


Abstract
Translation termination in eukaryotes is catalyzed by two release factors eRF1 and eRF3 in a cooperative manner. The precise mechanism of stop codon discrimination by eRF1 remains obscure, hindering drug development targeting aberrations at translation termination. By solving the solution structures of the wild-type N-domain of human eRF1 exhibited omnipotent specificity, i.e. recognition of all three stop codons, and its unipotent mutant with UGA-only specificity, we found the conserved GTS loop adopting alternate conformations. We propose that structural variability in the GTS loop may underline the switching between omnipotency and unipotency of eRF1, implying the direct access of the GTS loop to the stop codon. To explore such feasibility, we positioned N-domain in a pre-termination ribosomal complex using the binding interface between N-domain and model RNA oligonucleotides mimicking Helix 44 of 18S rRNA. NMR analysis revealed that those duplex RNA containing 2-nt internal loops interact specifically with helix α1 of N-domain, and displace C-domain from a non-covalent complex of N-domain and C-domain, suggesting domain rearrangement in eRF1 that accompanies N-domain accommodation into the ribosomal A site.
INTRODUCTION
One of the key steps in translation termination is to decode the stop codon within the ribosomal A site by class I polypeptide chain release factor (1,2). The two class I release factors in prokaryotes possess different decoding capability: RF1 recognizes exclusively UAA and UAG as stop codons, while RF2 terminates translation at UAA and UGA. The ‘tripeptide anticodon’ hypothesis was first proposed as a mechanism of stop codon recognition, in which the Pro-Ala/Val-Thr (P × T) loop in RF1 and the Ser-Pro-Phe (SPF) loop in RF2 decode the second and third stop codon positions via direct interactions (3,4). However, the high-resolution crystal structures of RF1-bound (5,6) and RF2-bound (7,8) ribosome, including their respective cognate stop codons, revealed a significantly more complex mode of stop codon recognition. These structures were then used in molecular dynamics free-energy calculations to elaborate on a potential mechanism of stop codon discrimination between RF1 and RF2 (9).
Eukaryotic and archaeal class I release factors, eRF1 and aRF1, are both omnipotent and share high sequence similarity with each other but do not possess any obvious sequence homology with their bacterial counterparts (2). Structurally, eRF1 is composed of three distinct protein domains (10–12) performing specific roles: N-domain recognizes the stop codon at the decoding site of the 40S subunit, M-domain triggers hydrolysis of the peptidyl-tRNA ester bond in the peptidyl transferase center (PTC), while C-domain forms a complex interface with class II release factor eRF3 (2). Despite availability of the crystal structure of full-length eRF1 (10), the mechanism of stop codon recognition remains obscure. Over the last decade, various experimental approaches led to different models. Among these models, the TASNIKS (13,14) and YxCxxxF motifs (15) in N-domain of eRF1 were proposed to play a role in stop codon recognition, forming the so-called non-linear model. In particular, experiments on photoactivatable cross-linking of a modified stop codon to N-domain in a pre-termination complex pinpointed the NIKS motif as being positioned in proximity to the first uridine, and the YxCxxxF motif in proximity to the purines in the second and third stop codon positions (16,17). Other models, known as the cavity models, were proposed based on a set of point mutations found to affect stop codon readthrough in yeast. In these models, individual nucleotides of the stop codon are accommodated into three defined cavities on the surface of N-domain (18,19). Although both the non-linear and cavity models share some residues implicated in stop codon recognition, they are not entirely compatible, thus requiring further experiments to resolve.
Investigation of stop codon recognition in eukaryotes has been making progress by studying eRF1 of variant-code organisms, in which one or two of the universal stop codons are reassigned to sense codon(s) (20,21). Chimeric eRF1s composed of N-domain from some of the variant-code organisms (ciliates) and the MC-domain of eRF1 from universal-code organisms (human or yeast) retain stop codon specificity typical for ciliate eRF1s, demonstrating unequivocally that N-domain of eRF1 directly decodes the stop codon (13,22–24). From these efforts, a mutant of human eRF1 with substitutions in N-domain corresponding to eRF1 of Stylonychia (ciliate) with UGA-only specificity (T122Q, S123F, L124M and L126F) was shown to exhibit strong UGA unipotency (23). By solving the solution structures of N-domain of both the wild-type (wt) eRF1 and the UGA-unipotent mutant (denoted as Q122FM(Y)F126 henceforth), we found that those point mutations, while preserving the global structure of N-domain, alter conformation of the strictly conserved GTS loop (positions 31–33) remote from the mutation sites. This indicates that switching between omnipotency and unipotency of eRF1 may be modulated by distinct conformations of the GTS loop.
Both N-domain and M-domain of eRF1 possess putative ribosomal binding sites (2). The genetic interactions between eRF1 and the decoding region of Helix 44 of 18S rRNA in the 40S subunit were reported (25). In the attempt to investigate whether direct interaction between N-domain and the decoding region of Helix 44 is possible, we utilized a 15-mer RNA oligonucleotide that contains internal loops, mimicking the decoding region of Helix 44 (26–29). By generating different mutants of the 15-mer RNA, we obtained constructs with different sizes of internal loop, and found that presence of a 2-nt internal loop is critical for strong binding to N-domain. These results indicate that helix α1 of N-domain potentially interacts with Helix 44 in the 40S subunit. Interestingly, the RNA-binding region is shielded partially in solution by C-domain, as observed from the crystal structure of full-length eRF1 (10). In solution, the 15-mer RNA is able to displace C-domain from the non-covalent complex of N-domain and C-domain, suggesting an imperative domain rearrangement in eRF1 during which N-domain accommodates itself into the ribosomal A site. A manually docked model of eRF1 N-domain onto the A site of the 40S subunit supports the structural plausibility of our findings.
MATERIALS AND METHODS
Expression and purification of protein samples
The DNA fragments encoding the wild-type and the Q122FM(Y)F126 mutant of N-domain (residue 1–142), the NM-domain (residues 1–275), and C-domain (residues 275–437) of human eRF1 with a C-terminal hexahistidine tag were cloned into pET23(+) vector (Novagen) under the phage T7 RNA-polymerase promoter (30,31), and were expressed in Escherichia coli Rosetta(DE3) host cells. Uniformly 13C, 15N-labeled wt N-domain and Q122FM(Y)F126 were produced in minimal media (M9) utilizing 15NH4Cl (1.0 g/l) and 13C6-glucose (2.0 g/l) as the sole nitrogen and carbon sources. Uniformly, 2H, 13C, 15N-labeled NM-domain was produced using 2H2O (99% d-enrichment) as the solvent. All recombinant proteins were purified from cell lysates utilizing a 5 ml HisTrap HP column (GE Healthcare), and further purified using three 5 ml HiTrap desalting columns (GE Healthcare) connected in series. NMR samples contained 0.1–1.0 mM protein in 20 mM MES, 100 mM KCl, 2 mM DTT at pH 6.0. For all experiments involving C-domain, 3 mM of β-mercaptoethanol was added to prevent oxidation of the cysteine thiol groups.
Cloning and point mutagenesis of chimeric Euplotes aediculatus/human and Stylonychia mytilus/human eRF1 genes, expression and purification of chimeric eRF1 proteins were described (23,24).
In vitro radio frequency (RF) activity assay
The eRF1 activity was measured in an in vitro system suggested by Caskey et al. (32). Rabbit reticulocyte ribosomes were isolated and purified as described (15).
NMR spectroscopy
All NMR spectra were acquired using 600, 700 or 900 MHz Bruker Avance II spectrometers. Chemical shifts were referenced to 2,2-dimethyl-2-silapentane-5-sulfonate (DSS) directly for 1H and indirectly for 13C and 15N spins. The NMR data were processed using TopSpin 2.0 (www.bruker-biospin.com) and analyzed using CARA (www.nmr.ch). 1H, 15N and 13C resonances of wt N-domain, Q122FM(Y)F126, and NM-domain were assigned using 3D TROSY-HNCA and TROSY-HNCACB. Side-chain 1H and 13C were assigned using iterative analysis of the 3D 15N-NOESY-HSQC and 13C-NOESY-HMQC spectra coupled with structure calculations. The assignment process was facilitated by comparison with chemical shifts deposited in the Biological Magnetic Resonance Data Bank (www.bmrb.wisc.edu) for individual domains (33–35). Reverse labeling of phenylalanine (36) and the dual amino acid-selective 13C–15N labeling technique (37,38) were employed to resolve ambiguous assignments in Q122FM(Y)F126. Residual dipolar couplings of wt N-domain and Q122FM(Y)F126 were abstracted from the chemical shifts of TROSY and anti-TROSY cross-peaks in isotropic and anisotropic solvent conditions, respectively. Partial alignment of the proteins was induced by addition of 10 mg/ml bacteriophage Pf1 (Hyglos GmbH). The axiality and rhombicity of the alignment tensor were calculated using PALES (39). Transverse relaxation time (T2) were measured with eight relaxation delays, i.e. 12.5, 25, 50, 62.5, 87.5, 112.5, 156.25 and 200 ms. The spectra measuring 1H–15N Nuclear Overhauser Effect (NOE) were acquired with a 2-s relaxation delay, followed by a 3-s period of proton saturation. In the absence of proton saturation, the spectra were recorded with a relaxation delay of 5 s. The exponential curve fitting and data analysis were carried out using Origin (Origin Lab).
Structure calculations
NOE distance restraints for the calculated structures of wt N-domain and Q122FM(Y)F126 were obtained from 15N-NOESY-HSQC and 13C-NOESY-HMQC spectra, respectively. Backbone dihedral angle restraints (φ and ψ) were derived from the backbone 13C′, 13Cα, 13Cβ, 1Hα and 1Hβ chemical shift values using TALOS (40). Structure calculations were performed using CYANA 3.0 (41,42) and visualized using MOLMOL (43) and PyMOL (Delano Scientific). Quality of the final structures was assessed using PROCHECK-NMR (44).
The homology models of the N-domains of Euplotes and Stylonychia were calculated by the I-TASSER server (45,46). The Template Modeling (TM)-score of both models as compared to the structure of wt N-domain were calculated by the TM-align server (47).
Structural characterization of RNA
All of the synthesized RNA oligonucleotides were purchased from 1st BASE. The lyophilized RNA was dissolved in diethylpyrocarbonate-treated water, and diluted into the NMR buffer supplemented with 2–5 mM MgCl2. The RNA samples were incubated at 80°C for 2 min followed by cooling to room temperature. Chemical shift assignments of 1H from the nucleobases of 15-mer-UAA were based on the standard 2D NOESY (mixing time 350 ms), DQF-COSY and natural abundance 13C-HSQC all acquired at 25°C, as well as 2D sofast-15N-HMQC (48) without 15N-decoupling measured at 20°C. Assignments of 1H from the other RNA constructs were based on their respective 2D NOESY spectra. H1′ protons were only partially assigned. 1D 31P-spectra with 1H-decoupling were used to distinguish between double-stranded helical and stem-loop structures. In the N-domain/RNA titration experiments, the 1H resonances of RNA were monitored as a function of added 13C, 15N-labeled wt N-domain to RNA, and vice versa. Suppression of 1H resonances stemming from 13C-, 15N-labeled protein was achieved using isotope filtering NMR experiment. Chemical shift perturbations were calculated by CSP = [(ΔδH)2 + (0.14 × ΔδN)2]1/2. Binding affinity was obtained based on the chemical shift perturbation of N-domain using the Scatchard plot method (49).
RESULTS
The GTS loop of N-domain adopts distinct conformations
To understand the structural effect of the quadruple mutations (i.e. T122Q, S123F, L124M and L126F) that lead to unipotency for UGA in the mutant eRF1, we determined the solution structures of wt N-domain and Q122FM(Y)F126, and compared them with the corresponding crystal structure of N-domain in full-length human eRF1 (10). The solution structures were verified against measured residual dipolar couplings, and the structure determination statistics are reported in Table 1. As expected, the NMR solution structure of wt N-domain matches closely with the crystal structure of N-domain in full-length eRF1 (Supplementary Figure S2A). The only observed significant deviations are positioning of the N-terminal part of helix α3 and conformation of the GTS loop.
Table 1.
Structural statistics for the final 20 conformers of Q122FM(Y)F126
| NMR restraints | |
| Total unambiguous distance restraints | 3081 |
| Intra residual | 824 |
| Sequential (|i – j| = 1) | 715 |
| Short-range (|i − j| ≤ 1) | 1539 |
| Medium (2 ≤ |i − j| ≤ 4) | 619 |
| Long range (|i − j| ≥ 5) | 923 |
| Dihedral angle restraints | 256 |
| Hydrogen bond restraints | 75 |
| RMSD from the average atomic coordinates (residues 6–140, Å) | |
| Backbone atoms | 0.26 ± 0.05 |
| All heavy atoms | 0.68 ± 0.03 |
| Ramachandran analysis (%) | |
| Residues in most favoured regions | 81.3 |
| Residues in additional allowed regions | 18.7 |
| Residues in generously allowed regions | 0.0 |
| Residues in disallowed regions | 0.0 |
None of the structures exhibits distance violations >0.2 Å or dihedral angle violations >2°.
The global structure of Q122FM(Y)F126 is well super-imposable with wt N-domain (Figure 1A). Their structural differences are confined to β-strand β4 that contains the point mutations, as well as the GTS loop. Likewise, helix α3 of Q122FM(Y)F126 is also repositioned relative to wt N-domain (Figure 1A). Although the four point mutations are spatially remote from the GTS loop, structural alteration that occurred to the GTS loop in Q122FM(Y)F126 is evident from the difference in amide chemical shift for the GTS loop and several other nearby residues including C97 and T99 (Figure 1B and Supplementary Figure S2B). Furthermore, different GTS loop conformations are also confirmed by clear differences in the NOEs patterns between wt N-domain and Q122FM(Y)F126 (Supplementary Figure S2C). The alternative conformation of the GTS loop in Q122FM(Y)F126 is maintained via an intricate propagation of hydrogen bonding perturbations from the mutation sites situated at the beginning of β-strand β4 that constitutes the hydrophobic core of N-domain (Figure 1A). In wt N-domain, this β-strand forms a well-defined network of hydrogen bonds with the adjacent β-strand starting from L124 to D128, as seen from the alternating directions of the side-chains in consecutive residues. In Q122FM(Y)F126, the regular hydrogen-bonding network is disrupted starting from M124. Mutation L126F is critical for breaking the regularity, since the phenylalanine aromatic ring is found flipped to the opposite side of β-strand β4 relative to the side-chain position of L126, thus inflicting position change of C127. As the side-chain of C127 is moved away from the hydrophobic core formed between the β-strand β4, helix α2 and helix α3, the N-terminal part of helix α3 is repositioned closer to the GTS loop. The phenylalanine substitution at position 126 in three of the ciliates that are unipotent for UGA suggests that similar structural features may have causal effect on the UGA unipotency in those organisms (Supplementary Figure S1). Nevertheless, the structural alteration in Q122FM(Y)F126 is likely to be an additive effect from all of the point mutations (23).
Figure 1.

Structural comparison between wt N-domain and Q122FM(Y)F126. (A) Superposition of the solution structures of wt N-domain (green) and Q122FM(Y)F126 (blue). Regions that are structurally distinct between the two are highlighted in cyan (wt N-domain) and magenta (Q122FM(Y)F126). (B) Differences in amide chemical shift between wt N-domain and Q122FM(Y)F126 are calculated by ΔδNH = [(ΔδH)2 + (0.14*ΔδN)2]1/2, and are mapped onto the structure of wt N-domain according to the colour scale. The four mutated residues are denoted in italics. (C) Conformations of the GTS loop (residues N30–S33) found in the crystal structure of wt N-domain (left panel), the solution structure of wt N-domain (middle panel), and the solution structure of Q122FM(Y)F126 (right panel). Positions of the residues that form the hydrophobic core above the GTS loop are shown by their side-chains only. Hydrogen bonding between the hydroxyl group of S70 and the carbonyl oxygen of S33 in Q122FM(Y)F126 mutant is denoted by a dashed line. The distance between the hydrogen donor and the acceptor is 2.67 ± 0.09 Å.
Remarkably, the GTS loop adopts distinct conformations in all three situations, namely the crystal and solution structures of wt N-domain as well as the solution structure of Q122FM(Y)F126 (Figure 1C). This implies that the GTS loop has the flexibility to adopt different conformations even within wt N-domain. In fact, the GTS loop in the solution structures has rather defined conformations, as backbone RMSD of the loop region (N30–M34) in wt N-domain and Q122FM(Y)F126 are 0.32 ± 0.28 Å and 0.05 ± 0.02 Å, respectively. This suggests that the observed GTS loop conformations might represent end-states in a complex equilibrium of different conformations. Close inspection reveals that side-chains of the individual residues have different solvent exposure in three structures, hinting at the possibility that alternative GTS loop conformations expose different functional chemical groups for interactions with the stop codons. Functional implication of the GTS loop in stop codon recognition had been reported (50), since T32A and T32V eRF1 mutants tended to UGA unipotency. Furthermore, two individual point mutations in the GTS loop of eRF1, i.e. T32A and S33A, were found to exhibit opposite effects on their release activity measured in vitro using fully reconstituted eukaryotic translation system, namely 32% UAA/30% UAG/75% UGA and 100% UAA/90% UAG/63% UGA, respectively (E. Alkalaeva, personal communication). Coincidentally, the side-chain of T32 in the structure of Q122FM(Y)F126 is hidden from the solvent, suggesting that T32 is not required for interacting with UGA (right panel in Figure 1C).
We found that in Q122FM(Y)F126, but not in wt N-domain, the resonance stemming from the hydroxyl proton of S70 can be observed (Supplementary Figure S2D), protected by potential hydrogen bonding to the carbonyl oxygen of S33 (Figure 1C). Hence, the structure of Q122FM(Y)F126 indicates that S70 might involve in stabilization of the GTS loop in an alternative conformation.
The 15N-relaxation rates of wt N-domain and Q122FM(Y)F126 have shown that the GTS loop is relatively more dynamic on the sub-nanosecond timescale than the bulk of N-domain (Figure 2). It was reported that the ligand-binding sites are often found at or close to the flexible regions in protein (51,52). It is interesting to note that, in spite of increased dynamics, the NOEs- and RDCs-derived averaged structure of the GTS loop is significantly different in wt N-domain and Q122FM(Y)F126, implying rather complex equilibrium between conformations conferring different functionality. We suggest that the observed the GTS loop ‘hotspot’ may provide necessary flexibility needed to switch between differently functionalized conformations upon interaction with the stop codon. Apparently, the dynamic properties of wt N-domain and Q122FM(Y)F126 do not differ significantly from each other (Figure 2). Hence, this led us to conclude that although the switching between omnipotency and unipotency of eRF1 can be sufficiently explained by the alteration of the GTS-loop conformation, it is not reflected by the fast dynamics.
Figure 2.

The dynamic properties of wt N-domain and Q122FM(Y)F126. Plots of the longitudinal relaxation rate R1 (A), the transverse relaxation rate R2 (B) and the heteronuclear 15N, 1H-steady-state NOE values (C) of the amide 15N-nuclei of wt N-domain and Q122FM(Y)F126 measured at 25°C. The GTS loop region (N30–M34) is highlighted. The standard error is indicated by the error bars, and the average values of the respective relaxation parameters (residues 16–142) are indicated by horizontal dashed lines.
RF activity of C127 mutants of eRF1 with omni-, bi- and uni-potent specificity
Although the distinct GTS loop conformation in Q122FM(Y)F126 compared to wt N-domain implicates a functional role of the GTS loop in stop codon recognition, one may argue that loss of UAA- and UAG-decoding capability in the mutant might be caused by the substituted residues directly. To prove the non-direct implication of the 122–128 region of eRF1 in stop codon decoding, the RF activity of the variant-code (Euplotes and Stylonychia) eRF1s with C127 mutations has been determined in an in vitro Caskey assay (32). C127 is an invariant residue in family of eRF1s and is located neighbour to F126 in the Q122FM(Y)F126 mutant that possesses the same stop codon specificity as Styloyichia eRF1 (Figure 1A). The sequence homology between human eRF1 and eRF1 of Euplotes and Stylonychia are 72.8 and 70.1%, respectively. Hence, their N-domains have very similar folds, as the models of Euplotes and Stylonychia’s N-domains derived by homology modeling possess a TM-score (between (0,1)] of 0.92 and 0.94, respectively.
As was shown earlier C127A and C127S mutants of human eRF1 exhibited tendency towards UGA unipotency (15), while C127S mutant of Euplotes eRF1-restored efficient recognition of UGA stop codon without changing of UAA and UAG stop codon decoding in the readthrough RF assay (53). We have shown that C127A and C127S mutants of Euplotes eRF1 also restored recognition of UGA stop codon but RF activity towards UAG was reduced for both mutants (Figure 3). However, insertion of the same C127A and C127S mutations into Stylonychia eRF1 with UGA-only specificity caused total abolishment of RF activity towards UGA (Figure 3).
Figure 3.

RF activity of C127 mutants of eRF1 with omni-, bi- and uni-potent specificity. In vitro RF activity of chimeric eRF1 constructs containing the whole N-terminal domains (positions 1–144) of Euplotes eRF1 (wt Eu-eRF1) or Stylonychia eRF1 (wt St-eRF1) and Eu-eRF1 and St-eRF1 mutants with C127 substitutions in the N-terminal domains. All eRF1 constructs contain MC-domain of human eRF1 (positions 145–437).
The different effects of the same C127 mutation on the recognition of UGA by human, Euplotes and Stylonychia eRF1s are a very hard argument in favour of suggestion that C127 does not participate directly in UGA recognition.
NMR structural characterization of 15-mer RNA mimicking the decoding region of Helix 44
To investigate potential interactions between N-domain and the A site of the 40S subunit, we designed a series of double-stranded RNA oligonucleotides, aiming to mimic the decoding region of Helix 44 in 18S rRNA (Figure 4A and B). The 15-mer RNA constructs are Ci- (inversion center) duplexes containing either two 2-nt asymmetric internal loops at the symmetrical positions or a single large internal loop (Figure 4B). It appears that symmetric structures provide sufficient thermodynamic stability to accommodate a significant number of mismatches in the central region, enabling studies of the impact of variability in the internal loop length found in different organisms (e.g. Tetrahymena thermophila and Thermomyces lanuginosus in Figure 4A) on binding to N-domain. The 7-mer RNA, which does not contain any internal loop, was used as a reference for RNA binding. In addition, in some constructs we incorporated 2′-O-methyl at C5 position exhibiting an easily detectable 1H-NMR line (54) acting as a structural probe for interactions near the internal loop. This modification has been shown to cause a negligible effect on the structure of double-stranded RNA (55).
Figure 4.

15-mer RNA constructs that mimic the decoding region of Helix 44. (A) Secondary structure of the highly conserved decoding region of Helix 44 in prokaryotic 30S and eukaryotic 40S ribosomal subunits. The internal loops are highlighted in bold; Watson-Crick and non-Watson-Crick base pairs are denoted by ‘–’ and ‘·’, respectively. (B) Secondary structure of the RNA constructs used in our studies. 15-mer-H44 contains two internal loops with the same sequence as the one in Tetrahymena thermophila 18S rRNA shown in (A). Four other RNA constructs are labeled according to their respective nucleotide substitutions, which are denoted in italics. The 7-mer RNA does not contain any internal loop. Paromomycin binds to the region of 15-mer-UAA designated by red dashed box as demonstrated in (D), while N-domain binds to the region of 15-mer-UGA designated by blue dashed box as demonstrated in Figures 5D and 5E. (C) 2D NOESY spectrum of 15-mer-UAA in H2O measured at 25°C with a mixing time of 350 ms, of which cross-peaks (in the right panel) to the imino protons (in the left panel) are labeled according to chemical shifts on the ω2 axis. Vertical dashed lines connect cross-peaks from the same protons. H22 of G3 in the minor conformation is denoted by caret, while H5 of Cm5 (denoted by plus) is attenuated by water suppression, which is otherwise observable on the other quadrant of the spectrum. (D) 1D proton spectra of 15-mer-UAA in H2O before and after addition of paromomycin. Three peaks stemming from paromomycin are indicated by caret.
Secondary structure of the 15-mer RNA constructs was determined using 2D-NOESY spectra. All mutant RNA constructs adopt the double-stranded helical structure (Supplementary Figure S3A). The canonical double-stranded helical structure can be clearly seen from the presence of three cross-peaks stemming from G2’s, G3’s and G10’s imino protons in the H1′–H5 region (5.0–6.2 ppm; right panel in Figure 4C) (56). In 15-mer-UAA, five Watson–Crick base pairs were detected using imino proton resonances protected from exchange with solvent (left panel in Figure 4C). Watson–Crick base pairing between A4 and U12 was not observed. Instead, a minor conformation associated with the closing base pair of A4–U12 in 15-mer-UGG and 15-mer-GAG was observed, indicating a slow ‘breathing’ action of the internal loop on the 1H chemical shift timescale (Figure 4C, Supplementary Figure S3B and C). This type of intra-molecular dynamics is expected in RNA-containing internal loops (57). The secondary structure of 15-mer-UGA is similar to 15-mer-UAA. The non-Watson–Crick base pair of U7–G8 is confirmed by detection of the corresponding imino proton resonances (Figure 5D) as well as strong NOEs between the two protons. For 15-mer-UGG and 15-mer-GAG, while an overall helical structure was maintained, both of the RNA constructs are deemed to form a large internal loop, due to the lack of observable stable base pairing from the nucleotides in the central region (Figure 5D). The 7-mer construct was expected to contain no internal loop, and this was confirmed by the observation of all expected base pairings (lower panel in Figure 5C).
Figure 5.

Specific interactions between N-domain and the different RNA constructs. (A) Chemical shift perturbations (CSP) of the backbone amide resonances of 15N-labeled N-domain upon titration with the 15-mer RNAs up to the RNA to protein molar ratio (R/P) as shown in (D). On the top of the plots, the secondary structure of N-domain is indicated. (B) The CSP values for wt N-domain + 15-mer-UGA from (A) are mapped onto the structure of wt N-domain according to the colour scale. Side-chains of the significantly perturbed residues upon RNA binding are shown and labeled. (C) CSP of the backbone amide resonances of 15N-labeled N-domain upon titration with the 7-mer RNA up to the level of R/P = 1.8 ± 0.2. (D) 1H spectra showing the imino protons of the 15-mer RNAs in free (blue) and N-domain-bound form (red). The 15N-split resonances stemming from 1HN E134 and 1HεN W15 of N-domain are indicated along with assignment of the imino protons of the RNAs. Transient base pairing between nucleotides in the central stem of 15-mer-UGG and 15-mer-GAG is observed as the minor resonances denoted by asterisk. (E) 1H spectra showing only resonances of 15-mer-UGA in free (blue) and N-domain-bound form (red) by selective suppression of amide and aromatic proton resonances stemming from the protein.
Aminoglycosides are a class of antibiotics that bind to the decoding region of Helix 44 in 16S rRNA (58). In particular, paromomycin binds much stronger to the prokaryotic than to the eukaryotic ribosome, owing to several differences in the rRNA sequence of the internal loop (25,53,59). Despite the fact that some of our selected 15-mer RNA constructs do not match exactly the oligonucleotide sequence of the decoding region of Helix 44 shown in Figure 4A, they interact with paromomycin at the internal loop as expected (Figure 4B and D). This indicates that the 15-mer RNA constructs possess some essential structural determinants sufficient for paromomycin binding. This clearly supports the structural relevance of the 15-mer RNA constructs as mimics of the decoding region of Helix 44.
The 15-mer RNAs bind to helix α1 of N-domain
Among the 15-mer RNA constructs, the strongest binding 15-mer-UGA interacts with N-domain with Kd of ∼150 μM as estimated from NMR titration data (Figure 5A). The binding interface spans from residues N11 to I21, covering most of helix α1 (Figure 5B). In particular, several lysine residues within the binding region, i.e. K16, K18 and K19, are highly conserved and are likely to contribute to the interactions with RNA (Supplementary Figure S1). In addition, the aromatic side-chain of the conserved W15 could be involved in a stacking interaction with the nucleic acid bases. These types of interactions are commonly found non-sequence-specific interactions in protein–RNA complexes (60,61).
Besides helix α1 some other residues of N-domain were also affected by RNA binding as detected by CSP (Figure 5A and B). All of these observations can be explained by allosteric propagation of the structural perturbations from helix α1 directly involved in RNA binding to other secondary structure elements located in its immediate spatial proximity. This effect can be seen as drastic CSP at F117, of which side-chain is in close contact with helix α1. Some residues with notable CSPs are located in helix α4, especially at and around T137, which is tightly packed against helix α1 with the side-chain of T137 facing I21 in helix α1. A site with rather subtle chemical shift perturbation is located on the opposite side of helix α1 at T58 (Figure 5B). Interestingly, in the crystal structure of eRF1 in complex with domain 2/3 of eRF3, an ATP molecule was found in proximity to T58 of eRF1, with N7 of adenine in contact with the threonine hydroxyl group (PDB ID: 3E1Y) (50). This indicates a possibility of interactions between T58 and the flipped out adenine in the internal loop of the 15-mer RNA.
Upon binding to N-domain, all tested 15-mer RNA constructs induced consistent CSP patterns, albeit with variable magnitudes reflecting different binding affinities (Figure 5A). Specifically, 15-mer-GAG exhibits significantly weaker response, which is likely due to its thermodynamically unstable central stem. This implies that binding of the 15-mer RNA to N-domain does not depend strongly on the sequence of the internal loop, but rather on the structural variability of the double-stranded RNA. To highlight the role of the internal loop in binding to N-domain, we tested the interaction using the 7-mer RNA, which comprises the central region of 15-mer-UGA but without the internal loops (Figure 5C). The significantly attenuated binding affinity shows that the internal loop is required for specific binding to helix α1 of N-domain. Furthermore, only a specific set of resonances of the 15-mer RNA was perturbed upon interaction with N-domain (Figure 5D and E). Based on that, the binding site can be mapped onto a region of the 15-mer RNA that includes the internal loop (Figure 4B). Close inspection revealed that nucleotides A9 and G10 seemed to experience a larger perturbation compared to other relevant nucleotides. We argue that if the central RNA stem was the only element required for the interaction, binding affinity of the 7-mer RNA to N-domain should be comparable to the 15-mer RNA, particularly 15-mer-UGA. Hence, we have established that the interaction between N-domain and RNA requires certain structural elements that include double-stranded helices and a 2-nt internal loop. These results strongly support the potential of N-domain to interact directly with the decoding region of Helix 44.
Binding of 15-mer RNAs displaces C-domain from a non-covalent complex of N-domain and C-domain
The 3D crystal structure of full-length eRF1 shows that its three protein domains extend outward from the center of mass to form a Y-shape tertiary structure, with C-domain exhibiting a considerable contact interface with N-domain (10). In solution, TROSY NMR spectra of full-length eRF1 showed fewer than expected resonances, with the majority of cross-peaks broadened by conformational exchange. We traced the source of this conformational jitter to the interactions between N-domain and C-domain, since the NM-domain construct corresponding to a C-domain-truncated eRF1 lacks significant structural interactions between N-domain and M-domain (Supplementary Figure S4). Interestingly, our model RNAs are interacting with helix α1 of N-domain that is found at the interface with C-domain in the crystal structure of eRF1 (Figure 6C). We hypothesized that the 15-mer RNA and C-domain may compete for binding to N-domain, and hence, set out to test it by competitive binding experiments.
Figure 6.

Interactions between N-domain, C-domain and the 15-mer RNA. (A) CSP of the backbone amide resonances of 15N-labeled N-domain upon addition of equimolar amount of unlabeled C-domain. The pink colour bars denote residues of which resonances are broadened beyond detection. (B) Reduction in signal intensity of the backbone amide resonances of 15N-labeled C-domain upon addition of equimolar amount of unlabeled N-domain. (C) The complex model of N-domain and C-domain is reconstructed by replacing C-domain in the crystal structure of full-length eRF1 (PDB ID: 1DT9) with the NMR solution structure of the individual C-domain (73)(PDB ID: 2KTV), since the minidomain of C-domain has not been resolved in the crystal structure. The crystal structure of eRF1 shows that C-domain contacts N-domain at the interface denoted by a dashed line. In solution, the binding interface between N-domain and C-domain can be inferred from the significantly affected residues upon complex formation: residues of N-domain (green) with CSP ≥ 0.03 ppm are coloured in red and those that were broadened beyond detection are coloured in magenta according to (A); residues of C-domain (blue) with intensity reduction ≥0.8 are coloured in pink and those that are not resolved are coloured in grey according to (B). (D) Overlay of TROSY [1H, 15N]-HSQC spectra measured at different stages of the competitive binding experiment: free 15N-labeled C-domain (blue), addition of equimolar amount of unlabeled N-domain to 15N-C-domain (red), addition of equimolar amount of 15-mer-UGA to the complex of unlabeled N-domain and 15N-labeled C-domain (black). Contour levels of the spectra are adjusted according to the cross-peak denoted by asterisk, which belongs to the His-tag of C-domain and is undisturbed at all three stages. Several examples of the shifted cross-peaks are highlighted by dashed boxes.
NMR titration experiments with 15N-labeled N-domain and unlabeled C-domain, and vice versa, show that N-domain indeed interacts with C-domain at the interface that includes the contact area observed in the crystal structure of eRF1 (Figure 6A–C). Majority of the amide resonances of C-domain were severely attenuated by line broadening upon N-domain binding (Figure 6B and D), while several amide resonances of N-domain were also disappeared when binding to C-domain (Figure 6A). This indicates that the kinetic rate of binding is in the fast to intermediate exchange regime on NMR timescale, and the binding of N-domain may induce additional conformational exchange to C-domain on another timescale. This low affinity or transient nature of binding between N-domain and C-domain is supported by thermodynamically weak, entropy-driven interactions observed in earlier isothermal scanning colourimetric studies (31). Nevertheless, CSP and extreme broadening of cross-peaks allowed us to map the binding interface on N-domain (Figure 6C). The binding interface seems to be more extensive than can be predicted from the crystal structure of eRF1, which suggests that there are other modes of complex formation between N-domain and C-domain. In fact, variations in domain orientation were observed in the different crystal structures of eRF1 in complex with domain 2/3 of eRF3, as the relative orientations between N-domain and C-domain differ by 15°, 16°, and 30° for human complex versus free eRF1, human complex versus Schizosaccharomyces pombe complex and S. pombe complex versus free eRF1, respectively (50). It is interesting to note that part of the GTS loop was also significantly affected by the presence of C-domain (Figure 6A and C).
Perturbations of the C-domain resonances were monitored in competitive binding between the two domains and 15-mer RNA. Initially, addition of N-domain attenuated majority of the cross-peaks and shifted some of them (left panel in Figure 6D). As 15-mer-UGA was titrated into the complex of N-domain and C-domain, the attenuated and shifted cross-peaks returned progressively to the initial state corresponding to free C-domain in solution (right panel in Figure 6D). As we have already shown the 15-mer RNA binds to N-domain, while C-domain does not interact with the 15-mer RNA at all (Supplementary Figure S5), this result clearly demonstrates that the 15-mer-RNA is able to displace C-domain from the non-covalent complex of N-domain and C-domain.
DISCUSSION
Selectivity of stop codon recognition is modulated by multiple GTS loop conformations
The strictly conserved GTS loop of N-domain is emerging as being implicated in decoding or in direct contact with the stop codon (17,62,63). The most significant insight from the finding of distinct GTS loop conformations in wt N-domain and Q122FM(Y)F126 is that the shift in stop codon selectivity might be determined by the structural changes, that are critical for interactions of amino acid(s) with the stop codon. Having the same amino acid sequence as the wild-type at the positions 122–126, eRF1 with a single point mutation T32A had been shown to exhibit tendency towards UGA unipotency (50, E. Alkalaeva, personal communication). Why does T32A mutant show similar attribute as Q122FM(Y)F126? The same question can be asked about the stop codon decoding by various eRF1 mutants investigated in previous studies; since; in many cases; the point mutations scattered across a large part of N-domain resulted in the same bias of stop codon selectivity. This paradox can be explained in two ways: (i) eRF1 mutant, which has lost interaction with one or two out of three nucleotides of a stop codon, can still support the peptide release for that particular stop codon but with reduced efficiency, probably because, one amino acid substitution is not enough to destroy completely the direct interaction with stop codon(s) (this might be the case for T32A mutant) and (ii) the part of described earlier point mutations, which have given rise to altered stop codon specificity, are responsible for structural modulation of N-domain regions that actually interact with the stop codon(s) (this might be the case for Q122FM(Y)F126).
Similar mutations of the highly conserved C127 in human, Euplotes and Stylonychia eRF1s evoke different responses: the decreased recognition for UAA and UAG for human eRF1, the appearance of UGA recognition for Euplotes eRF1, and its disappearance for Stylonychia eRF1. This indicates that C127 does not participate directly in stop codon recognition as well as very likely the same for the other amino acid residues in the region 122–126. It also shows that residue nearby to the stop codon recognition site can critically affect stop codon selectivity.
In the context of our proposed mechanism, point mutation that alters selectivity of stop codon recognition is likely to modulate the structure of the GTS loop, or even its capacity to switch between different conformations. Indeed, a few point mutations were found to hit on the residues that constitute the hydrophobic core right above the GTS loop, e.g. I35, V71, V78 and C127 (18,50,53). Remarkably, the width of this hydrophobic core is directly related to the differential positioning of helix α3 as observed in the structures of wt N-domain and Q122FM(Y)F126 (Figure 1A and Supplementary Figure S2A). The width, measured as the distance between the amides of M34 and V71, is reduced from 7.68 Å and 7.38 ± 0.18 Å in the crystal and solution structures of wt N-domain, respectively, to 6.56 ± 0.13 Å in Q122FM(Y)F126. In light of these observations, it is attractive to hypothesize that repositioning of helix α3 in N-domain occurs during stop codon recognition as the GTS loop is sampling different configurations. Interestingly, S70 on helix α3 was found to involve in stabilization of the GTS loop in Q122FM(Y)F126 (Figure 1C). Residue S70 is critical for UGA-decoding, as a point mutation S70A restricts human eRF1 to recognize UAA and UAG only. At the same time, the single A70S substitution in Euplotes eRF1 changes stop codon recognition from UAR-only specificity to omnipotent one (24). These data verify the assumption that S70A substitution is associated with UGA reassignment (64).
Besides helix α3, helix α2 could also play a role in modulating the selectivity of stop codon recognition. First, M51 and E55 on helix α2 are able to alter stop codon recognition patterns (18,65). Secondly, the TASNIKS motif was found to confer distinct requirement of eRF3 upon eRF1 on decoding UAA/UAG and UGA (53). As T58 in the TASNIKS motif was observed to interact with the 15-mer RNA (Figure 5A and B) and the NIKS motif had also been implicated in ribosome binding (14), interactions between helix α2 and the ribosome is highly possible. Furthermore, P41 and P89, which may be critical for the formation of the β-turns that connect the core β-sheet to helices α2 and α3, were also found to affect stop codon recognition (18,25). This hints at a higher than anticipated degree of complexity in the stop codon decoding mechanism of eRF1.
Interactions between N-domain and mimics of the decoding region of Helix 44
With the knowledge of the GTS loop being implicated in stop codon recognition, we seek to explore possible orientations of N-domain within the ribosomal A site by investigating the interactions between N-domain and 18S rRNA. As genetic interactions between eRF1 and the decoding region of Helix 44 of 18S rRNA were reported (25), we decided to test if there is any direct interaction between them. Our model 15-mer RNA intended to mimic the decoding region of Helix 44 outside of the ribosome does interact specifically with helix α1 of N-domain, and its binding affinity is significantly reduced in the absence of the internal loop. How far has this somewhat reductionist’s approach achieved? Our results are well-supported by previous studies and are able to provide novel insights. A truncated mutant of eRF1, eRF121–437, was shown to reduce significantly its own release activity, as well as the stimulating activity towards eRF3 GTPase, indicating that N-terminal deletion of eRF1 until residue I21 is enough to affect its binding to the ribosome (11).
Aminoglycosides are known to reduce the fidelity of both elongation and termination of protein translation (66). Although they bind much stronger to the prokaryotic than to the eukaryotic ribosome, the latter is still susceptible to the nonsense suppression effect of various aminoglycosides (67,68). Furthermore, those drugs have been used effectively in alleviating diseases caused by premature termination codon (69). Most importantly, it was shown that the nonsense suppression induced by paromomycin in yeast is likely to be caused by the interference to the termination process, instead of compromising the selection of cognate tRNA (70). All these results support the hypothesis of direct interaction between N-domain and the decoding region of Helix 44.
A1752G (rdn15), a point mutation in Helix 44 of Saccharomyces cerevisiae 18S rRNA (refer to 18S rRNA of Thermomyces lanuginosus in Figure 4A for the nucleotide position), is able to rescue cell lethality caused by a mutant eRF1, Sup45p-P86A, at 37°C (25). The point mutation P86A was thought to reduce Sup45p’s efficiency in stop codon recognition, rather than inhibiting it completely. The rescue mechanism by rdn15 is far from clear, but was postulated to prolong the residence time of the mutant eRF1 at the ribosomal A site. Interestingly, we have found that 15-mer-GAG, having a larger internal loop than the rest, binds significantly weaker to N-domain. As rdn15 effectively reduces the native 3-nt to a 2-nt internal loop, the inverse correlation between RNA-N-domain binding affinity and the size of the internal loop provides a possible explanation for the complementary between rdn15 and Sup45p-P86A.
A model of N-domain bound in the pre-termination complex
Prior to solving the high-resolution structures of eRF1-bound pre-termination complex (pre-TC), understanding of the mechanism of translation termination in eukaryotes will have to rely on piercing together biochemical, structural and genetics data from different studies. Our structural study of N-domain led us to suggest that eRF1 might decode different stop codons by adopting distinct GTS loop conformations, thus implying direct access of the GTS loop to the stop codon. In addition, the data on N-domain-RNA interactions have shown that N-domain potentially interacts with Helix 44 of 18S rRNA. Based on these results, we propose a structural model that encompasses currently known interactions between N-domain of eRF1 and the A site of eukaryotic ribosome (Figure 7).
Figure 7.
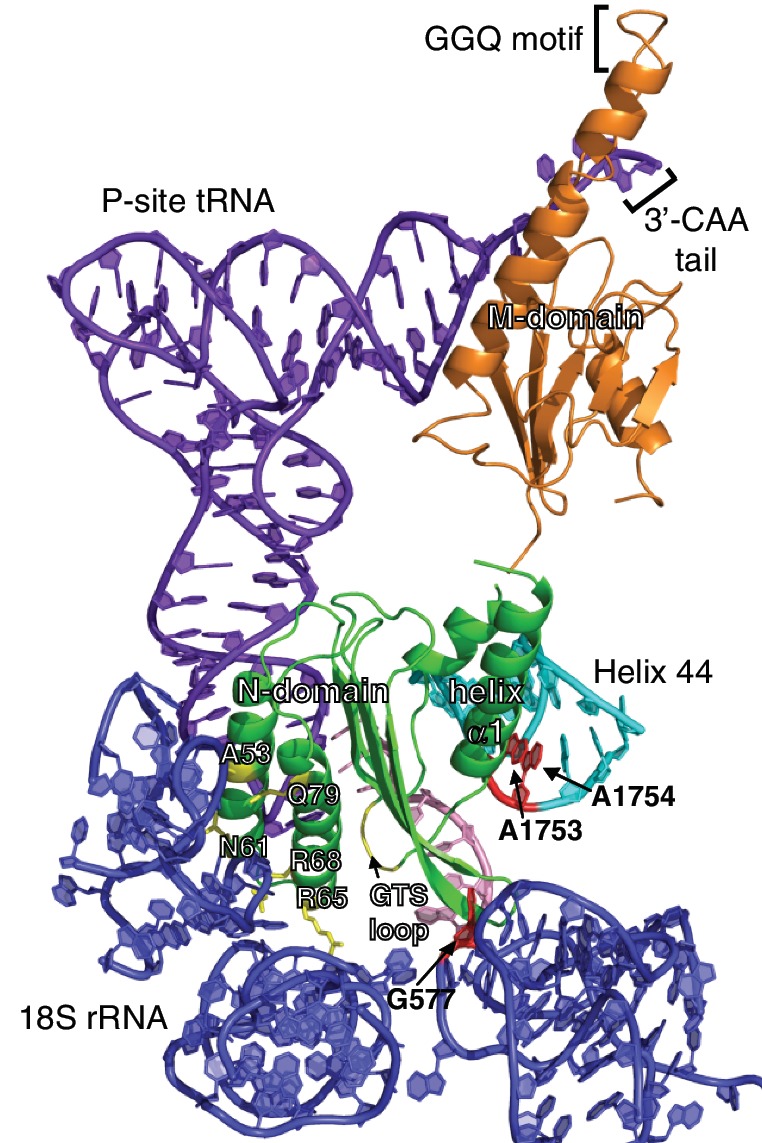
A model of eRF1 bound to the A site of eukaryotic ribosome. NM-domain of eRF1 (green and orange) was docked onto the A site of 18S rRNA (blue) with P-site bound tRNA (purple) and mRNA (pink; PDB ID: 3IZ7), based on the insights derived from the interactions between helix α1 of N-domain and the decoding region of Helix 44 (cyan), as well as the putative role of the GTS loop in stop codon recognition. The corresponding nucleotides critical for tRNA selection in prokaryotic ribosome and selected residues of eRF1 are highlighted in red and yellow, respectively.
The model shows that it is possible for the GTS loop to contact the stop codon while helix α1 is positioned next to the decoding region of Helix 44. Although helix α1 is not in the exact position to interact with Helix 44, a slight forward movement of the stop codon towards the P site will compensate for this discrepancy. Interestingly, it was reported that 2-nt toe-print shift occurs when the eRF1·eRF3·GTP complex binds to the pre-TC (71). On the other side of N-domain, the side-chains of A53, N61, R65, R68 and Q79 are facing 18S rRNA. Residues R65 and R68 affect the binding of eRF1 to the ribosome (14), while each of the point mutants, A53K, N61K and Q79K/R, was shown to substantially reduce the level of stop codon readthrough in comparison to wild-type, indicating enhanced ribosome binding due to the lysine or arginine substitution (72). In our model, the GTS loop is close enough to the stop codon to allow photoactivatable cross-linking with the second and third stop codon positions (17). In the eRF1/pre-TC cross-linking experiments, the KSR loop (positions 63–65) and V66 were suggested to be in contact with the first stop codon position (16,17). Although within margins of cross-linking experiments, in our model these residues are not located in the direct proximity of the uridine of the stop codon, thereby requiring further experiments to resolve.
With the orientation of N-domain in our model, a hinge motion between N-domain and M-domain would allow the GGQ motif to reach the 3′-CAA tail of P-site tRNA as had been suggested earlier (17,50), while C-domain would be required to move away from helix α1 (Figure 7). The latter is well demonstrated by the competitive binding experiments, albeit in a reductionist’s approach (Figure 6D). Hence, a major domain rearrangement between N-domain and C-domain is likely to occur in translation termination during which N-domain accommodates itself into the A site.
ACCESSION NUMBERS
The structure of Q122FM(Y)F126 has been deposited into PDB with ID 2LGT, and the assigned chemical shifts have been deposited into BMRB with ID 17822.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Figures 1–5 and Supplementary Reference [74].
FUNDING
Singapore Ministry of Education AcRF grant (T207B3205 to K.P.); Russian Foundation for Basic Research (grant 11-04-00840 to L.F.); Program ‘Molecular and Cell Biology’ of the Presidium of the Russian Academy of Sciences (grant to L.F.). In addition, we acknowledge funding of several graduate scholarships by the Singapore Ministry of Education. Funding for open access charge: Singapore Ministry of Education AcRF grant T207B3205.
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
The work on comparison of the N-domain structures of wild-type human eRF1 and its mutant with UGA unipotency by means of NMR was initiated by Lev Kisselev. We thank Sergey Lekomtsev for the constructs of C127 mutants of Euplotes and Stylonychia eRF1s and Elena Alkalaeva for making her results available before publication. We are grateful for the access of SON NMR Large Scale Facility in Utrecht, The Netherlands, funded by the European Union (contract number RII3-026145). We also appreciate Dr. Alistair Irvine for reading the manuscript.
REFERENCES
- 1.Capecchi MR. Polypeptide chain termination in vitro: isolation of a release factor. Proc. Natl Acad. Sci. USA. 1967;58:1144–1151. doi: 10.1073/pnas.58.3.1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kisselev L, Ehrenberg M, Frolova L. Termination of translation: interplay of mRNA, rRNAs and release factors? EMBO J. 2003;22:175–182. doi: 10.1093/emboj/cdg017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ito K, Uno M, Nakamura Y. A tripeptide ‘anticodon’ deciphers stop codons in messenger RNA. Nature. 2000;403:680–684. doi: 10.1038/35001115. [DOI] [PubMed] [Google Scholar]
- 4.Nakamura Y, Ito K. A tripeptide discriminator for stop codon recognition. FEBS Lett. 2002;514:30–33. doi: 10.1016/s0014-5793(02)02330-x. [DOI] [PubMed] [Google Scholar]
- 5.Laurberg M, Asahara H, Korostelev A, Zhu J, Trakhanov S, Noller HF. Structural basis for translation termination on the 70S ribosome. Nature. 2008;454:852–857. doi: 10.1038/nature07115. [DOI] [PubMed] [Google Scholar]
- 6.Korostelev A, Zhu J, Asahara H, Noller HF. Recognition of the amber UAG stop codon by release factor RF1. EMBO J. 2010;29:2577–2585. doi: 10.1038/emboj.2010.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Korostelev A, Asahara H, Lancaster L, Laurberg M, Hirschi A, Zhu J, Trakhanov S, Scott WG, Noller HF. Crystal structure of a translation termination complex formed with release factor RF2. Proc. Natl Acad. Sci. USA. 2008;105:19684–19689. doi: 10.1073/pnas.0810953105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Weixlbaumer A, Jin H, Neubauer C, Voorhees RM, Petry S, Kelley AC, Ramakrishnan V. Insights into translational termination from the structure of RF2 bound to the ribosome. Science. 2008;322:953–956. doi: 10.1126/science.1164840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sund J, Ander M, Aqvist J. Principles of stop-codon reading on the ribosome. Nature. 2010;465:947–950. doi: 10.1038/nature09082. [DOI] [PubMed] [Google Scholar]
- 10.Song H, Mugnier P, Das AK, Webb HM, Evans DR, Tuite MF, Hemmings BA, Barford D. The crystal structure of human eukaryotic release factor eRF1-mechanism of stop codon recognition and peptidyl-tRNA hydrolysis. Cell. 2000;100:311–321. doi: 10.1016/s0092-8674(00)80667-4. [DOI] [PubMed] [Google Scholar]
- 11.Frolova LY, Merkulova TI, Kisselev LL. Translation termination in eukaryotes: Polypeptide release factor eRF1 is composed of functionally and structurally distinct domains. RNA. 2000;6:381–390. doi: 10.1017/s135583820099143x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ito K, Ebihara K, Nakamura Y. The stretch of C-terminal acidic amino acids of translational release factor eRF1 is a primary binding site for eRF3 of fission yeast. RNA. 1998;4:958–972. doi: 10.1017/s1355838298971874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ito K, Frolova L, Seit-Nebi A, Karamyshev A, Kisselev L, Nakamura Y. Omnipotent decoding potential resides in eukaryotic translation termination factor eRF1 of variant-code organisms and is modulated by the interactions of amino acid sequences within domain 1. Proc. Natl Acad. Sci. USA. 2002;99:8494–8499. doi: 10.1073/pnas.142690099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Frolova L, Seit-Nebi A, Kisselev L. Highly conserved NIKS tetrapeptide is functionally essential in eukaryotic translation termination factor eRF1. RNA. 2002;8:129–136. doi: 10.1017/s1355838202013262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Seit-Nebi A, Frolova L, Kisselev L. Conversion of omnipotent translation termination factor eRF1 into ciliate-like UGA-only unipotent eRF1. EMBO Rep. 2002;3:881–886. doi: 10.1093/embo-reports/kvf178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chavatte L, Seit-Nebi A, Dubovaya V, Favre A. The invariant uridine of stop codons contacts the conserved NIKSR loop of human eRF1 in the ribosome. EMBO J. 2002;21:5302–5311. doi: 10.1093/emboj/cdf484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bulygin KN, Khairulina YS, Kolosov PM, Ven’yaminova AG, Graifer DM, Vorobjev YN, Frolova LY, Kisselev LL, Karpova GG. Three distinct peptides from the N domain of translation termination factor eRF1 surround stop codon in the ribosome. RNA. 2010;16:1902–1914. doi: 10.1261/rna.2066910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bertram G, Bell HA, Ritchie DW, Fullerton G, Stansfield I. Terminating eukaryote translation: domain 1 of release factor eRF1 functions in stop codon recognition. RNA. 2000;6:1236–1247. doi: 10.1017/s1355838200000777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Inagaki Y, Blouin C, Doolittle WF, Roger AJ. Convergence and constraint in eukaryotic release factor 1 (eRF1) domain 1: the evolution of stop codon specificity. Nucleic Acids Res. 2002;30:532–544. doi: 10.1093/nar/30.2.532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Inagaki Y, Doolittle WF. Class I release factors in ciliates with variant genetic codes. Nucleic Acids Res. 2001;29:921–927. doi: 10.1093/nar/29.4.921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lozupone CA, Knight RD, Landweber LF. The molecular basis of nuclear genetic code change in ciliates. Curr. Biol. 2001;11:65–74. doi: 10.1016/s0960-9822(01)00028-8. [DOI] [PubMed] [Google Scholar]
- 22.Salas-Marco J, Fan-Minogue H, Kallmeyer AK, Klobutcher LA, Farabaugh PJ, Bedwell DM. Distinct paths to stop codon reassignment by the variant-code organisms Tetrahymena and Euplotes. Mol. Cell Biol. 2006;26:438–447. doi: 10.1128/MCB.26.2.438-447.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lekomtsev S, Kolosov P, Bidou L, Frolova L, Rousset JP, Kisselev L. Different modes of stop codon restriction by the Stylonychia and Paramecium eRF1 translation termination factors. Proc. Natl Acad. Sci. USA. 2007;104:10824–10829. doi: 10.1073/pnas.0703887104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Eliseev B, Kryuchkova P, Alkalaeva E, Frolova L. A single amino acid change of translation termination factor eRF1 switches between bipotent and omnipotent stop-codon specificity. Nucleic Acids Res. 2010;39:599–608. doi: 10.1093/nar/gkq759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Velichutina IV, Hong JY, Mesecar AD, Chernoff YO, Liebman SW. Genetic interaction between yeast Saccharomyces cerevisiae release factors and the decoding region of 18 S rRNA. J. Mol. Biol. 2001;305:715–727. doi: 10.1006/jmbi.2000.4329. [DOI] [PubMed] [Google Scholar]
- 26.Fourmy D, Recht MI, Blanchard SC, Puglisi JD. Structure of the A site of Escherichia coli 16S ribosomal RNA complexed with an aminoglycoside antibiotic. Science. 1996;274:1367–1371. doi: 10.1126/science.274.5291.1367. [DOI] [PubMed] [Google Scholar]
- 27.Lynch SR, Puglisi JD. Structure of a eukaryotic decoding region A-site RNA. J. Mol. Biol. 2001;306:1023–1035. doi: 10.1006/jmbi.2000.4419. [DOI] [PubMed] [Google Scholar]
- 28.Lynch SR, Gonzalez RL, Puglisi JD. Comparison of X-ray crystal structure of the 30S subunit-antibiotic complex with NMR structure of decoding site oligonucleotide-paromomycin complex. Structure. 2003;11:43–53. doi: 10.1016/s0969-2126(02)00934-6. [DOI] [PubMed] [Google Scholar]
- 29.Kondo J, Urzhumtsev A, Westhof E. Two conformational states in the crystal structure of the Homo sapiens cytoplasmic ribosomal decoding A site. Nucleic Acids Res. 2006;34:676–685. doi: 10.1093/nar/gkj467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dubovaya VI, Kolosov PM, Alkalaeva EZ, Frolova LY, Kisselev LL. Influence of individual domains of the translation termination factor eRF1 on induction of the GTPase activity of the translation termination factor eRF3. Mol. Biol. 2006;40:310–316. [PubMed] [Google Scholar]
- 31.Kononenko AV, Mitkevich VA, Dubovaya VI, Kolosov PM, Makarov AA, Kisselev LL. Role of the individual domains of translation termination factor eRF1 in GTP binding to eRF3. Proteins. 2008;70:388–393. doi: 10.1002/prot.21544. [DOI] [PubMed] [Google Scholar]
- 32.Caskey CT, Beaudet AL, Tate WP. Mammalian release factor: in vitro assay and purification. Methods Enzymol. 1974;30:293–303. doi: 10.1016/0076-6879(74)30032-8. [DOI] [PubMed] [Google Scholar]
- 33.Oda Y, Muramatsu T, Yumoto F, Ito M, Tanokura M. Backbone (1)H, (13)C and (15)N resonance assignment of the N-terminal domain of human eRF1. J. Biomol. NMR. 2004;30:109–110. doi: 10.1023/B:JNMR.0000042944.14441.d6. [DOI] [PubMed] [Google Scholar]
- 34.Ivanova EV, Kolosov PM, Birdsall B, Kisselev LL, Polshakov VI. NMR Assignments of the Middle Domain of Human Polypeptide Release Factor eRF1. J. Biomol. NMR. 2006;36(Suppl. 1):8. doi: 10.1007/s10858-005-4741-1. [DOI] [PubMed] [Google Scholar]
- 35.Mantsyzov AB, Ivanova EV, Birdsall B, Kolosov PM, Kisselev LL, Polshakov VI. NMR assignments of the C-terminal domain of human polypeptide release factor eRF1. Biomol. NMR Assign. 2007;1:183–185. doi: 10.1007/s12104-007-9050-z. [DOI] [PubMed] [Google Scholar]
- 36.Kelly MJS, Krieger C, Ball LJ, Yu YH, Richter G, Schmieder P, Bacher A, Oschkinat H. Application of amino acid type-specific H-1- and N-14-labeling in a H-2-, N-15-labeled background to a 47 kDa homodimer: Potential for NMR structure determination of large proteins. J. Biomol. NMR. 1999;14:79–83. doi: 10.1023/a:1008351606073. [DOI] [PubMed] [Google Scholar]
- 37.Kainosho M, Tsuji T. Assignment of the three methionyl carbonyl carbon resonances in Streptomyces subtilisin inhibitor by a carbon-13 and nitrogen-15 double-labeling technique. A new strategy for structural studies of proteins in solution. Biochemistry. 1982;21:6273–6279. doi: 10.1021/bi00267a036. [DOI] [PubMed] [Google Scholar]
- 38.Yabuki T, Kigawa T, Dohmae N, Takio K, Terada T, Ito Y, Laue ED, Cooper JA, Kainosho M, Yokoyama S. Dual amino acid-selective and site-directed stable-isotope labeling of the human c-Ha-Ras protein by cell-free synthesis. J. Biomol. NMR. 1998;11:295–306. doi: 10.1023/a:1008276001545. [DOI] [PubMed] [Google Scholar]
- 39.Zweckstetter M. NMR: prediction of molecular alignment from structure using the PALES software. Nat. Protoc. 2008;3:679–690. doi: 10.1038/nprot.2008.36. [DOI] [PubMed] [Google Scholar]
- 40.Cornilescu G, Delaglio F, Bax A. Protein backbone angle restraints from searching a database for chemical shift and sequence homology. J. Biomol. NMR. 1999;13:289–302. doi: 10.1023/a:1008392405740. [DOI] [PubMed] [Google Scholar]
- 41.Guntert P, Mumenthaler C, Wuthrich K. Torsion angle dynamics for NMR structure calculation with the new program DYANA. J. Mol. Biol. 1997;273:283–298. doi: 10.1006/jmbi.1997.1284. [DOI] [PubMed] [Google Scholar]
- 42.Herrmann T, Guntert P, Wuthrich K. Protein NMR structure determination with automated NOE assignment using the new software CANDID and the torsion angle dynamics algorithm DYANA. J. Mol. Biol. 2002;319:209–227. doi: 10.1016/s0022-2836(02)00241-3. [DOI] [PubMed] [Google Scholar]
- 43.Koradi R, Billeter M, Wuthrich K. MOLMOL: a program for display and analysis of macromolecular structures. J. Mol. Graph. 1996;14:51–55. doi: 10.1016/0263-7855(96)00009-4. [DOI] [PubMed] [Google Scholar]
- 44.Laskowski RA, Rullmannn JA, MacArthur MW, Kaptein R, Thornton JM. AQUA and PROCHECK-NMR: programs for checking the quality of protein structures solved by NMR. J. Biomol. NMR. 1996;8:477–486. doi: 10.1007/BF00228148. [DOI] [PubMed] [Google Scholar]
- 45.Roy A, Kucukural A, Zhang Y. I-TASSER: a unified platform for automated protein structure and function prediction. Nat. Protoc. 2010;5:725–738. doi: 10.1038/nprot.2010.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang Y. I-TASSER server for protein 3D structure prediction. BMC Bioinformatics. 2008;9:40. doi: 10.1186/1471-2105-9-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang Y, Skolnick J. TM-align: a protein structure alignment algorithm based on the TM-score. Nucleic Acids Res. 2005;33:2302–2309. doi: 10.1093/nar/gki524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schanda P, Kupce E, Brutscher B. SOFAST-HMQC experiments for recording two-dimensional heteronuclear correlation spectra of proteins within a few seconds. J. Biomol. NMR. 2005;33:199–211. doi: 10.1007/s10858-005-4425-x. [DOI] [PubMed] [Google Scholar]
- 49.Fielding L. NMR methods for the determination of protein-ligand dissociation constants. Curr. Top. Med. Chem. 2003;3:39–53. doi: 10.2174/1568026033392705. [DOI] [PubMed] [Google Scholar]
- 50.Cheng Z, Saito K, Pisarev AV, Wada M, Pisareva VP, Pestova TV, Gajda M, Round A, Kong C, Lim M, et al. Structural insights into eRF3 and stop codon recognition by eRF1. Genes Dev. 2009;23:1106–1118. doi: 10.1101/gad.1770109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ishima R, Torchia DA. Protein dynamics from NMR. Nat. Struct. Biol. 2000;7:740–743. doi: 10.1038/78963. [DOI] [PubMed] [Google Scholar]
- 52.Jarymowycz VA, Stone MJ. Fast time scale dynamics of protein backbones: NMR relaxation methods, applications, and functional consequences. Chem. Rev. 2006;106:1624–1671. doi: 10.1021/cr040421p. [DOI] [PubMed] [Google Scholar]
- 53.Fan-Minogue H, Bedwell DM. Eukaryotic ribosomal RNA determinants of aminoglycoside resistance and their role in translational fidelity. RNA. 2008;14:148–157. doi: 10.1261/rna.805208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Clore GM, Gronenborn AM, Piper EA, McLaughlin LW, Graeser E, van Boom JH. The solution structure of a RNA pentadecamer comprising the anticodon loop and stem of yeast tRNAPhe: a 500 MHz 1H-n.m.r. study. Biochem. J. 1984;221:737–751. doi: 10.1042/bj2210737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Popenda M, Biala E, Milecki J, Adamiak RW. Solution structure of RNA duplexes containing alternating CG base pairs: NMR study of r(CGCGCG)2 and 2′-O-Me(CGCGCG)2 under low salt conditions. Nucleic Acids Res. 1997;25:4589–4598. doi: 10.1093/nar/25.22.4589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Heus HA, Pardi A. Novel proton NMR assignment procedure for RNA duplexes. J. Am. Chem. Soc. 1991;113:4360–4361. [Google Scholar]
- 57.Popenda L, Adamiak RW, Gdaniec Z. Bulged adenosine influence on the RNA duplex conformation in solution. Biochemistry. 2008;47:5059–5067. doi: 10.1021/bi7024904. [DOI] [PubMed] [Google Scholar]
- 58.Moazed D, Noller HF. Interaction of antibiotics with functional sites in 16S ribosomal RNA. Nature. 1987;327:389–394. doi: 10.1038/327389a0. [DOI] [PubMed] [Google Scholar]
- 59.Recht MI, Douthwaite S, Puglisi JD. Basis for prokaryotic specificity of action of aminoglycoside antibiotics. EMBO J. 1999;18:3133–3138. doi: 10.1093/emboj/18.11.3133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Morozova N, Allers J, Myers J, Shamoo Y. Protein-RNA interactions: exploring binding patterns with a three-dimensional superposition analysis of high resolution structures. Bioinformatics. 2006;22:2746–2752. doi: 10.1093/bioinformatics/btl470. [DOI] [PubMed] [Google Scholar]
- 61.Allers J, Shamoo Y. Structure-based analysis of protein-RNA interactions using the program ENTANGLE. J. Mol. Biol. 2001;311:75–86. doi: 10.1006/jmbi.2001.4857. [DOI] [PubMed] [Google Scholar]
- 62.Wang Y, Chai B, Wang W, Liang A. Functional characterization of polypeptide release factor 1b in the ciliate Euplotes. Biosci. Rep. 2010;30:425–431. doi: 10.1042/BSR20090154. [DOI] [PubMed] [Google Scholar]
- 63.Bulygin KN, Khairulina YS, Kolosov PM, Ven’yaminova AG, Graifer DM, Vorobjev YN, Frolova LY, Karpova GG. Adenine and guanine recognition of stop codon is mediated by different N domain conformations of translation termination factor eRF1. Nucleic Acids Res. 2011;39:7134–7146. doi: 10.1093/nar/gkr376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Liang H, Wong JY, Bao Q, Cavalcanti AR, Landweber LF. Decoding the decoding region: analysis of eukaryotic release factor (eRF1) stop codon-binding residues. J. Mol. Evol. 2005;60:337–344. doi: 10.1007/s00239-004-0211-8. [DOI] [PubMed] [Google Scholar]
- 65.Kolosov P, Frolova L, Seit-Nebi A, Dubovaya V, Kononenko A, Oparina N, Justesen J, Efimov A, Kisselev L. Invariant amino acids essential for decoding function of polypeptide release factor eRF1. Nucleic Acids Res. 2005;33:6418–6425. doi: 10.1093/nar/gki927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Rospert S, Rakwalska M, Dubaquie Y. Polypeptide chain termination and stop codon readthrough on eukaryotic ribosomes. Rev. Physiol. Biochem. Pharmacol. 2005;155:1–30. doi: 10.1007/3-540-28217-3_1. [DOI] [PubMed] [Google Scholar]
- 67.Palmer E, Wilhelm JM, Sherman F. Phenotypic suppression of nonsense mutants in yeast by aminoglycoside antibiotics. Nature. 1979;277:148–150. doi: 10.1038/277148a0. [DOI] [PubMed] [Google Scholar]
- 68.Singh A, Ursic D, Davies J. Phenotypic suppression and misreading Saccharomyces cerevisiae. Nature. 1979;277:146–148. doi: 10.1038/277146a0. [DOI] [PubMed] [Google Scholar]
- 69.Rowe SM, Clancy JP. Pharmaceuticals targeting nonsense mutations in genetic diseases: progress in development. BioDrugs. 2009;23:165–174. doi: 10.2165/00063030-200923030-00003. [DOI] [PubMed] [Google Scholar]
- 70.Salas-Marco J, Bedwell DM. Discrimination between defects in elongation fidelity and termination efficiency provides mechanistic insights into translational readthrough. J. Mol. Biol. 2005;348:801–815. doi: 10.1016/j.jmb.2005.03.025. [DOI] [PubMed] [Google Scholar]
- 71.Alkalaeva EZ, Pisarev AV, Frolova LY, Kisselev LL, Pestova TV. In vitro reconstitution of eukaryotic translation reveals cooperativity between release factors eRF1 and eRF3. Cell. 2006;125:1125–1136. doi: 10.1016/j.cell.2006.04.035. [DOI] [PubMed] [Google Scholar]
- 72.Hatin I, Fabret C, Rousset JP, Namy O. Molecular dissection of translation termination mechanism identifies two new critical regions in eRF1. Nucleic Acids Res. 2009;37:1789–1798. doi: 10.1093/nar/gkp012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mantsyzov AB, Ivanova EV, Birdsall B, Alkalaeva EZ, Kryuchkova PN, Kelly G, Frolova LY, Polshakov VI. NMR solution structure and function of the C-terminal domain of eukaryotic class 1 polypeptide chain release factor. FEBS J. 2010;277:2611–2627. doi: 10.1111/j.1742-4658.2010.07672.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Legault P, Pardi A. 31P chemical shift as a probe of structural motifs in RNA. J. Magn. Reson. B. 1994;103:82–86. doi: 10.1006/jmrb.1994.1012. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.