed] [Google Scholar]
- 29.Schauer N, et al. Mode of inheritance of primary metabolic traits in tomato. Plant Cell. 2008;20:509–523. doi: 10.1105/tpc.107.056523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Keurentjes JJB, et al. The genetics of plant metabolism. Nat Genet. 2006;38:842–849. doi: 10.1038/ng1815. [DOI] [PubMed] [Google Scholar]
- 31.Kim S, et al. Recombination and linkage disequilibrium in Arabidopsis thaliana. Nat Genet. 2007;39:1151–1155. doi: 10.1038/ng2115. [DOI] [PubMed] [Google Scholar]
- 32.Chan EK, Rowe HC, Kliebenstein DJ. Understanding the evolution of defense metabolites in Arabidopsis thaliana using genome-wide association mapping. Genetics. 2010;185:991–1007. doi: 10.1534/genetics.109.108522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Illig T, et al. A genome-wide perspective of genetic variation in human metabolism. Nat Genet. 2010;42:137–141. doi: 10.1038/ng.507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Suhre K, et al. A genome-wide association study of metabolic traits in human urine. Nat Genet. 2011;43:565–569. doi: 10.1038/ng.837. [DOI] [PubMed] [Google Scholar]
- 35.Fu J, et al. System-wide molecular evidence for phenotypic buffering in Arabidopsis. Nat Genet. 2009;41:166–167. doi: 10.1038/ng.308. [DOI] [PubMed] [Google Scholar]
- 36.Elshire RJ, et al. A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS ONE. 2011;6:e19379. doi: 10.1371/journal.pone.0019379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bonawitz ND, Chapple C. The genetics of lignin biosynthesis: Connecting genotype to phenotype. Annu Rev Genet. 2010;44:337–363. doi: 10.1146/annurev-genet-102209-163508. [DOI] [PubMed] [Google Scholar]
- 38.Humphreys JM, Chapple C. Rewriting the lignin roadmap. Curr Opin Plant Biol. 2002;5:224–229. doi: 10.1016/s1369-5266(02)00257-1. [DOI] [PubMed] [Google Scholar]
- 39.Endler A, Persson S. Cellulose synthases and synthesis in Arabidopsis. Mol Plant. 2011;4:199–211. doi: 10.1093/mp/ssq079. [DOI] [PubMed] [Google Scholar]
- 40.Kulma A, Szopa J. Catecholamines are active compounds in plants. Plant Sci. 2007;172:433–440. [Google Scholar]
- 41.Chen F, Dixon RA. Lignin modification improves fermentable sugar yields for biofuel production. Nat Biotechnol. 2007;25:759–761. doi: 10.1038/nbt1316. [DOI] [PubMed] [Google Scholar]
- 42.Simmons BA, Loqué D, Ralph J. Advances in modifying lignin for enhanced biofuel production. Curr Opin Plant Biol. 2010;13:313–320. doi: 10.1016/j.pbi.2010.03.001. [DOI] [PubMed] [Google Scholar]
- 43.Vanholme R, Demedts B, Morreel K, Ralph J, Boerjan W. Lignin biosynthesis and structure. Plant Physiol. 2010;153:895–905. doi: 10.1104/pp.110.155119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Leplé JC, et al. Downregulation of cinnamoyl-coenzyme A reductase in poplar: Multiple-level phenotyping reveals effects on cell wall polymer metabolism and structure. Plant Cell. 2007;19:3669–3691. doi: 10.1105/tpc.107.054148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chabannes M, et al. Strong decrease in lignin content without significant alteration of plant development is induced by simultaneous down-regulation of cinnamoyl CoA reductase (CCR) and cinnamyl alcohol dehydrogenase (CAD) in tobacco plants. Plant J. 2001;28:257–270. doi: 10.1046/j.1365-313x.2001.01140.x. [DOI] [PubMed] [Google Scholar]
- 46.Weng JK, Li X, Bonawitz ND, Chapple C. Emerging strategies of lignin engineering and degradation for cellulosic biofuel production. Curr Opin Biotechnol. 2008;19:166–172. doi: 10.1016/j.copbio.2008.02.014. [DOI] [PubMed] [Google Scholar]
- 47.Matsuda F, et al. Metabolic flux analysis of the phenylpropanoid pathway in elicitor-treated potato tuber tissue. Plant Cell Physiol. 2005;46:454–466. doi: 10.1093/pcp/pci042. [DOI] [PubMed] [Google Scholar]
- 48.Grieder C, et al. Determination of methane fermentation yield and its kinetics by near infrared spectroscopy and chemical composition in maize. JNIRS. 2011;19:463–477. [Google Scholar]
- 49.Cuadros-Inostroza A, et al. TargetSearch—a Bioconductor package for the efficient preprocessing of GC-MS metabolite profiling data. BMC Bioinformatics. 2009;10:428. doi: 10.1186/1471-2105-10-428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Piepho H. Data transformation in statistical analysis of field trials with changing treatment variance. Agron J. 2009;101:865–869. [Google Scholar]
- 51.Aulchenko YS, Ripke S, Isaacs A, van Duijn CM. GenABEL: An R library for genome-wide association analysis. Bioinformatics. 2007;23:1294–1296. doi: 10.1093/bioinformatics/btm108. [DOI] [PubMed] [Google Scholar]
- 52.Yu J, et al. A unified mixed-model method for association mapping that accounts for multiple levels of relatedness. Nat Genet. 2006;38:203–208. doi: 10.1038/ng1702. [DOI] [PubMed] [Google Scholar]
- 53.Chen WM, Abecasis GR. Family-based association tests for genomewide association scans. Am J Hum Genet. 2007;81:913–926. doi: 10.1086/521580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sun G, et al. Variation explained in mixed-model association mapping. Heredity (Edinb) 2010;105:333–340. doi: 10.1038/hdy.2010.11. [DOI] [PubMed] [Google Scholar]
- 55.Browning BL, Browning SR. A unified approach to genotype imputation and haplotype-phase inference for large data sets of trios and unrelated individuals. Am J Hum Genet. 2009;84:210–223. doi: 10.1016/j.ajhg.2009.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.