

Abstract
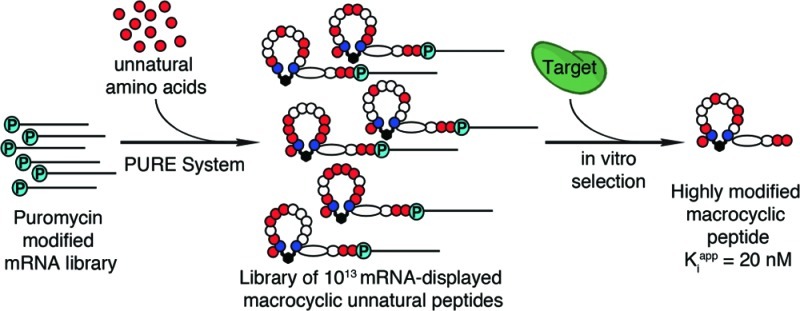
There is a great demand for the discovery of new therapeutic molecules that combine the high specificity and affinity of biologic drugs with the bioavailability and lower cost of small molecules. Small, natural-product-like peptides hold great promise in bridging this gap; however, access to libraries of these compounds has been a limitation. Since ribosomal peptides may be subjected to in vitro selection techniques, the generation of extremely large libraries (>1013) of highly modified macrocyclic peptides may provide a powerful alternative for the generation and selection of new useful bioactive molecules. Moreover, the incorporation of many non-proteinogenic amino acids into ribosomal peptides in conjunction with macrocyclization should enhance the drug-like features of these libraries. Here we show that mRNA-display, a technique that allows the in vitro selection of peptides, can be applied to the evolution of macrocyclic peptides that contain a majority of unnatural amino acids. We describe the isolation and characterization of two such unnatural cyclic peptides that bind the protease thrombin with low nanomolar affinity, and we show that the unnatural residues in these peptides are essential for the observed high-affinity binding. We demonstrate that the selected peptides are tight-binding inhibitors of thrombin, with Kiapp values in the low nanomolar range. The ability to evolve highly modified macrocyclic peptides in the laboratory is the first crucial step toward the facile generation of useful molecular reagents and therapeutic lead molecules that combine the advantageous features of biologics with those of small-molecule drugs.
Introduction
Two classes of molecules dominate drug discovery efforts: small molecules and biologics.1 Therapeutic biological macromolecules have become one of the most successful classes of therapeutics due to their high affinity and specificity toward a specific antigen and their predictable nature in the clinic.2 However, the high cost and complexity of production of biologics might be prohibitive for their use in the treatments of chronic diseases.3 In addition, since biologics cannot penetrate cells, ∼90% of drug targets are not accessible for targeting.1 Most small molecules that satisfy Lipinski’s rule of five4 are able to penetrate cells and can be chemically synthesized in large amounts. However, small molecules have much higher attrition rates in the clinic than biologic drugs, and only ∼25% of recently approved small-molecule drugs can be defined as new molecular entities, compared to ∼90% of biologic drugs.5 In addition, since ∼10% of proteins encoded in the genome are estimated to be amenable to small-molecule targeting and another 10% by biologic drugs, 80% of proteins are currently termed as “undruggable”.1,6,7 Thus, it is clear that new classes of molecules are urgently needed for drug discovery.
Small constrained peptides comprise a large class of molecules that could combine the high specificity of biologic drugs with the bioavailability of small molecules. Organisms have evolved the ability to produce an arsenal of macrocyclic peptides such as lantibiotics, peptide hormones, toxins, and non-ribosomal peptides (NRPs) that show a wide range of biological activities as a result of their ability to bind and interact with a diverse range of targets.8 Importantly, these macrocyclic peptides rarely function by binding to the active site of an enzyme, but rather modulate key macromolecular interactions that are difficult to target using small molecules.9 In addition, synthetic structurally constrained peptides (stapled-helices) have had great success in combining the surface-recognition properties of biologics with the bioavailability and synthetic manipulability of small molecules.10,11 The common feature of these molecules is the structural constraint afforded by cyclization, often in combination with the presence of unnatural amino acids with side-chain and backbone modifications.
However, the isolation, screening, identification, and preparative synthesis of many naturally occurring macrocyclic peptides has been very challenging, and thus this structural class of molecules has been underexploited.9 Although the chemical diversity of building blocks that can be used for the synthesis of peptide libraries is large, synthetic peptide libraries tend to be small, thus decreasing the chance of finding potent and selective binders. Stapled peptides are very attractive modulators of protein–protein interactions and have shown promising properties. However, a significant amount of knowledge about the particular protein target is required for the synthesis and screening of stapled peptides for the desired activity.10,11 Ribosomal peptides are amenable to powerful in vitro selection technologies such as phage, yeast, or mRNA-display, allowing the screening of trillions of molecules with the desired properties. However, the poor bioavailability of proteinogenic peptides has limited their use as therapeutics.
The drug-like properties of ribosomal peptides could be enhanced by increasing the chemical diversity of the building blocks in conjunction with macrocyclization of the unnatural peptides analogous to naturally occurring cyclic peptides. Since the resulting highly modified peptides are templated by mRNA, in vitro selections could allow isolation of therapeutic lead molecules from large, unexplored libraries for a wide range of important biological targets. However, few in vitro selection experiments have used unnatural amino acids.12−14 One of the major challenges in the field is that unnatural amino acids are often incorporated into peptides very inefficiently, resulting in a bias against peptides that include them. For this reason, only selections in which the unnatural amino acid provided a strong selection advantage (e.g., a biotinylated amino acid with a streptavidin target, covalent modification) have been successful. In other cases, none of the surviving peptide sequences contained the unnatural amino acid.15 This bias against unnatural amino acids is magnified when one attempts to synthesize peptide libraries that contain multiple, different unnatural amino acids. Thus, for our goal of selecting highly modified peptides from large, unbiased libraries, we needed a system that would allow us to carefully adjust experimental conditions such that sequences containing unnatural amino acids would not be eliminated from the pool.
We thought that the bias against peptides that contain many unnatural amino acids might be overcome by combining the PURE translation system (Protein Synthesis Using Recombinant Elements) with mRNA-display for generating libraries of highly modified peptides. mRNA-display is a robust and completely in vitro selection technique that covalently links individual peptides with their corresponding mRNA, creating large peptide libraries with 1013 or more members that are suitable for in vitro selection experiments.16,17 The PURE system reconstitutes the ribosomal translational machinery in vitro from purified components.18 Recently, several groups, including ours, have used mRNA-templated peptide synthesis to incorporate unusual amino acids into peptides for the generation of highly modified linear and cyclic peptides, using the PURE translation system.19−28 We showed that over 50 unnatural amino acids can be incorporated into peptides by the ribosomal translational machinery. This approach allowed us to produce peptides containing as many as 13 different unnatural amino acids using optimized mRNA templates.22 In addition, we have shown that the system can be manipulated so that the mis-incorporations resulting from competition with near-cognate aminoacyl-tRNAs are minimized,23 leading to improved incorporation of up to three N-methyl amino acids into one peptide. Subsequently, Suga’s group used a modified PURE system for the in vitro selection and isolation of cyclic N-methyl peptides with sub-nanomolar binding affinities. In this system, 5 of 16 codons were reassigned to 4 N-methyl amino acids and the start codon was reassigned to chloroacetyl-d-tryptophan using precharged tRNAs and a limited set of natural amino acids.29 Here we describe the optimization of a combined PURE/mRNA-display system that generates the tRNAs charged with many different unnatural amino acids in situ. Using this system, we reassign 12 of the 20 natural amino acids to unnatural amino acids with a diverse set of side-chain and backbone modifications, to discover highly modified macrocyclic peptides with antibody-like binding affinities. In addition we show that these peptides act as potent enzyme inhibitors. This work lays the foundation for a powerful platform for the in vitro selection and evolution of drug-like molecules that can bridge the gap between small-molecule and biologic drugs.
Results and Discussion
For our selection we used a DNA library (Figure 1A) that was designed for the mRNA-display of short peptides consisting of 10 random amino acids flanked by Cys residues. We chose the unnatural amino acid building blocks on the basis of the following criteria: The building blocks had to be compatible with each other, serve as efficient substrates for only one aminoacyl-tRNA synthetase (AARS), be translated with high fidelity and yield using mRNAs transcribed from our DNA library, and possess interesting functional groups. We decided not to include any unnatural amino acids that provide a strong selection (binding) advantage in our library. No previous selections produced winners from naïve peptide libraries that contained a majority of unnatural amino acids, and we wanted to show that our selection platform could yield highly modified peptides with high binding affinity from such peptide libraries. Using a set of sequences cloned from our library, we tested several combinations of amino acid analogues, searching for a set that resulted in high translational fidelity and good peptide yields. Initially we observed significant mistranslated and truncation products and low yields using unnatural amino acids with our library (Figure 2).
Figure 1.

In vitro selection of macrocyclic peptides using mRNA-display. (A) General scheme for the selection and amplification of cyclic peptides. The DNA library encodes peptides with 10 random amino acids flanked by two Cys residues. Following transcription and cross-linking of the RNA to a 3′-puromycin oligonucleotide, the library was translated in a completely reconstituted translation system with either all natural or replacing 12 natural amino acids with 12 unnatural amino acids to form two separate libraries of mRNA-peptide fusions. The 12 unnatural amino acids used are shown in panel B. Fusions were immobilized on an oligo-dT column and cyclized via bis-alkylation of the Cys residues, reverse transcribed (RT), and purified on a Ni-NTA resin to yield ∼2 × 1013 cyclic peptides. Each library was incubated separately with biotinylated thrombin in solution. Complexes were captured on streptavidin beads, and unbound material was washed away. Active mRNA–peptide fusions were eluted with a 2-fold excess of hirudin to thrombin, followed by RT-PCR amplification to generate the input material for the next round of selection/amplification. (B) Unnatural amino acids used in the unnatural peptide selection. Differences between the natural and unnatural residues are highlighted in red. The single-letter code refers to the corresponding natural amino acid that was replaced by the unnatural amino acid, with a subscript for analogue. The natural amino acids used were C, A, G, S, I, N, Q, and H.
Figure 2.

Optimizing translation with unnatural amino acids. MALDI-TOF spectra of in vitro translation reactions with unoptimized and optimized concentrations of unnatural amino acids. (A) Reaction directed by mRNA encoding the sequence MaCVaFaGNRaGTaQPaFaCGSGSLaGHHHHHHRaLa (unnatural amino acids are labeled with the subscript a) shows several minor incorrect peaks and a major incorrect peak (×) at 3124.5 Da (most likely corresponding to −L truncation; exp −127.1 Da, obs −127). The peak at 3251.5 Da corresponds to the correct product (expected mass of 3251.4 Da). (B) In vitro translation of the same templates after optimization of the amino acid concentrations, showing improved incorporation. (C) Unoptimized in vitro translation reaction directed by mRNA encoding the sequence MaCEaFaFaDaKaKaILaAPaCGSGSLaGHHHHHHRaLa shows several minor undesired peaks and a major incorrect peak (×) at 3249.7 Da (most likely corresponding to −L truncation). The peak at 3377.9 Da corresponds to the correct product. (D) In vitro translation of the same template after optimization of amino acid concentrations, showing improved incorporation.
After several iterations of adjusting concentrations of the amino acid building blocks and eliminating problematic amino acids, we were able to find 12 unnatural amino acids that met our criteria and effectively replaced 12 of the natural amino acids (Figures 1B and 2; Table S1), retaining 8 natural amino acids for the remaining coding blocks. The 12 unnatural amino acids display functional groups not found in the standard 20 amino acids, including an alkyne (Ma), a thiazolidine (Pa), two aryl halides (Ya, Fa), an alkene (Ka), two unusual heterocycles (Ea, Wa), and the tert-butyl group (La). The Ra and Ya residues have altered pKa’s relative to their natural counterparts. Other unique properties include the blue-shifted fluorescence of Wa and the metal chelating ability of Da. This set also includes an α,α-disubstituted amino acid (Va), which would lead to a local conformational constraint on the peptide backbone. Half of the unnatural amino acids used to build our library are racemic mixtures. Although we do not know with certainty, we believe that incorporation of d-amino acids is unlikely (discussed further in the SI).
Target specificity and proteolytic stability are important features of therapeutically active peptides30,31 and natural products such as NRPs.32 These advantageous pharmacological properties are thought to result in part from peptide cyclization: the afforded structural rigidity locks the peptide into a target specific conformation, thereby increasing specificity while also increasing protease resistance by reducing protease accessible conformations.9 We recently showed that peptides containing unnatural amino acids can be cyclized efficiently using a previously described bis-alkylation reaction to cross-link two Cys residues.20,33 This alkylation chemistry has also been used in a phage-display selection to afford bicyclic peptides with target-specific binding.34 We examined the cyclization of peptides from our library that were translated with either natural amino acids or multiple unnatural amino acids. Incubation of individual pure natural peptides, a pool of natural peptides, or a pool of unnatural peptides with dibromoxylene resulted in efficient cyclization (Figure S1). By applying the same procedure to a library of mRNA-displayed unnatural peptides (generated by in vitro translation of mRNAs conjugated to an oligonucleotide with a 3′-puromycin residue), we were able to generate a library of ∼1013 unique macrocyclic peptides, each containing multiple unnatural amino acids and displayed on its own mRNA.
Once we were confidant that most, if not all, random sequence unnatural peptides would be efficiently translated and cyclized, we proceeded to attempt to isolate target-specific ligands by in vitro selection from the library. We chose the protease thrombin as our initial target since RNA, DNA, and peptide aptamers that bind to this enzyme have been isolated and would serve as interesting points of comparison.35−37 Thrombin is part of the blood coagulation cascade, catalyzing the cleavage of fibrinogen to fibrin, and new thrombin inhibitors are still being pursued for the prevention and treatment of thrombosis.38
Beginning with our unnatural macrocyclic peptide library, we performed successive rounds of in vitro selection for thrombin binding followed by amplification by RT-PCR and in vitro translation (Figure 1A). In order to compare the results of selection from unnatural and natural peptide libraries, we performed a parallel selection using the same DNA library to produce cyclic peptides composed solely of natural amino acids. In each round, peptide-mRNA fusions were allowed to bind biotinylated thrombin in solution, followed by capture of the complexes on streptavidin beads. Immobilized complexes were then washed extensively and selectively eluted with hirudin, a small protein (65 amino acids) that contains a sulfated Tyr residue at position 6339 that contributes its femtomolar inhibition constant to thrombin.40 Since we wanted to evolve novel peptide sequences, we did not use sulfated Tyr in our set of unusual amino acids, even though it is a substrate for the translational machinery and has been used with phage-display.12 After six rounds of selection and amplification, both the natural and unnatural pools were enriched with thrombin binders (Figure 3A). We cloned and sequenced the eluted fusions after the seventh round. The natural peptide selection had converged on one dominant sequence, but the unnatural peptide selection yielded a highly diverse set of sequences (Figure S2). In order to select for peptides with the best possible binding, we continued the selection for three additional rounds of increased stringency. We selected for both natural and unnatural peptides with a very slow off-rate by amplifying only those peptide–mRNA fusions that remained bound to thrombin after 1 h in the presence of hirudin and were subsequently eluted during an overnight incubation with hirudin.
Figure 3.
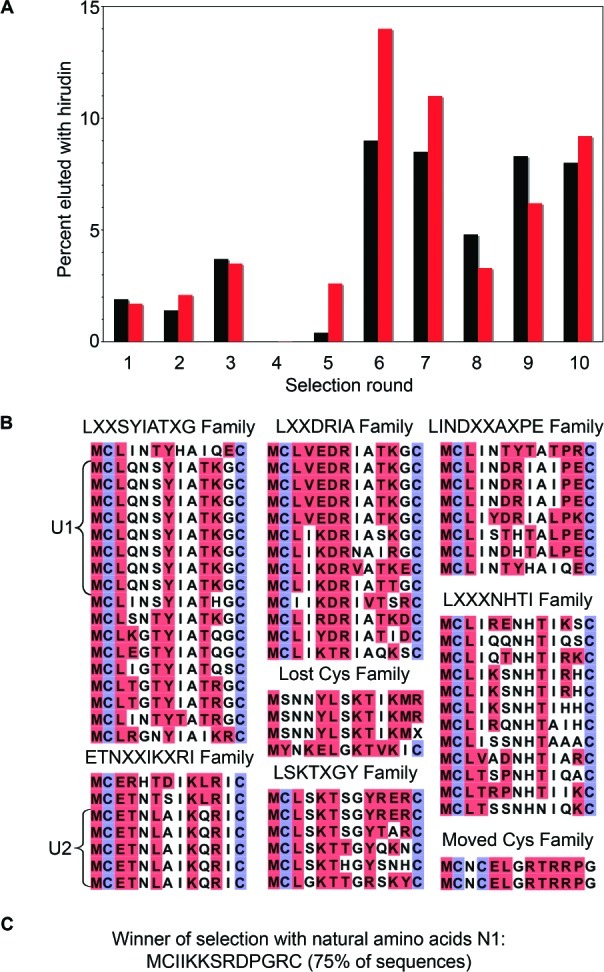
Selection progress and sequences of selected peptides. (A) The fraction of 35S-labeled peptide that bound to thrombin and eluted with hirudin at each round of selection is shown. Starting in round four, complexes were washed more rigorously, and in round seven the selection pressure was increased further by only amplifying mRNA–peptide fusions that remained bound after 1 h of incubation with hirudin and were subsequently eluted. Red and black bars show the progress of the unnatural and natural peptide selections, respectively. (B) Sequences of unnatural peptides after round 10. Unnatural peptide sequences U2 and U1 are labeled. Unnatural amino acids are highlighted in red, and the Cys residues used for cyclization are shown in blue; residues following the second Cys are not shown. (C) Sequence of the natural peptide winner N1 after round 10.
The sequences we obtained after a total of 10 rounds of selection for the unnatural and natural peptides are shown in Figure 3B,C. Sequences for the unnatural peptides were assigned on the basis of the aaRS/tRNA pair that was responsible for incorporation of the corresponding unnatural building block into the peptides. The unnatural peptide selection yielded eight families from which two or more copies of identical sequences were recovered. The largest family contained eight copies of a sequence coding for a peptide, named U1, followed by two other families containing five copies of peptide sequences. Sequence analysis showed that ∼50% of the amino acids encoded within the random region are unnatural. We expected the peptides to contain ∼50% unnatural amino acids, based on the set of unnatural amino acids and the DNA library we used. These results suggest that these amino acids are well tolerated and not selected against during in vitro translation and mRNA display.
The natural cyclic peptide that we selected contains the same amino acid motif, DPGR, that had been previously identified and shown to be critical for binding in an independent study using mRNA-display.35 As expected, this motif is absent in our unnatural peptide sequences, since three of the four residues in this motif were replaced with chemically distinct analogues. None of the selected natural or unnatural sequences show any similarity to hirudin. Interestingly, the macrocyclic thrombin inhibitor Cyclotheonamide A,41 a natural product, also shares no similarity to any of our selected cyclic peptides. The fact that the natural and unnatural sequences are unrelated strongly suggests that the unnatural amino acids sample a different functional and chemical space than their natural counterparts.
We chose the two most abundant unnatural (U1 and U2) and the dominant natural (N1) peptide sequences for further studies (Figure 3B,C). We translated these sequences as free peptides (i.e., not linked to their mRNA) and then cyclized and purified the resulting peptides on Ni-NTA resin (Figures 4A,B and S3–S5). To determine the mass of the peptides, we performed high-resolution LC/MS analysis on the cyclized full-length peptides U1, U2, and N1 (Figures 4B and S4,5). The observed masses of the cyclic peptides are in excellent agreement with the calculated values. To confirm the correct site-specific incorporation of the unnatural amino acids, we sequenced our two unnatural peptide winners by MS/MS (Figures 4C and S6). The sequencing unambiguously demonstrated that each unnatural amino acid was present at the expected site in the unnatural peptides.
Figure 4.

Verification of sequence of unnatural peptide U1. (A) Cyclization of unnatural peptide U1. Unnatural amino acids are shown in red. Cys residues used for cyclization are shown in blue. (B) LC-MS confirms formation of full-length cyclic unnatural peptide U1 (calcd [M]4+m/z = 791.62226 (4.4 ppm), [M]5+m/z = 633.49937 (0.9 ppm), [M]6+m/z = 528.08411 (0.5 ppm) (A.U., arbitrary units). (C) Peptide lacking the His6-tag was translated and the two Cys residues modified with iodoacetamide to prevent disulfide formation. LC-MS/MS analysis confirms site-specific incorporation of each unnatural amino acid into peptide U1 in the correct order. Predicted and observed ions are summarized in Tables S3–S5.
We tested the peptides derived from the natural and unnatural peptide libraries for target binding. We measured the affinity of these in vitro translated cyclic peptides, labeled with 35S-Cys, for thrombin using an equilibrium ultrafiltration binding assay.42 Unnatural peptides U1 and U2 bind with Kd = 4.5 and 20, nM respectively, and the natural peptide winner N1 binds with Kd = 1.5 nM (Figures 5 and S7–S9; Table 1). To test the role of the unnatural residues in target recognition, we substituted the unnatural amino acids with their natural counterparts. As expected, the peptides lose all measurable binding activity when the unnatural amino acids are replaced with their natural counterparts (Figures 5, S3, S4, and S8; Table 1). Thus, the unnatural amino acids are critical for activity and must result in target recognition through a different set of interactions than are made by the natural peptide. It is possible that the higher affinity of the natural peptide N1 reflects binding at a site on thrombin that has evolved to recognize a natural peptide ligand. Future biochemical and structural studies will allow a more detailed comparison of the binding modes of the natural and unnatural peptides.
Figure 5.

Binding of selection winners to thrombin. Binding curves for unnatural cyclic peptide U1 (closed circles), unnatural linear peptide U1 (open circles), and cyclic peptide U1 translated with natural amino acids (closed triangles). 35S-Cys-labeled peptides were incubated with varying concentrations of thrombin for 1 h. Cyclization via disulfide formation of the linear unnatural peptide U1 was prevented by the addition of 0.2 mM TCEP in the binding buffer. Bound and free peptides were separated using a 30 kDa MW cutoff spin-filter. The fraction of bound peptide fa (see Materials and Methods) was plotted against the concentration of thrombin and fit to a simple hyperbola to obtain the Kd values.
Table 1. Summary of Binding and Inhibition Constants of Selection Winners and Variants to Thrombin.
|
Kd (nM) |
Kiapp (nM) | |||
|---|---|---|---|---|
| peptide | cyclic | linear | natural | cyclic |
| U1 | 4.5 ± 0.8 | 350 ± 270 | >500 | 23 ± 3.3 |
| U2 | 20 ± 7 | >500 | >500 | 35 ± 19 |
| N1 | 1.5 ± 0.2 | 17 ± 8 | N.A. | 6.3 ± 3.8 |
Macrocyclization is believed to increase the affinity of peptides to their target by decreasing the entropic cost of binding.31 To determine the effect of macrocyclization of the peptides on binding affinity, we compared the Kd valus of the linear and cyclic translated 35S-labeled peptides (Figures S3–S5). Cyclization of peptides U1, U2, and N1 contributes significantly to their affinity to thrombin, as demonstrated by an 11–80-fold increase in the Kd values for the linear peptides relative to the corresponding cyclic peptides (Figures 5, S8, and S9; Table 1). Interestingly, the affinities of the unnatural peptides show a larger dependence on cyclization than that of the natural peptide. Thus, structural constraint is more important for the activity of the selected unnatural peptides U1 and U2 than for that of N1, suggesting that the linear unnatural peptides might be more disordered and less likely to form well-defined structures than the linear natural peptide.
Next, we determined the ability of the selected peptides to inhibit the enzymatic activity of thrombin. Peptides U1, U2, and N1 were in vitro translated in the presence of 35S-Cys, followed by cyclization and purification. Thrombin was preincubated with varying concentrations of macrocycles U1, U2, or N1 (as determined by scintillation counting) and assayed for enzymatic activity using a small, internally quenched FRET substrate of thrombin (AnaSpec). Inhibition is observed for all macrocyclic peptides. Fitting of the data to the Morrison equation for tight-binding inhibitors yielded Kiapp values of 23, 35, and 6.3 nM for peptides U1, U2, and N1, respectively (Figure 6; Table 1). These values are in excellent agreement with the observed Kd values. Although we did not select for enzyme inhibitors, we obtained potent tight-binding inhibitors of thrombin. It will be interesting to determine where these inhibitors bind to inhibit enzymatic activity. Since we expect target recognition of the unnatural peptides through a different set of interactions than are made by the natural peptide, the mechanism of enzymatic inhibition also might be distinct.
Figure 6.
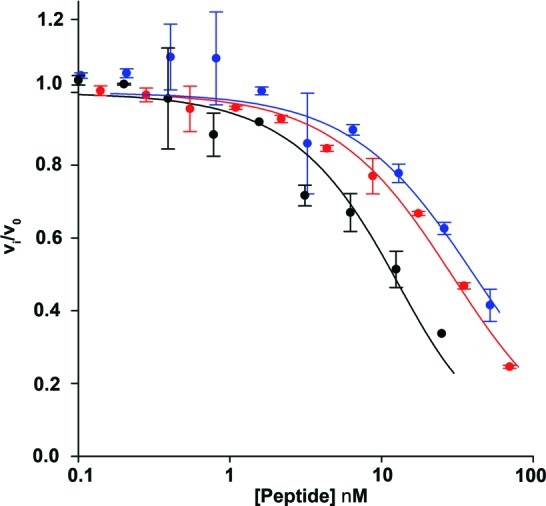
Inhibition of thrombin activity. Inhibition curves for unnatural cyclic peptide U1 (red), unnatural cyclic peptide U2 (blue), and natural cyclic peptide N1 (black). Thrombin was preincubated with varying concentrations of 35S-Cys labeled peptides for 1 h. The reaction was started by the addition of a 10mer peptide substrate (AnaSpec), and the increase in fluorescence was monitored for 1 h. Initial rates were determined, and the fraction of remaining enzymatic activity was plotted against the concentration of peptide and fit to the Morrison equation for tight-binding inhibitors to obtain Kiapp. All experiments were done at least in duplicate.
Conclusions
Our work shows that in vitro selections can be used to evolve small macrocyclic unnatural peptides that bind with high affinity to a target and inhibit its enzymatic activity from very large unbiased libraries (1013). Our work, in conjunction with the recently published selection of N-methyl amino acid containing cyclic peptides, shows that selections of natural-product-like molecules that contain a wide range of side-chain and backbone modifications should be feasible. An important advantage of our system is that very large libraries of bioactive molecules can be constructed and screened in the laboratory with basic molecular biology and biochemistry tools. Since these selections are affinity based, they require only small amounts of material and moderate amount of labor. This is in stark contrast to traditional small-molecule high-throughput screening methods where only small libraries can be constructed and screened. In addition, most laboratories are restricted to commercially available libraries, sampling a rather narrow chemical space. To our knowledge, the only other successful approach for the discovery of novel bioactive macrocycles using in vitro selections used DNA-encoded libraries,43 from which the most potent isolated molecule was found to have a Ki of 700 nM. Again, this method is not available to most laboratories since these libraries have to be chemically synthesized. In addition, since these DNA encoded libraries are chemically synthesized, they tend to be small (1.4 × 104).
Based on the known activities of structurally constrained peptides such as NRPs, it is likely that such highly modified peptides will be useful in modulating challenging targets that have been recalcitrant to small-molecule inhibition or activation. A major question for the future is whether the structural and functional diversity offered by unnatural amino acids will ultimately allow for sufficient bioavailability of cyclic unnatural peptides to enable therapeutic applications. The known resistance of cyclic peptides to protease digestion may be further enhanced by the replacement of natural amino acids with analogues that decrease recognition by proteases, while backbone modifications such as N-methylation may enhance cell permeability. Future advances in the technology of unnatural peptide synthesis by ribosomal translation will lead to the ability to incorporate a much larger array of modifications into peptides. The flexizyme system,44 a highly evolved ribozyme that is used to charge unnatural amino acids onto tRNAs, has already facilitated the incorporation of α-hydroxy acids, N-methyl amino acids, and other unusual building blocks into peptides by ribosomal peptide synthesis.25,45 Ultimately the engineering of EF-Tu and the ribosome itself46 may allow the direct incorporation of even more diverse building blocks into modified peptides by in vitro translation. In the meantime, many modifications can be introduced by post-translational derivatization. For example, peptides can be simultaneously cyclized and labeled with a wide range of Cys-alkylating agents to attach fluorophores, lipids, and other small molecules.47 Amino acids with electrophilic side-chain warheads such as dehydroalanine, which is often found in NRPs, can be used to covalently attach new functional groups or to cyclize the peptide in a manner analogous to lantibiotic biosynthesis.20,48−50 Peptides containing alkynes and azides can also be cyclized or modified by the incorporation of virtually any functional group,19,46,51 such as pegylation, which results in lower renal clearance, superior stability in vivo, and lower immunogenicity.52
As more selections with diverse building blocks are completed, it may become possible to discern whether certain amino acids are generally beneficial for the directed evolution of drug-like molecules, or if the choice of amino acids should be target specific. Ultimately it may be possible to design libraries of unnatural cyclic peptides that are predisposed to favor oral availability, good pharmacokinetics, and low toxicity, so that molecules with good drug-like properties are already present in the starting pool. If this potential can be realized, the ability to evolve macrocyclic peptide libraries will be a powerful method for the discovery of novel therapeutics.
Materials and Methods
Unnatural Amino Acid Optimization on mRNA Library
The PURE system and standard translation assays were previously described, and detailed methods can be found in the Supporting Information (SI).19,22 Based on previous work in which we identified amino acid analogues that could be incorporated into peptides,22 we selected a set of 12 unnatural amino acids along with 8 natural amino acids for optimization. Two templates from the library (see Figure 2) were chosen for the optimization reactions. The concentrations of the unnatural amino acids were varied and the results analyzed using the peptide translation assay. Peptide yields were determined by 35S-Cys incorporation, and purity was assessed by mass spectrometry on a Applied Biosystems Voyager MALDI-TOF with delayed extraction operated in the positive mode (Figure 2). The purity of the unnatural peptides produced improved markedly after optimization; in addition, the yield of the translated mRNA library improved from 10% of the natural amino acid translation to 50% after optimization. In particular, the proline analogue Pa was a translation inhibitor at concentrations above 20 μM, and the leucine analogue La required addition at a high concentration (6.6 mM) to prevent premature translation truncation. Optimized concentrations are summarized in Table S1.
Cyclization of Individual Library Members
Several members of the naïve library were chosen at random from sequences that had no stop codons and no cysteines in the random region. In vitro translated and Ni-NTA purified (see SI) peptides were desalted by Zip-Tip and eluted with 6 μL of 1:1 CH3CN:0.1% TFA. The resulting peptides (3 μL) were used in cyclization reactions (10 μL) containing 20 mM NH4CO3 pH 8.6, 200 μM tris-carboxyethylphosphine (TCEP), 1.1 mM dibromoxylene, and 25% CH3CN. Each reaction also contained 10 mM TRH-SH Pro (American Peptide Co.) as a positive control for cyclization and as a MALDI-MS standard. After 60 min, 1 μL of the reaction mixture was mixed with 1 μL of 10 mg/mL CHCA matrix 1:1 CH3CN:0.1% TFA. This mixture was spotted directly onto a MALDI plate for MS analysis.
Cyclization of the Peptide Library
In vitro translations were performed as described above with natural amino acids (250 μL) or unnatural amino acids (500 μL) using the mRNA CX10C library as a template. The peptide products were purified using Ni-NTA and eluted with 50 μL of 1% TFA. After Zip-tip desalting and eluting in 4 μL of 1:1 CH3CN:0.1% TFA, 0.5 μL (natural) or 1 μL (unnatural) of these solutions were analyzed by MALDI-TOF MS. The Zip-tipped peptide libraries were added to a reaction mixture containing final concentrations of 20 mM NH4CO3 pH 8.6, 200 μM TCEP, 1.1 mM dibromoxylene (Fluka), and 25% CH3CN. After 25 min, the reaction mixture was combined with CHCA matrix solution 1:9 and analyzed by MALDI-TOF MS.
mRNA Display: In Vitro Selection and Evolution
Materials used for mRNA display can be found in the SI. The peptide–mRNA fusions were produced by translating mRNA photochemically ligated to a puromycin linker (0.57 μM cross-linked mRNA, 1.14 μM total mRNA) in 5 or 10 mL standard translation reactions with either natural or unnatural amino acids respectively for 1 h at 37 C.19,53 Following the translation reaction, the KCl and Mg(OAc)2 concentrations were adjusted to 550 and 50 mM, incubated an additional 15 min at room temperature, and then frozen overnight at −30 °C. The resulting mRNA–puromycin–peptide fusions were diluted 10-fold for the natural peptides and 5-fold for the unnatural peptides into oligo(dT) cellulose binding buffer (10 mM EDTA, 1 M NaCl, 0.5 mM TCEP, 20 mM Tris, pH 8, 0.2% w/v Triton X-100), and this mixture was incubated with 10 mg mL–1 oligo(dT) cellulose (New England Biolabs) for 15 min at 4 °C with rotation. The mixture was transferred into several 20 mL Econo-Pac chromatography columns (BioRad). The eluate was passed through the columns two more times, and then each column was washed with 2 × 10 mL of wash buffer (0.3 M NaCl, 0.5 mM TCEP, 20 mM Tris, pH 8, 0.1% w/v Triton X-100). The immobilized fusions were then cyclized by the addition of 3 mL of cyclization buffer (660 mM NaCl, 10 mM Tris pH 8, 3.3 mM dibromoxylene, 0.5 mM TCEP, 33% CH3CN) to each column. The columns were washed twice with 4 mL of wash buffer (20 mM Tris pH 8, 300 mM NaCl, 0.5 mM TCEP) and eluted with 8 × 375 μL of water. Fractions with the highest amount of radioactivity (3–5) were combined, spin-filtered, and ethanol precipitated using 0.1 vol of 3 M KOAc, pH 5.2, 0.002 vol of glycogen (5 mg/mL), and 3 vol of ethanol. Pellets were resuspended in 800 μL of water and reverse transcribed in a final volume of 2 mL of RT-mix (0.5 μM RT-primer 5′-TTTTTTTTTTTTTTTGTGATGGTGATGGTGGCCTAAGC-3′, 0.5 mM dNTPs, 5 μM DTT, 2 U/μL RNaseOUT, 5 U/μL Superscript III) in RT buffer at 55 °C for 15 min. The reaction mixture was then diluted with 1.5 mL of denaturing binding buffer (100 mM NaH2PO4, 10 mM Tris, 6 M guanidinium hydrochloride, 0.2% Triton X-100, 5 mM BME, pH 8.0) and combined with 1.25 mL of Ni-NTA beads (bead volume) and an additional 5.5 mL of denaturing binding buffer in a 20 mL BioRad column. The column was tumbled for 1 h at 4 °C, washed with 2 × 10 mL of wash buffer (100 mM NaH2PO4, 300 mM NaCl, 0.2% Triton X-100, 5 mM BME, pH 8.0), and eluted with native elution buffer (50 mM NaH2PO4, 300 mM NaCl, 250 mM imidazole, 0.2% Triton X-100, 5 mM BME, pH 8.0) in 500 μL fractions. Fractions with the highest amount of fusions (3–6) were combined and ethanol precipitated with 3 vol of ethanol. The pellets were resuspended in 1.5 mL of 1× selection buffer (50 mM Tris, pH 7.8, 100 mM NaCl, 4 mM MgCl2, 0.25%Triton X-100) to yield 31 pmol of natural and 39 pmol of unnatural purified mRNA-displayed peptides, equivalent to 1.9 × 1013 natural and 2.3 × 1013 unnatural peptides. These were used for the first round of selection.
Next, 200 pmol (23 NIH units) of biotinylated thrombin (Novagen) was added to the peptide fusions and allowed to bind for 1 h at room temperature. Complexes were captured on agarose streptavidin beads (Pierce Ultralink streptavidin beads) for 2.5 min and washed with 3 × 2 mL of selection buffer. Thrombin-bound sequences were eluted with 2 mL of selection buffer containing 43 units of hirudin (Sigma) by incubating for 1 h and collecting the flow-through. The beads were washed 2 more times with 150 μL of buffer. The flow-through and first wash were combined, yielding 0.74 pmol of natural and 0.8 pmol of unnatural peptide fusions in 2.15 mL. This material was diluted 2-fold with water and PCR amplified, using 19.8 μL of fusions in each 100 μL PCR reaction. PCR reactions were chloroform phenol extracted, concentrated with 1-butanol, and ethanol precipitated using 0.1 vol of 3 M KOAc, 2 vol of ethanol, and 0.001 vol of glycogen (5 mg/mL). The pellets were dissolved in 1× transcription buffer to give a final concentration of ∼1.5 mg/mL of PCR product. T7 transcription reactions (0.5 mL) were set up overnight at 37 °C with a final concentration of 0.6 mg/mL PCR product. Transcription reactions were treated with 50 units of turbo DNase (Ambion) for 15 min at 37 °C, followed by gel purification on an 8% polyacrylamide gel. Purified RNA was processed as described above for the preparation of mRNA-displayed peptides for the next round of selection and amplification.
Every round was assayed by scintillation counting of the 35S-cysteine-labeled peptides to measure the efficiencies of the various steps. These data were then used to determine the number of purified individual peptide sequences introduced into the round 1 selection and subsequent rounds. This procedure was repeated for 10 rounds except for the following changes: in round 2 and in all subsequent rounds the volume of the translation reaction was 0.25 mL for the natural and 0.5 mL for the unnatural peptide selection. Reactions and purifications were scaled accordingly. Magnetic beads (Dynabeads M-280 Streptavidin, Invitrogen) were used for streptavidin capture of complexes. Starting in round 4 complexes were washed five times, and in round 7 the selection pressure was increased further by only amplifying mRNA–peptide fusions that remained bound to thrombin after 1 h of incubation with hirudin and were subsequently eluted during an overnight incubation with additional hirudin.
To analyze the results of the selection, cDNAs were cloned into the pCR-TOPO vector (TOPO TA Cloning, Invitrogen). Colonies were screened using X-gal and sent out to SeqWright for plasmid isolation and sequencing. To generate peptides from individual clones, plasmids (isolated from SeqWright) were amplified with primers CX10 FWD and CX10 REV and the PCR products used for T7 in vitro transcription. The resulting mRNAs were gel purified and used in translation reactions.
Sequence Analysis
For DNA and peptide sequence alignments, Jalview was used.54,55 Unnatural amino acids were assigned on the basis of the tRNA/aaRS pairs responsible for their incorporation into peptides.
Ribosomal Synthesis, Cyclization and Purification of Selection Winners, and LC-MS Analysis
Sequences were amplified from plasmids using PCR primers CX10 FWD and CX10 REV, the PCR product used for T7 transcription, and the resulting mRNAs gel purified. Translation reactions were set up as above, with minor modifications as described in the SI. Peptides were cyclized on the Ni-NTA resin in cyclization buffer (20 mM Tris pH 8.0, 100 mM NaCl, 0.2 mM TCEP, 5 mM dibromoxylene, 50% CH3CN) for 30 min at 25 °C and eluted with a 1:1 mixture of 0.1% TFA and CH3CN in fractions. Fractions with the highest amount of peptide were combined, spin-filtered through a YM-10 spin-filter, and concentrated. Peptides were analyzed by MALDI-TOF-MS, and spectra were calibrated using P14R (1532.8582 Da) and insulin B chain (3493.6513 Da) as the internal standards (Sigma).
For high-resolution MS analysis, the peptide solution was concentrated in a YM-3 spin-filter to ∼10 μL. The samples were reconstituted in 0.1% formic acid (FA), 0.2% CH3CN, and 10 mM EDTA to give a final volume of 40 μL. Peptides were analyzed by LC-MS on a 6520 Accurate-Mass Q-TOF LC/MS (Agilent) using a 2.1 mm × 100 mm, 3.5-μm ZORBAX 300SB-C18 column (Agilent). Samples were injected at 200 μL/min and eluted with a 10 min gradient of 2–100% CH3CN, 0.1% FA at 0.6 mL min–1. The mass spectrometer was operated in positive mode and calibrated with reference masses 121.050873 and 922.009798 Da.
Ribosomal Synthesis, Iodoacetamide Modification and Purification of Selection Winners with His Tag Deletions, and LC-MS/MS Analysis
Sequences were amplified from the plasmids containing the full-length transcripts for U2 and U1 using PCR primers CX10 FWD and the corresponding reverse primer for the His deletion (U1-HisR, 5′-CTACTAGCCTAAGCTACCGGAGCCGCATCC-3′; U2-HisR, 5′-CTACTAGCCTAAGCGACCGGAGCCGC-3′). The PCR product was gel purified and used for T7 transcription. The resulting mRNAs were gel purified and used in subsequent translation reactions. Translations were set up as before and purified with minor modifications as described in the SI. Cys residues were modified with iodoacetamide, purified, and analyzed by LC-MS/MS. Samples were run as described above. The mass spectrometer was operated in positive mode, and ions were selected for MS/MS.
Kd Determination of Translated Peptides
Kd values for thrombin were determined by equilibrium ultrafiltration42 as described in the SI.
Kiapp Determination of Translated Peptides
Kiapp values for the inhibition of thrombin by the selected peptides were determined using a fluorescence-based assay (AnaSpec) as described in the SI.
Acknowledgments
This research was supported by the Howard Hughes Medical Institute. J.W.S. is an investigator of the Howard Hughes Medical Institute. We thank Florian Seebeck, Buckhard Seelig, Chi-Wang Lin, Doug Treco, Alonso Ricardo, Craig Blain, Matt Simon, Jason Schrum, Christian Hentrich, and Katarzyna Adamala for helpful discussions.
Supporting Information Available
Detailed materials and methods; supplemental figures, tables, and schemes. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare the following competing financial interest(s): Employment: K. Josephson is employed by Ra Pharmaceuticals, which is further developing the technology described in this paper. Ra Pharmaceuticals may benefit from the publication of this paper. Personal financial interests: J. W. Szostak, K. Josephson, and M. C. T. Hartman hold shares in Ra Pharmaceuticals; J. W. Szostak and M. C. T. Hartman receive consulting fees from Ra Pharmaceuticals.
Author Present Address
† Department of Chemistry and Massey Cancer Center, Virginia Commonwealth University, Richmond, VA 23284-2006
Author Present Address
‡ Ra Pharmaceuticals, Suite B14301, One Kendall Square, Cambridge, MA 02139
Supplementary Material
References
- Verdine G. L.; Walensky L. D. Clin. Cancer Res. 2007, 13, 7264. [DOI] [PubMed] [Google Scholar]
- Reichert J. M.; Rosensweig C. J.; Faden L. B.; Dewitz M. C. Nat. Biotechnol. 2005, 23, 1073. [DOI] [PubMed] [Google Scholar]
- Malik N. N. Nat. Rev. Clin. Oncol. 2009, 6, 550. [DOI] [PubMed] [Google Scholar]
- Lipinski C. A. J. Pharmacol. Toxicol. Methods 2000, 44, 235. [DOI] [PubMed] [Google Scholar]
- Swinney D. C.; Anthony J. Nat. Rev. Drug Discov. 2011, 10, 507. [DOI] [PubMed] [Google Scholar]
- Hopkins A. L.; Groom C. R. Nat. Rev. Drug Discov. 2002, 1, 727. [DOI] [PubMed] [Google Scholar]
- Russ A. P.; Lampel S. Drug Discov. Today 2005, 10, 1607. [DOI] [PubMed] [Google Scholar]
- Koehn F. E.; Carter G. T. Nat. Rev. Drug Discov. 2005, 4, 206. [DOI] [PubMed] [Google Scholar]
- Driggers E. M.; Hale S. P.; Lee J.; Terrett N. K. Nat. Rev. Drug Discov. 2008, 7, 608. [DOI] [PubMed] [Google Scholar]
- Walensky L. D.; Kung A. L.; Escher I.; Malia T. J.; Barbuto S.; Wright R. D.; Wagner G.; Verdine G. L.; Korsmeyer S. J. Science 2004, 305, 1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moellering R. E.; Cornejo M.; Davis T. N.; Del Bianco C.; Aster J. C.; Blacklow S. C.; Kung A. L.; Gilliland D. G.; Verdine G. L.; Bradner J. E. Nature 2009, 462, 182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C. C.; Mack A. V.; Tsao M. L.; Mills J. H.; Lee H. S.; Choe H.; Farzan M.; Schultz P. G.; Smider V. V. Proc. Natl. Acad. Sci. U.S.A. 2008, 105, 17688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C. C.; Mack A. V.; Brustad E. M.; Mills J. H.; Groff D.; Smider V. V.; Schultz P. G. J. Am. Chem. Soc. 2009, 131, 9616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S.; Millward S.; Roberts R. J. Am. Chem. Soc. 2002, 124, 9972. [DOI] [PubMed] [Google Scholar]
- Millward S. W.; Fiacco S.; Austin R. J.; Roberts R. W. ACS Chem. Biol. 2007, 2, 625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts R. W.; Szostak J. W. Proc. Natl. Acad. Sci. U.S.A. 1997, 94, 12297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson D. S.; Keefe A. D.; Szostak J. W. Proc. Natl. Acad. Sci. U.S.A. 2001, 98, 3750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu Y.; Inoue A.; Tomari Y.; Suzuki T.; Yokogawa T.; Nishikawa K.; Ueda T. Nat. Biotechnol. 2001, 19, 751. [DOI] [PubMed] [Google Scholar]
- Josephson K.; Hartman M. C. T.; Szostak J. W. J. Am. Chem. Soc. 2005, 127, 11727. [DOI] [PubMed] [Google Scholar]
- Seebeck F. P.; Szostak J. W. J. Am. Chem. Soc. 2006, 128, 7150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartman M. C. T.; Josephson K.; Szostak J. W. Proc. Natl. Acad. Sci. U.S.A. 2006, 103, 4356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartman M. C.; Josephson K.; Lin C. W.; Szostak J. W. PLoS One 2007, 2, e972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subtelny A. O.; Hartman M. C.; Szostak J. W. J. Am. Chem. Soc. 2008, 130, 6131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forster A. C.; Tan Z.; Nalam M. N.; Lin H.; Qu H.; Cornish V. W.; Blacklow S. C. Proc. Natl. Acad. Sci. U.S.A. 2003, 100, 6353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawakami T.; Murakami H.; Suga H. J. Am. Chem. Soc. 2008, 130, 16861. [DOI] [PubMed] [Google Scholar]
- Kawakami T.; Murakami H.; Suga H. Chem. Biol. 2008, 15, 32. [DOI] [PubMed] [Google Scholar]
- Ohta A.; Murakami H.; Higashimura E.; Suga H. Chem. Biol. 2007, 14, 1315. [DOI] [PubMed] [Google Scholar]
- Kawakami T.; Ohta A.; Ohuchi M.; Ashigai H.; Murakami H.; Suga H. Nat. Chem. Biol. 2009, 5, 888. [DOI] [PubMed] [Google Scholar]
- Yamagishi Y.; Shoji I.; Miyagawa S.; Kawakami T.; Katoh T.; Goto Y.; Suga H. Chem. Biol. 2011, 18, 1562. [DOI] [PubMed] [Google Scholar]
- Sato A. K.; Viswanathan M.; Kent R. B.; Wood C. R. Curr. Opin. Biotechnol. 2006, 17, 638. [DOI] [PubMed] [Google Scholar]
- Adessi C.; Soto C. Curr. Med. Chem. 2002, 9, 963. [DOI] [PubMed] [Google Scholar]
- Cane D. E.; Walsh C. T.; Khosla C. Science 1998, 282, 63. [DOI] [PubMed] [Google Scholar]
- Timmerman P.; Beld J.; Puijk W. C.; Meloen R. H. Chembiochem 2005, 6, 821. [DOI] [PubMed] [Google Scholar]
- Heinis C.; Rutherford T.; Freund S.; Winter G. Nat. Chem. Biol. 2009, 5, 502. [DOI] [PubMed] [Google Scholar]
- Raffler N. A.; Schneider-Mergener J.; Famulok M. Chem. Biol. 2003, 10, 69. [DOI] [PubMed] [Google Scholar]
- Kubik M. F.; Stephens A. W.; Schneider D.; Marlar R. A.; Tasset D. Nucleic Acids Res. 1994, 22, 2619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tasset D. M.; Kubik M. F.; Steiner W. J. Mol. Biol. 1997, 272, 688. [DOI] [PubMed] [Google Scholar]
- Di Nisio M.; Middeldorp S.; Buller H. R. N. Engl. J. Med. 2005, 353, 1028. [DOI] [PubMed] [Google Scholar]
- Bagdy D.; Barabas E.; Graf L.; Petersen T. E.; Magnusson S. Methods Enzymol. 1976, 45, 669. [DOI] [PubMed] [Google Scholar]
- Stone S. R.; Hofsteenge J. Biochemistry 1986, 25, 4622. [DOI] [PubMed] [Google Scholar]
- Maryanoff B. E.; Qiu X.; Padmanabhan K. P.; Tulinsky A.; Almond H. R. Jr.; Andrade-Gordon P.; Greco M. N.; Kauffman J. A.; Nicolaou K. C.; Liu A.; Brungs P. H. Proc. Natl. Acad. Sci. U.S.A. 1993, 90, 8048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenison R. D.; Gill S. C.; Pardi A.; Polisky B. Science 1994, 263, 1425. [DOI] [PubMed] [Google Scholar]
- Kleiner R. E.; Dumelin C. E.; Tiu G. C.; Sakurai K.; Liu D. R. J. Am. Chem. Soc. 2010, 132, 11779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami H.; Ohta M.; Ashigai H.; Suga H. Nat. Methods 2006, 3, 357. [DOI] [PubMed] [Google Scholar]
- Ohta A.; Yamagishi Y.; Suga H. Curr. Opin. Chem. Biol. 2008, 12, 159. [DOI] [PubMed] [Google Scholar]
- Neumann H.; Wang K.; Davis L.; Garcia-Alai M.; Chin J. W. Nature 2010, 464, 441. [DOI] [PubMed] [Google Scholar]
- Dewkar G. K.; Carneiro P. B.; Hartman M. C. Org. Lett. 2009, 11, 4708. [DOI] [PubMed] [Google Scholar]
- Levengood M. R.; van der Donk W. A. Nat. Protoc. 2006, 1, 3001. [DOI] [PubMed] [Google Scholar]
- Goto Y.; Iwasaki K.; Torikai K.; Murakami H.; Suga H. Chem. Commun. (Camb.) 2009, 3419. [DOI] [PubMed] [Google Scholar]
- Seebeck F. P.; Ricardo A.; Szostak J. W. Chem. Commun. (Camb.) 2011, 47, 6141. [DOI] [PubMed] [Google Scholar]
- Deiters A.; Cropp T. A.; Summerer D.; Mukherji M.; Schultz P. G. Bioorg. Med. Chem. Lett. 2004, 14, 5743. [DOI] [PubMed] [Google Scholar]
- Harris J. M.; Chess R. B. Nat. Rev. Drug Discov. 2003, 2, 214. [DOI] [PubMed] [Google Scholar]
- Kurz M.; Gu K.; Lohse P. A. Nucleic Acids Res. 2000, 28, E83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterhouse A. M.; Procter J. B.; Martin D. M.; Clamp M.; Barton G. J. Bioinformatics 2009, 25, 1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clamp M.; Cuff J.; Searle S. M.; Barton G. J. Bioinformatics 2004, 20, 426. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.