


Abstract
Determining transcriptional regulator activities is a major focus of systems biology, providing key insight into regulatory mechanisms and co-regulators. For organisms such as Escherichia coli, transcriptional regulator binding site data can be integrated with expression data to infer transcriptional regulator activities. However, for most organisms there is only sparse data on their transcriptional regulators, while their associated binding motifs are largely unknown. Here, we address the challenge of inferring activities of unknown regulators by generating de novo (binding) motifs and integrating with expression data. We identify a number of key regulators active in the metabolic switch, including PhoP with its associated directed repeat PHO box, candidate motifs for two SARPs, a CRP family regulator, an iron response regulator and that for LexA. Experimental validation for some of our predictions was obtained using gel-shift assays. Our analysis is applicable to any organism for which there is a reasonable amount of complementary expression data and for which motifs (either over represented or evolutionary conserved) can be identified in the genome.
INTRODUCTION
A common theme of systems biology is to understand transcriptional regulation, i.e., to identify regulators driving a particular function and to determine the activities of those transcription factors (TFs). A key problem, however, is that TFs are typically regulated (activated) at the protein level; thus their expression (mRNA) is not a reliable surrogate for their activity. Although there are methods to directly ascertain protein modifications, e.g., iTRAQ (1,2), these methods are unable to give a global coverage and typically only a few samples are available. Further, the targets of these TFs are often unknown; TF binding sites can be ascertained directly by ChIP-Seq/chip, but the proportion of functional binding sites is low at 50–58% (3,4), most likely due to the absence of (unexpressed) codependents under the experimental conditions. Because of these problems, computational techniques have emerged to infer both the regulatory network and the associated activity of the specified regulators/TFs (3–9). A comparison of some of these methods were performed in (10). These methods combine gene expression data with TF binding site information, either predicted from a known motif or using ChIP-Seq/chip data. Essentially by restricting the network to the implied binding targets, expression data can be used to infer the activity of the associated TF. These methods perform best with high volumes of expression data, being able to utilize both steady state and time-series data. As such they are ideal methods for integration of data sets. However, all these methods are restricted to highly studied organisms such as Escherichia coli, Schizosaccharomyces pombe and Drosophila. To address TF/regulator activity in poorly studied organisms we present a generic method to infer regulator activities designed for when neither the regulators nor their binding motifs are known. The method integrates expression data with the genome sequence. It generalizes the above methods, inferring activities of de novo identified motifs from their target gene expression profiles. The requirements are a sequenced genome for which a de novo search of motifs is successful and a reasonable quantity of gene expression data (e.g. >50 arrays). Motifs can be detected either because of their over representation in the genome or because they are evolutionarily conserved. These motifs are used to define potential regulatory connections between the (unknown) TFs/regulators and their targets; the expression data is then used to trim the network into active and inactive (or false) binding sites with a concurrent inference of the activity of the associated regulators.
Our analysis is based on a modification of the model of Sabatti and James (6); itself based on a commonly used statistical model for determining explanatory variables through pattern identification, specifically a factor model. Gene expression is modelled as a linear regression on the activity of a small number of unknown factors—in our case the motif activities—
| (1) |
where eit is the expression of gene i, pjt the (unknown) activity of TF j, both at time t, aij the control strength of the transcriptional regulator through motif j on gene i, and γit Gaussian noise, assumed homogeneous in time but possibly gene specific. Connectivity is given by the connectivity matrix zij with zij = 1 implying that the TF associated with motif j regulates gene i; this is only possible if motif j lies in the upstream region of gene i. Then aij is zero if zij = 0. We implemented our model using a Markov chain Monte Carlo (MCMC) methodology, see Supplementary Data.
We applied our method to an important soil bacterium Streptomyces coelicolor, a model organism in the actinomycetes, a phylum that is responsible for the production of most of the antibiotics currently in use. Streptomyces coelicolor initiates production of antibiotics under nutrient depletion (11,12), undergoing a so-called metabolic switch (13) from primary metabolism (and growth), to secondary metabolism, producing a rich array of metabolites including up to four antibiotics. Despite decades of research the complex regulatory mechanisms responsible for the metabolic switch are largely unknown. This organism has 66 sigma factors and over 700 potential DNA binding regulators (Supplementary Table 8), while very few binding motifs are known. The list of known binding motifs includes a PHO box of the phosphate response regulator PhoP (SCO4230) (14–16), a key regulator in the response to limited phosphate; the DasR binding motif, a global regulator of carbon utilization (17); ARE-sequence binding sites of key regulators such as ScbR (18) that regulate secondary metabolism pathways; binding sites for pathway-specific regulators of the SARP family (19,20); an inferred binding motif for the sigma factor σR (SCO5216) (21), a key regulator in sensing oxidative stress; and binding sites for GlnR (SCO4159) (22). In our study we examined computational predictions for both evolutionary conserved motifs within the actinomycetes and over represented sequences in the genome. We found that over represented binding sites of the dyad type, i.e., two conserved sequences with a variable spacer between them, [a common pattern in bacterial genomes (23–26)], to be the most informative and only report on these in the following.
This article is organized as follows. In ‘Materials and Methods’ section, we describe the data sets, motif prediction, enrichment methodology, and factor model for integration of sequence and expression datasets. Also the experimental verification method is outlined. In ‘Results’ section, we enumerate the number of motif hits and implement a filtering/enrichment process to determine the informative motifs. On applying the factor model we find 10 distinct motif activity profiles/clusters supporting 61% of the target binding sites. We examine the overlap between target sets among motifs with similar activity profiles, detecting cases at both extremes—specifically, cases where the motifs model the same binding site, and where there is no target or motif correlation. In ‘Discussion’ section, we discuss the key motifs and their profiles. To determine the identity of the regulator we examine if there is any obvious homology of the motif to known bacterial motifs or if there are any regulators with expression profiles similar to the inferred activity profile.
MATERIALS AND METHODS
Gene expression data, differentially expressed genes
Gene expression data from three time series were used, TS1: wild-type under phosphate depletion (27), TS3: a phoP knock-out under phosphate depletion (M. Juarez et al., submitted for publication), and TS5: the wild-type under glutamate depletion (28). There were 94 times points in total. Normalization was performed using RMA. For each of the time series we determined the differentially expressed (DE) genes using BATS software (29) and clustered the DE genes in each TS using splinecluster (30).
Motif prediction and enrichment analysis
Upstream regions of genes (up to 300 bp) in the S. coelicolor genome (NCBI, http://www.ncbi.nlm.nih.gov/) (31) were used to search for over represented dyads using the software pipeline of (24). Using the operon definitions of ref. (32), we associate a motif located in the upstream regions of any of the genes within an operon with all genes in the operon. We used a hyper-geometric test to determine significant enrichment of dyad motifs in the DE genes and the expression profile clusters. To correct for multiple testing we used a Benjamini and Hochberg correction. Further details are given in Supplementary Data.
Factor model: integrating motif and expression data
We modified the hierarchical factor model of (6) and the associated MCMC algorithm for use on S. coelicolor. The underlying network model is a two-layered bipartite network where edges are between regulators and their target genes as shown in Supplementary Figure S2. We use a Bayesian methodology for the inference of the model parameters, i.e., matrices A, P, noise variance 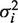 as well as network structure (connectivity matrix Z), Equation 1 (we use lowercase to denote elements of a matrix). Gibbs variable selection is used to infer the significant links (those with zij = 1) in the network topology. The motif predictions are used as prior information restricting the possible regulatory links as (6), i.e., zij is a random variable with zij ∈ {0, 1} iff motif j lies upstream of gene i (or is in the operon of i), otherwise zij = 0. We implemented both an MCMC and Metropolis Coupled MCMC algorithm to sample the posterior distribution of the model parameters. The latter overcame the slow mixing of the network topology variables Z which was a particular issue in this high GC organism. Convergence was assessed using multiple chains. Matlab code for the factor model is available from MI on request.
as well as network structure (connectivity matrix Z), Equation 1 (we use lowercase to denote elements of a matrix). Gibbs variable selection is used to infer the significant links (those with zij = 1) in the network topology. The motif predictions are used as prior information restricting the possible regulatory links as (6), i.e., zij is a random variable with zij ∈ {0, 1} iff motif j lies upstream of gene i (or is in the operon of i), otherwise zij = 0. We implemented both an MCMC and Metropolis Coupled MCMC algorithm to sample the posterior distribution of the model parameters. The latter overcame the slow mixing of the network topology variables Z which was a particular issue in this high GC organism. Convergence was assessed using multiple chains. Matlab code for the factor model is available from MI on request.
Electromobility shift assays
DNA fragments of genes of 100–250 bp upstream were amplified by PCR using genomic DNA of S. coelicolor M145 (genes and primers listed in Supplementary Table 7) and used for electromobility shift assay (EMSA) with S. coelicolor cell lysate under various culture conditions, see Supplementary Data.
RESULTS
We used the factor model, Equation 1 on data for the bacterium S. coelicolor, strain M145 (33), to determine the key regulator activity profiles and their associated regulatory motifs during the metabolic switch (13). Our expression data comprises three extended high-resolution longitudinal time series, TS1, TS3, TS5 that map the transcriptome over the switch from primary to secondary metabolism under nutrient depletion (phosphate or glutamate, see ‘Materials and Methods’ section). Between the three time series there is a total of 1620 genes that we considered DE in at least one time-series; these DE gene profiles and the motifs located in the genome by a de novo search were used in the factor model analysis.
Motif search:dyads
We searched the S. coelicolor genome for statistically over represented dyad type motifs, i.e., binding sites with two conserved sequences and a variable non-conserved spacer between them using the method described in (24,25). Within the upstream 300 bp we identified 2120 potential motifs (with conserved sequences of length 4 and 5 and a spacer length lying between 4 and 20 bp) across the 7769 genes [Uniprot-GOA annotation (34)]. A further subtlety arises here because of the high GC content of S. coelicolor; this reduces the information content in the motifs against more balanced nt genomes, especially for high GC motifs. Thus, the motifs with high GC content have a huge number of genome hits, much more than is realistic for a bacterial TF, Supplementary Figure S7. Thus, in our final motif shortlist we removed any motifs that have a very high number of genome hits (more than 300 operons) and high GC content (>75%). We filtered out motifs that obviously had no explanatory power for the transcriptome data, i.e., we restricted to motifs that were highly enriched in at least one of (i) the differentially expressed gene sets of TS1, 3 or 5, (ii) in the dynamic gene clusters of TS1, 3, or 5, or (iii) in a set of PhoP dependant genes obtained by a comparative analysis of TS1 and TS3 (i.e., genes which have a significant change in their time series variance between the two time series). This enrichment analysis was done at the level of operons (see ‘Methods’ section) using the operon definitions of (32). This left 55 motifs with an approximately Poisson distribution for the number of motifs per gene, Supplementary Figure S9 with mean 0.525, the main deviation occurring at high motif counts where we find a few targets have a higher number of motifs than the Poisson distribution. Motif logos (a qualitative representation of the sequence signature) for all the motifs are shown in the Supplementary Figures S17–S26.
Modelling motif activity: a factor model
The motif search defined a regulatory network comprising 55 motifs and 551 potential target genes with 855 links, i.e., an average connectivity of 1.55 motifs per gene, each motif occurring on average 16 times, Supplementary Figure S8. However, all binding sites may not be real, i.e., there may be false positives or sites may be inactive under our experimental conditions. Thus, to refine the network we used expression data to identify the active binding sites and their associated motifs. This was accomplished by using the factor model, Equation 1 constrained by the motif-binding site network above. Our application is more challenging than the original study on E. coli (6). We therefore had to modify the original model and MCMC inference algorithm to improve its performance on our data, see Supplementary Data. The analysis is Bayesian, computing the (posterior) probability P(zij = 1| Data) of each motif–gene interaction being present based on the expression data, shortened to P(zij = 1) in the following, while also inferring the activity profiles pjt of the potential regulator associated with each motif. In our model the prior probability on the connectivity is itself considered a variable (i.e., the indicator variable for a link between gene i, motif j, zij ∼ Binomial(ρj) is parametrized by ρj (motif specific), a random variable, with prior ρj ∼ Beta(2,2); this contrasts to (6) who fixed ρj at 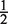 ). Throughout the article, significant targets for a given motif j will mean those targets for which the posterior link probability is greater than the prior, i.e., P(zij = 1) ≥ ρj. Inference of the model parameters was performed concurrently across all model parameters using an MCMC methodology (Supplementary Data).
). Throughout the article, significant targets for a given motif j will mean those targets for which the posterior link probability is greater than the prior, i.e., P(zij = 1) ≥ ρj. Inference of the model parameters was performed concurrently across all model parameters using an MCMC methodology (Supplementary Data).
The motif–gene links supported by our experimental time series were determined, Supplementary Table 1. The average number of significant targets per motif is 0.958, compared to the original 1.55 targets per motif, Supplementary Figure S8. A few of the motifs are even switched off altogether, e.g., Motif 51, Supplementary Figure S16, indicating that our selection of motifs was probably sufficiently broad. The posterior values of ρj for each motif j are shown in Supplementary Figure S10, again indicating that there is heterogeneity among the motifs in their explanatory power. Thus, the expression data introduces a strong selection on the potential binding sites with only 61% of the potentially informative computationally determined binding sites being active in our data. This is despite the fact that we initially selected only strong matches in the genome-wide search of binding sites (using a high threshold, HMMER score = 16).
Regulator activity profiles
The factor model predicts the activity profiles of the regulator associated with each motif, identifying when they are active in the respective experiments, Figure 1. There are some highly variable patterns, but also some commonality. For instance, there are a number of motifs with oscillations at the start of the time series and a class with gradual decay in all the time series, most of the latter also showing a slight dip in TS5 around the time of glutamate depletion suggesting that these may be related to the stringent response. The stringent response is very pronounced in TS5. A global decay pattern is not unexpected since the fermentation environment becomes more hostile over time as nutrients deplete and toxins accumulate. We grouped the activity profiles into clusters using hierarchical clustering on the profile correlations, see Supplementary Figure S11. A cut-off of r2 = 0.6 gave a good separation of patterns, grouping the profiles into 10 motif activity clusters, Figure 2. Some of the activity clusters comprise only single motifs (activity clusters 1, 6, 8 and 9), i.e., they have unique profiles across the time points while other groups (activity clusters 2, 3, 4, 5, 7 and 10) contain more than one motif indicating that their associated regulators have similar activity profiles. For example, activity cluster 5 contains all the regulators that have oscillations in their profiles prior to nutrient depletion; these oscillations are more pronounced in TS3 than TS1 and TS5 possibly because of the slower growth in TS3 (not shown), while activity cluster 7 includes all regulators which have an activity profile matching the gene expression profile of phoP (phosphate response regulator), (27), i.e., their activity is significantly increased at the time of phosphate depletion in TS1 while showing no significant activity in TS3 (where phoP is knocked-out) and TS5 (where phoP is inactive, data not shown). Motif 22 in this activity cluster is the PhoP motif, comprising directed repeats of the PHO box (14–16).
Figure 1.

Activity profiles of selected motifs (those having > 50% correlation with their targets). Shown in red is the (mean) predicted activity of the motif using the factor model while in blue is the average expression profile of the (at most) top five significant targets. The shaded area (light blue) shows the range of the gene profiles of these top significant targets. Vertical lines (grey) separate the three time series, TS1, TS3 and TS5 respectively. Activity profiles of all motifs are given in Supplementary Data (Supplementary Figures S14 and S15).
Figure 2.

Predicted motif activity profiles grouped into 10 activity clusters. Individual motif patterns are distinguished by colour. In each of the plots, vertical solid lines (Brown) separate the three times series, TS1, TS3 and TS5 respectively, while the vertical dashed lines (Magenta) correspond to the nutrient depletion time in each time series.
Although the activity clusters look compact, Figure 2, the correlation among their target sets is very variable. For example, for the PhoP-like activity cluster 7, the profile is very similar for all four motifs of the activity cluster but they do not share targets among each other except for 1 between motifs 40 and 47, Table 1. Further, their motif signatures do not share much similarity as shown in Figure 3, although all have a common pattern—a dyad with a repeated nt in each word of the dyad.
Table 1.
Proportion of targets shared by motifs in the PhoP activity cluster 7 (15 targets)
| Motifs | 21 | 22 | 40 | 47 | Targets |
|---|---|---|---|---|---|
| 21 | 1 | 0.0 | 0.0 | 0.0 | 5 |
| 22 | 0.0 | 1 | 0.0 | 0.0 | 4 |
| 40 | 0.0 | 0.0 | 1 | 0.2 | 5 |
| 47 | 0.0 | 0.0 | 0.5 | 1 | 2 |
Each entry corresponds to the proportion of common targets (defined as those having posterior P(zij = 1) > ρj)) among the motifs k (row) and j (column) relative the number of targets of motif k.
Figure 3.

Motif Sequence logos for activity cluster 7 (15 target genes, 4 motifs). (a–d) Motif logos. Motif 22 (b) is the S. coelicolor directed repeat PHO box, albeit with the missing nt G at the start of the first conserved sequence}. Motif logos generated using (35), http://weblogo.berkeley.edu/.
In contrast, activity cluster 5 comprises motifs that not only share similar activity profiles but share most of their targets. Further, the motifs overlap in the upstream regions, Figure 4 and there is high level of similarity in the motif sequences, Supplementary Figure S21. Motif 3 within this activity cluster contains all the significant targets within the motifs of cluster 5, Table 2, suggesting there is in fact only one binding motif.
Figure 4.

(a) Cluster 5 consensus logo created using the following procedure: The MEME suite (36) was used to construct a common underlying motif among the combined targets in this motif cluster. The logo in (a) was created from the multiple alignment of the sequences corresponding to the MEME output motifs plus some flanking sequences. This extended logo better represents the different overlapping motifs (Supplementary Figure S21) in this cluster than the MEME consensus logo generated from all targets in this activity cluster. (b) Locations of cluster 5 motifs in the upstream regions of the target genes (showing only those targets with at least two motif binding sites).
Table 2.
Proportions of significant targets (having posterior P(zij = 1) > ρj)) shared by motifs in activity cluster 5 (7 targets)
| Motifs | 3 | 39 | 41 | 42 | 43 | 44 | 46 | 50 | Targets |
|---|---|---|---|---|---|---|---|---|---|
| 3 | 1 | 0.14 | 0.14 | 0.14 | 0.42 | 0.14 | 0.14 | 0.14 | 7 |
| 39 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| 41 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| 42 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| 43 | 1 | 0.33 | 0.33 | 0.33 | 1 | 0.33 | 0.33 | 0.33 | 3 |
| 44 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| 46 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| 50 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
Each entry corresponds to the proportion of common targets among the motifs k (row) and j (column) relative the number of targets of motif k.
Members of the activity clusters 3, 4 and 10 do not have any common targets amongst themselves while the remaining activity clusters, except cluster 2, are only single motif activity clusters. Although cluster 2 contains most of the motifs (32 in total) there are a number of non-zero target overlaps in this activity cluster, Supplementary Table 5; i.e., the proportion of common targets is predominantly low. The motifs in this activity cluster which do share targets, e.g., motifs 16 and 19, 4 and 5 as well as motifs 7, 28 and 29, suggest that this activity cluster comprises of the order of 28 distinct motifs, and associated regulators, that regulate the response to the degrading environment. Locations of the upstream binding sites for motifs in cluster 2 are shown in Supplementary Figure S27 demonstrating that multiple motifs in the upstream regions are often well separated and thus distinct.
Sensitivity analysis
The original motif search required a significance threshold to be chosen (P or q-value), thereby affecting how many motifs we utilize in the analysis. Of concern is whether we have included sufficient motifs as explanatory variables or used too stringent a criterion such that the results are strongly dependent on the thresholds. Thus, we carried out a sensitivity analysis altering the enrichment threshold (from 9% to 10%) to increase the number of motifs to 72 from the previous 55, giving 688 genes with at least one motif. All other thresholds (HMMER threshold score of 16, GC content (<=75%), number of genome wide hits <=300) were identical. Examining the (posterior) probability of common links between the two analyses gave a high level of correlation (r = 0.82), Figure 5. Out of a total of 855 a priori common links, there is agreement on the majority of the links in both cases, i.e., 448 significant links in both cases and 283 links which are switched off in both while there are only 124 links which are significant in one case while switched off in the other, Figure 5. Further, the average number of posterior significant links per motif for the motifs in this analysis is 52% as compared to 56% in the original analysis. Out of the 55 common motifs, the number of inactive motifs (for which no posterior link is significant) was 2, respectively 3 in the larger/smaller runs. Of the additional 17 motifs of the larger run, one is inactive. We also clustered the predicted activity profiles of the 72 motifs into 9 clusters (r2 = 0.5), Supplementary Figure S12, reproducing all the dominant patterns of the smaller run.
Figure 5.

Robustness analysis. The posterior probabilities P(zij = 1) for all common motifs j and genes i are plotted for the two enrichment thresholds, 9% (Original) and 10%. All common motifs/genes between the two cases are plotted. Four subcategories are shown: blue asterisks for links which are ON (Significant) in First run and OFF (Not significant) in second, red squares show links which are OFF in first run and ON in second, green circles represent those links which are ON in both experiments while black point markers correspond to the links which are switched off in both cases. The right panel shows the number of links in each of the four categories of links in this comparison.
DISCUSSION: COMPARISON WITH THE KNOWN REGULATORS
In order to determine possible identities of the regulators associated with each motif we used two methods. Firstly, we searched for regulators that are transcriptionally regulated under our experimental conditions by comparing our inferred motif activities against the expression profiles of a list of potential regulators in S. coelicolor, Supplementary Table 8. We calculated the distance between the activity profile for each motif against all the regulators in this list. Matches include a range of bacterial regulators—transcriptional/response regulators, two component systems, DNA binding proteins etc. In Figure 6, we show a few of these matches. The top ten matches are given in Supplementary Table 3. Secondly, we compared our motifs against known bacterial regulator motifs reported in the literature (through RegulonDB), and with the motif comparison tool STAMP (37). STAMP uses multiple databases for motif comparison including DPInteract for E. coli regulators (http://arep.med.harvard.edu/dpinteract/index.html). Each hit was then examined for a corresponding regulator in S. coelicolor. These analyses are discussed below for some individual motifs (also see Supplementary Data for Motif 51 (Zur/Fur homolog) and Motif 27 (PhoB homolog)).
Figure 6.

Comparison of six of our predicted motifs (red) with their 5 best matching known regulators (blue) in S. coelicolor (vertical lines separate the three times series, TS1, TS3 and TS5 respectively). Matching profiles are ascertained by using an Euclidean distance metric. These top hits are, (a) motif 3: XRE family DNA binding protein (SCO6770 which contains HTH3 Helix-turn-helix domain), putative TetR family regulatory protein (SCO4313) and a TetR family transcriptional regulator (SCO0622), a Lacl family transcriptional regulator (SCO0062) as well as a sig15 sigma factor (SCO3068), (b) motif 8: SARP family regulator ActII-ORF4 (actinorhodin cluster regulator SCO5085), Afsr2 sigma like protein (SCO4425), ActII-1 putative TetR family transcriptional regulatory protein (SCO5082), an AraC family regulatory protein (SCO5017), and LuxR (absR2) regulatory protein (SCO6993), (c) motif 22: a response regulator PhoP (SCO4230) and AraC family transcriptional regulator (SCO0266), and MarR family regulatory protein (SCO0940), among others, (d) motif 23: DNA binding protein BldD (SCO1489), AraC family transcriptional regulator (SCO2792), Glk glucokinase (SCO2126) as well as transcriptional regulatory protein GlnR (SCO4159), (e) motif 34: MarR, MerR regulatory proteins (SCO5405, SCO2105) and TetR (SCO3129) family transcriptional regulators and a conserved hypothetical protein(SCO4639) as well as an anti Sigma factor (SCO7324), (f) motif 36: Matches to this motif include important transcriptional and response regulators like (SARP family) RedD (SCO5877) and RedZ (SCO5881) and a LuxR2comp two component regulator (SCO5785).
Motif 22: phosphate response regulator PhoP
Motif 22 (cluster 7 in Figure 2) has one of the most distinctive activity profiles with a dramatic transient increase in activity in TS1 at the time of nutrient (phosphate) depletion, while it is almost inactive (constant) in TS3 and TS5, see Supplementary Figure S13. The motif is similar to the binding motif of PhoP comprising directed repeats of the PHO box (essentially GTTCA) (14–16), (although the first letter ‘G’ is lost/weak in each repeat, Figure 3). Together this suggests that the regulator associated with motif 22 is PhoP. This is confirmed by it's high confidence targets that are previously identified members of the PhoP regulon (14–16); SCO4142 which encodes a phosphate binding protein precursor within the high affinity phosphate transporter pst operon (SCO4139-42), SCO1393 which encodes for an acetoacetyl-CoA synthetase, SCO3790 a conserved hypothetical protein and SCO1906 a secreted protein. The activity profile has a high correlation to the three regulators PhoP (SCO4230), PhoU (SCO4228), AraC (SCO0466), (r = 0.83, 0.84 and 0.67, respectively). It is known that PhoP is transcriptionally activated under phosphate depletion (11,15,16) and regulates PhoU; hence explaining this high correlation.
We previously noted that three other motifs (21,39,46) in cluster 7 have very similar activity profiles but there are negligible common targets. Examination of the targets of motif 21 in activity cluster 7 reveals that one of the binding sites is within the upstream region of the diverging genes SCO4873 and SCO4874, i.e., within the genomic cluster (SCO4873-4882) of the pho regulon involved in the phosphate-free biosynthesis of secondary polymers of the cell wall (27), a known target of PhoP. Motif 40 has SCO4228, SCO4229 as targets, also known to be in the PhoP regulon (the phosphate two component system phoURP itself). However, these are the only targets that can be linked to the PhoP regulon reported in; i.e., the other nine targets are not known to be in the PhoP regulon. These targets also show occasional dynamics in TS3 and TS5 distinguishing them from those of motif 22, Supplementary Figure S13. Further, a search for common motifs using MEME (36) within the combined targets of the motifs in this cluster found a PHO box as a consensus motif in the upstream regions of 6 of the 15 targets of cluster 7, Supplementary Table 6, which include three out of four targets of motif 22, two out of five targets (SCO4873 and SCO4884) of motif 21 and SCO4229 (encoding the sensor kinase PhoR). Despite these differences in the performance of computational motif prediction methods, this analysis gives support to the suggestion that PhoP does not regulate all these targets and our original motif decomposition indicating multiple regulators is essentially correct, Table 1. This is consistent with an independent analysis of this data that suggests that there is a PhoP-independent phosphate response (M. Juarez et al., submitted for publication).
Cluster 5: the siderophores
The second rich dynamic pattern comprises eight motif activity profiles that group into activity cluster 5, Figure 2, activity profiles that oscillate prior to nutrient depletion with TS3 and TS5 exhibiting the strongest oscillations. There is high overlap in their significant targets (all having motif 3) while the motifs have high levels of similarity, Supplementary Figure S21. Further, motifs are closely clustered in the upstream regions of all targets (for genes having more than one binding site), Figure 4. This indicates that these motifs are different overlapping versions of same underlying motif. We created a consensus sequence logo for this cluster using MEME (36), Figure 4.
The target profile strongly suggests that this motif is the regulatory binding site for the siderophores and other iron-related genes. Among the 7 significant targets in this cluster we find 3 of the 18 siderophore genes, i.e., SCO0498, SCO2780 (putative secreted protein), and SCO7400. However, the siderophores are organized into operons, specifically SCO0494-97 and SCO2783-85 (32); the targets SCO0498 and SCO2780 lie upstream of these two operons respectively, indicating that our targets probably include more than 1/2 of the siderophore genes. In fact SCO0498 (cchB) encodes an acyl transferase peptide monooxygenase of the coelichelin biosynthesis cluster (38); in S. coelicolor coelichelin acts as a siderophore. Further, cchB is regulated by PhoP under phosphate-limited conditions (16) and is a target of the σR regulon (39). The other four targets are SCO0499 (putative formyltransferase), SCO5999 (aconitase), SCO1926 (putative DNA-binding protein) and SCO1623 (conserved hypothetical protein). The Streptomyces aconitases belong to the Iron-Regulatory-Protein/AcnA family, while the aconitase AcnA of S. viridochromogenes possesses regulatory function within iron metabolism (40). Thus, there is a high density of iron-related genes among the targets of cluster 5.
Transcriptional oscillations have been previously reported in iron homeostasis in E. coli, including the observation of damped oscillations in the siderophores, (41). The hypothesized mechanism is through a coupling of iron transport regulating Fur binding. If the mechanism of the oscillations is similar, i.e., exponential growth in our fermentors leads to an iron concentration down shift that induces oscillation in the iron sensing circuit, then this suggests that this motif is that for the Fur homologue in S. coelicolor, furS [SCO0561, (42)]. Of note, we failed to find a putative regulator with similar dynamics, consistent with regulation through iron binding. Further, the motif is unknown, although we did find a Zur/Fur motif homologue (Motif 51), see Supplementary Data.
Motif 6: CRP family
Motif 6, a palindrome in activity cluster 2, looks identical to a bacterial motif for the CRP family of regulators (RegulonDB), Figure 7. These proteins play a key role in bacterial stress responses (43). A putative CRP transcriptional regulator in S. coelicolor is SCO7543 (http://strepdb.streptomyces.org.uk). Since the activity profile has little distinguishing structure there were many hits with expression profiles of putative regulators, Supplementary Table 3, we are therefore unable to identify potential transcriptionally regulated CRP family regulators associated with motif 6. The 10 targets of this motif include the acetyltransferase gene (SCO1864), the cytochrome oxidase gene (SCO3945), the ferredoxin gene (SCO5135) and some conserved hypothetical proteins. We used a gel-shift assay to obtain preliminary experimental confirmation for the most promising targets of this motif. We detected strong gel-shifts in SCO3320, SCO3945, SCO4562, see Supplementary Figure S5, while there was no shift in corresponding control sequences.
Figure 7.

Motif 6 (CRP): (a) Sequence logo for motif 6. (b) Sequence logos for two bacterial CRP family regulators which are very similar to motif 6 with P-values of 1.89e−10 and 6.1e−08. (c) Predicted Activity profile (red) along with the expression range (filled area with mean in blue) of the target genes. Vertical solid lines (Brown) separate three times series, TS1, TS3 and TS5 respectively while the vertical dashed lines (Magenta) correspond to the nutrient depletion time in each time series.
Motif 35: analogues of E. coli regulator LexA
Motif 35 of cluster 2 (8 targets), a directed repeat motif, has a predicted generic decay of activity over all three time series with a sharp inhibition after nutrient depletion in TS1 and at depletion in TS5, recovering temporarily in the latter. Alternatively, the patterns after depletion may be viewed as evidence of oscillations, especially in TS3 and TS5. Thus, it is distinct from the gradual decay in the majority of motifs in activity cluster 2. There was a significant match of the motif (using STAMP) with that of LexA, Figure 8, a known transcriptional repressor in E. coli, (P–value = 6.24e−04). The analogue in S. coelicolor is also called LexA by sequence homology. We conclude that motif 35 is the LexA binding motif, while the correlation between the inferred motif activity profile and lexA expression is 0.60 indicating some degree of transcriptional control. The eight targets of motif 35 are SCO5085 (ActII-ORF4, actinorhodin cluster activator protein), SCO5646 (probable solute binding lipoprotein), SCO5737 (gpsI guanosine pentaphosphate synthetase), SCO4662 (tuf1 elongation factor TU-1), SCO3961 (serS Seryl-tRNA synthase), SCO5169 (putative ATP-binding protein), SCO5899 (hypothetical protein), and SCO3089 (putative ABC transporter ATP-binding protein).
Figure 8.

Motif 35 (LexA). (a) Sequence logo for motif 35 (b) Sequence logo for bacterial regulator lexA which is found as a best match to motif 35. (c) Predicted Activity profile (red). Annotation as Figure 7.
Motifs 8 and 36: antibiotic synthesis
Motifs 8 and 36 are similar directed repeat motifs, but the former has a strongly structured activity profile, the latter a substantially weaker one, Figure 9. The activity profile for motif 8 shows a distinct pattern with delayed activation relative to nutrient depletion, an activation that is slower in TS5. The targets include part of the actinorhodin synthesis cluster suggesting that this motif corresponds to the associated SARP (ActII-ORF4, SCO5085); this is consistent with the high correlation of the activation profile with this gene, r = 0.949. Specifically, we have as targets two neighbouring genes (SCO5071 and SCO5072) on opposite strands, while SCO5072 is upstream of the genes SCO5073-5080 on the same strand and constitutes part of ACT cluster (27). Other targets in the ACT cluster are SCO5086 (ketoacyl reductase), SCO5087 (actinorhodin polyketide beta-ketoacyl synthase alpha subunit), and SCO5091 (cyclase). For motif 36, one of its target genes SCO5888 (redP) is upstream of a large cluster of genes (SCO5889-SCO5898) which comprise part of the undercylprodigiosin (Red) cluster. Further, redD, redZ are within the top 5 regulatory genes that have an expression profile which correlates with the activity profile, (r = 0.75, 0.72 for redD and redZ respectively), Figure 6, although the average correlation of motif 36 with its targets is poor (r=0.0245), Figure 9. This suggests that motif 36 may be the motif for RedD (SCO5877), the regulatory cascade occurring through Z → D → red biosynthesis (SCO5886-5897), the pathway from afsS to redZ being unknown (44). Other targets of motif 36 which are not part of Red cluster are SCO4947 (nitrate reductase alpha chain NarG3), SCO3928 (putative thiamine biosynthesis protein), SCO1245 (adenosylmethionine-8-amino-7-oxononanoate aminotransferase), SCO7403 (putative membrane protein), and SCO0902 (hypothetical protein). The structure of the motifs also supports the suggestion that these correspond to binding sites for SARPs; they have a periodicity of 11 nt corresponding to a complete turn of the DNA, while motif 8 has the previously identified distinctive TCGA pattern (19).
Figure 9.

Antibiotic synthesis: (a) Sequence logo for motif 8. (b) Sequence logo for motif 36. (c) Predicted Activity profile (red) for motif 8 and 36. Annotation as Figure 7.
CONCLUSION
We present a new methodology for the analysis of transcriptomic data in poorly studied organisms that integrates expression data with the genome sequence. The requirements for using our technique are low, only requiring a sequenced genome in which motifs can be detected, and a sufficiently informative expression data set comprising either multiple time series and/or steady state data. In contrast to many other integration methods, e.g., (45), a training set is not required. We applied our method to S. coelicolor, a model organism in the actinomycetes examining expression data during nutrient depletion (27,28 and M. Juarez et al., Submitted for publication). We found 10 distinct patterns of ‘motif’ activity among the 55 motifs analysed (based on 94 arrays, 3 time series), Figure 2. The dominant pattern (activity cluster 2) was a gradual decay comprising 32 motifs, although there were some differences in the detail of the profiles within this activity cluster, Figures 2, 7 and 8, particularly at nutrient depletion. Given the lack of target overlaps we suggest that there are multiple regulators within this cluster with similar activity; separating these regulators would require additional data under conditions in which they are differentially activated. There were three highly distinctive activity patterns that are similar to the phoP expression profile (activity cluster 7), Figure 2, which included the PHO box directed repeat motif (and three others), a secondary metabolism biosynthesis profile (activity cluster 8 with targets that include the act cluster), Figures 2 and 9 and an oscillatory activity profile (activity cluster 5, that includes the siderophores in their targets), Figure 2. We made tentative predictions of the identity of the associated regulators. Our method successfully detected some known regulators, in particular PhoP and its PHO box, and the antibiotic SARPS ActII-ORF4, RedD/Z. Further, we identified the possible motif of the CRP regulator SCO7543. The correlation of the inferred activity of these motifs with the regulator's gene expression indicates whether it is transcriptionally regulated or is predominantly post-translationally regulated.
Preliminary experimental confirmation of some of our results was obtained by using gel-shift assays. These assays only provide evidence that there is a protein bound to the upstream region tested, and can not directly prove that the predicted motif is responsible. We found strong evidence of protein binding for targets containing motifs 6, 19, 20 and 25, Supplementary Figure S5 and Supplementary Table 7. We also found evidence of binding for SCO0079 with S. coelicolor M145 cell lysate from S-medium (48 h); SCO0079 was a prediction under an earlier analysis for binding Motif 6 but removed later during the enrichment filtering step. Thus, altogether from 26 DNA sequences that were tested in EMSAs, five were identified to be specifically retarded (under a 500-fold excess dilution of unlabelled DNA), while no retardation occured for any of the negative controls. The reason why the success rate is so low may be because of the experimental conditions. In such assays, purified binding protein is normally applied in appropriate concentrations to the respective target DNA but since we do not know the binding protein, we used cell lysate. Thus, the concentration of the binding protein in the S. coelicolor cell lysates may be low or the effector absent under the current conditions. Therefore, although the confirmation rate was low, we consider this as sufficient evidence to demonstrate that the analysis method is able to detect biologically relevant targets, and sufficient to motivate further analysis using more sophisticated, and challenging techniques that are able to identify the bound protein.
Our analysis identified key regulatory profiles and potential regulators in each of the experimental time-series. The wt response to phosphate depletion (TS1) is dominated by the exceptional strong activity cluster 7 pattern, the associated regulators being specific to this TS. This cluster includes the PhoP directed repeat PHO box binding site (motif 22), although this motif was present in only 1/2 the targets of this cluster. Our analysis therefore suggests that there are multiple regulators besides PhoP with similar profiles, which may be either downstream of the signalling cascade initiated by PhoP, or in parallel. Under glutamate depletion in the wt (TS5) we found a number of motifs with activity localized to nutrient depletion, and weak, or unresponsive to phosphate depletion in TS1/3. This suggests that motifs 6 (homologous to the CRP motif), 12, 13, 15, 25, 38, 54 correspond to specific cascades for carbon or nitrogen limitation, while a number of motifs have an inferred localized activity at/around both phosphate and glutamate depletion in wt suggesting a common stress response: this includes motifs 26, 27 and 35 (homology to E. coli LexA motif). Finally, we found a couple of activity profiles localized at phosphate depletion in the phoP KO: including motifs 11, 29, 33, motif 36 showing a phosphate response in TS1 & 3, while motifs 1, 14, 53 show activity in all time-series at depletion. This analysis indicates that the wt response to phosphate depletion is highly coordinated, primarily through PhoP, while response to glutamate depletion has considerable diversity with a rich range of activity profiles. The weakest signals were found in the phoP KO under phosphate depletion; a small number specific to this case were found, and a couple in common with glutamate depletion. This indicates that in absence of a PhoP response to deal with low phosphate, both a new specific response and a common response to glutamate depletion are triggered.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Tables 1–8, Supplementary Figures S1–S28, Supplementary Methods, Supplementary Results and Supplementary References [46–53].
FUNDING
Biotechnology and Biological Sciences Research Council (BBSRC), UK, grant number BB/FF003498/1, awarded through the ERA-NET SysMO initiative. (to M.I.); Higher Education Commission of Pakistan in collaboration with Dow University of Health Sciences, Karachi, Pakistan, and the ERA-IB Immunotech project (0315931) to R.A. Experimental data was generated under STREAM, an international consortium funded under the ERA-NET SysMO initiative (Systems Biology of Microorganisms) http://www.sysmo.net. Funding for open access charge: Biotechnology and Biological Sciences Research Council (BBSRC).
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
Names and contacts of the members of the STREAM consortium can be found at https://www.wsbc.warwick.ac.uk/groups/sysmopublic. We thank J. Moore for bioinformatic support.
REFERENCES
- 1.Ross PL, Huang YN, Marchese JN, Williamson B, Parker K, Hattan S, Khainovski N, Pillai S, Dey S, Daniels S, et al. Multiplexed protein quantitation in Saccharomyces cerevisiae using amine-reactive isobaric tagging reagents. Mol. Cell. Proteomics. 2004;3:1154–1169. doi: 10.1074/mcp.M400129-MCP200. [DOI] [PubMed] [Google Scholar]
- 2.Shadforth IP, Dunkley TPJ, Lilley KS, Bessant C. i-Tracker: for quantitative proteomics using iTRAQ. BMC Genomics. 2005;6 doi: 10.1186/1471-2164-6-145. 145, doi:10.1186/1471-2164-6-145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gao F, Foat BC, Bussemaker HJ. Defining transcriptional networks through integrative modeling of mRNA expression and transcription factor binding data. BMC Bioinformtics. 2004;5 doi: 10.1186/1471-2105-5-31. doi:10.1186/1471-2105-5-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ucar D, Beyer A, Parthasarathy S, Workman CT. Predicting functionality of protein-DNA interactions by integrating diverse evidence. Bioinformatics. 2009;25:i137–i144. doi: 10.1093/bioinformatics/btp213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liao J, Boscolo R, Yang Y, Tran LM, Sabatti C, Roychowdhury VP. Network component analysis: reconstruction of regulatory signals in biological systems. PNAS. 2003;100:15522–15527. doi: 10.1073/pnas.2136632100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sabatti C, James GM. Bayesian sparse hidden components analysis for transcription regulation networks. Bioinformatics. 2006;22:739–746. doi: 10.1093/bioinformatics/btk017. [DOI] [PubMed] [Google Scholar]
- 7.Sanguinetti G, Lawrence ND, Rattray M. Probabilistic inference of transcription factor concentrations and gene-specific regulatory activities. Bioinformatics. 2006;22:2775–2781. doi: 10.1093/bioinformatics/btl473. [DOI] [PubMed] [Google Scholar]
- 8.Honkela A, Girardot C, Gustafson EH, Liu YH, Furlong EE, Lawrence ND, Rattray M. Model-based method for transcription factor target identification with limited data. Proc. Natl Acad. Sci. USA. 2010;107:7793–7798. doi: 10.1073/pnas.0914285107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Khanin R, Vinciotti V, Mersinias V, Smith CP, Wit E. Statistical reconstruction of transcription factor activity using Michaelis-Menten kinetics. Biometrics. 2007;63:816–823. doi: 10.1111/j.1541-0420.2007.00757.x. [DOI] [PubMed] [Google Scholar]
- 10.Pournara I, Wernisch L. Factor analysis for gene regulatory networks and transcription factor activity profiles. BMC Bioinformatics. 2007;8:61. doi: 10.1186/1471-2105-8-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Martín JF, Liras P. Engineering of regulatory cascades and networks controlling antibiotic biosynthesis in Streptomyces. Curr. Opin. Microbiol. 2010;13:263–273. doi: 10.1016/j.mib.2010.02.008. [DOI] [PubMed] [Google Scholar]
- 12.Rokem JS, Lantz AE, Nielsen J. Systems biology of antibiotic production by microorganisms. Natural Prod. Rep. 2007;24:1262–1287. doi: 10.1039/b617765b. [DOI] [PubMed] [Google Scholar]
- 13.Novotna J, Vohradsky J, Berndt P, Gramajo H, Langen H, Li XM, Minas W, Orsaria L, Roeder D, Thompson CJ. Proteomic studies of diauxic lag in the differentiating prokaryote Streptomyces coelicolor reveal a regulatory network of stress-induced proteins and central metabolic enzymes. Mol Microbiol. 2003;48:1289–1303. doi: 10.1046/j.1365-2958.2003.03529.x. [DOI] [PubMed] [Google Scholar]
- 14.Sola-Landa A, Rodríguez-García A, Apel AK, Martín JF. Target genes and structure of the direct repeats in the DNA-binding sequences of the response regulator PhoP in Streptomyces coelicolor. Nucleic Acids Res. 2008;36:1358–1368. doi: 10.1093/nar/gkm1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rodríguez-García A, Sola-Landa A, Apel AK, Santos-Beneit F, Martín JF. Phosphate control over nitrogen metabolism in Streptomyces coelicolor: direct and indirect negative control of glnR, glnA, glnII and amtB expression by the response regulator PhoP. Nucleic Acids Res. 2009;37:3230–3242. doi: 10.1093/nar/gkp162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rodríguez-García A, Barreiro C, Santos-Beneit F, Sola-Landa A, Martín JF. Genome-wide transcriptomic and proteomic analysis of the primary response to phosphate limitation in Streptomyces coelicolor M145 and in a ΔphoP mutant. Proteomics. 2007;7:2410–2429. doi: 10.1002/pmic.200600883. [DOI] [PubMed] [Google Scholar]
- 17.Rigali S, Titgemeyer F, Barends S, Mulder S, Thomae AW, Hopwood DA, van Wezel GP. Feast or famine: the global regulator DasR links nutrient stress to antibiotic production by Streptomyces. EMBO Rep. 2008;9:670–675. doi: 10.1038/embor.2008.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Folcher M, Gaillard H, Nguyen LT, Nguyen KT, Lacroix P, Bamas-Jacques N, Rinkel M, Thompson CJ. Pleiotropic functions of a Streptomyces pristinaespiralis autoregulator receptor in development, antibiotic biosynthesis, and expression of a superoxide dismutase. J. Biol. Chem. 2001;276:44297–44306. doi: 10.1074/jbc.M101109200. [DOI] [PubMed] [Google Scholar]
- 19.Wietzorrek A, Bibb M. A novel family of proteins that regulates antibiotic production in streptomycetes appears to contain an OmpR-like DNA-binding fold. Mol. Microbiol. 1997;25:1177–1184. doi: 10.1046/j.1365-2958.1997.5421903.x. [DOI] [PubMed] [Google Scholar]
- 20.Sheldon PJ, Busarow SB, Hutchinson CR. Mapping the DNA-binding domain and target sequences of the Streptomyces peucetius daunorubicin biosynthesis regulatory protein, DnrI. Mol. Microbiol. 2002;44:449–460. doi: 10.1046/j.1365-2958.2002.02886.x. [DOI] [PubMed] [Google Scholar]
- 21.Paget MS, Kang JG, Roe JH, Buttner MJ. σRan RNA polymerase sigma factor that modulates expression of the thioredoxin system in response to oxidative stress in Streptomyces coelicolor A3(2) EMBO J. 1998;17:5776–5782. doi: 10.1093/emboj/17.19.5776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Reuther J, Wohlleben W. Nitrogen metabolism in Streptomyces coelicolor: transcriptional and post-translational regulation. J. Mol. Microbiol. Biotechnol. 2007;12:139–146. doi: 10.1159/000096469. [DOI] [PubMed] [Google Scholar]
- 23.Touzain F, Schbath S, Debled-Rennesson I, Aigle B, Kucherov G, Leblond P. SIGffRid: a tool to search for sigma factor binding sites in bacterial genomes using comparative approach and biologically driven statistics. BMC Bioinformatics. 2008;9:73. doi: 10.1186/1471-2105-9-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Studholme DJ, Bentley SD, Kormanec J. Bioinformatic identification of novel regulatory DNA sequence motifs in Streptomyces coelicolor. BMC Microbiology. 2004;4:14. doi: 10.1186/1471-2180-4-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li H, Rhodius V, Gross C, Siggia ED. Identification of the binding sites of regulatory proteins in bacterial genomes. Proc. Natl. Acad. Sci. USA. 2002;99:11772–11777. doi: 10.1073/pnas.112341999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Robison K, McGuire AM, Church GM. A comprehensive library of DNA-binding site matrices for 55 proteins applied to the complete Escherichia coli K-12 genome. J. Mol. Biol. 1998;284:241–254. doi: 10.1006/jmbi.1998.2160. [DOI] [PubMed] [Google Scholar]
- 27.Nieselt K, Battke F, Herbig A, Bruheim P, Wentzel A, Jakobsen ØM, Sletta H, Alam MT, Merlo ME, Moore J, et al. The dynamic architecture of the metabolic switch in Streptomyces coelicolor. BMC Genomics. 2010;11 doi: 10.1186/1471-2164-11-10. doi:10.1186/1471-2164-11-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Waldvogel E, Herbig A, Battke F, Amin R, Nentwich M, Nieselt K, Ellingsen TE, Wentzel A, Hodgson DA, Wohlleben W, et al. The PIIprotein GlnK is a pleiotropic regulator for morphological differentiation and secondary metabolism in Streptomyces coelicolor. Appl. Microbiol. Biotechnol. 2011;92:1219–1236. doi: 10.1007/s00253-011-3644-1. [DOI] [PubMed] [Google Scholar]
- 29.Angelini C, Cutillo L, De Canditiis D, Mutarelli M, Pensky M. BATS: a Bayesian user-friendly software for Analyzing Time Series microarray experiments. BMC Bioinformatics. 2008;9:145. doi: 10.1186/1471-2105-9-415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Heard NA, Holmes CC, Stephens DA. A quantitative study of gene regulation involved in the immune response of Anopheline mosquitoes: an application of Bayesian hierarchical clustering of curves. J. Amer. Stat. Assoc. 2006;101:18–29. [Google Scholar]
- 31.Bentley SD, Chater KF, Cerdeño-Tárraga AM, Challis GL, Thomson NR, James KD, Harris DE, Quail MA, Kieser H, Harper D, et al. Complete genome sequence of the model actinomycete Streptomyces coelicolor A3(2) Nature. 2002;417:141–147. doi: 10.1038/417141a. [DOI] [PubMed] [Google Scholar]
- 32.Charaniya S, Mehra S, Lian W, Jayapal KP, Karypis G, Hu W. Transcriptome dynamics-based operon prediction and verification in Streptomyces coelicolor. Nucleic Acids Res. 2007;35:7222–7236. doi: 10.1093/nar/gkm501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kieser T, Bibb MJ, Buttner MJ, Chater KF, Hopwood DA. Practical Streptomyces Genetics. Norwich: John Innes Foundation; 2000. [Google Scholar]
- 34.Barrell D, Dimmer E, Huntley RP, Binns D, O'Donovan C, Apweiler R. The GOA database in 2009–an integrated Gene Ontology Annotation resource. Nucleic Acids Res. 2009;37:D396–D403. doi: 10.1093/nar/gkn803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Crooks GE, Hon G, Chandonia JM, Brenner SE. WebLogo: a sequence logo generator. Genome Res. 2004;14:1188–1190. doi: 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bailey TL, Elkan C. Proceedings of the Second International Conference on Intelligent Systems for Molecular Biology. Menlo Park, California: AAAI Press; 1994. Fitting a mixture model by expectation maximization to discover motifs in biopolymers; pp. 28–36. [PubMed] [Google Scholar]
- 37.Mahony S, Benos PV. STAMP: a web tool for exploring DNA-binding motif similarities. Nucleic Acids Res. 2007;35:W253–W258. doi: 10.1093/nar/gkm272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Barona-Gómez F, Lautru S, Francou FX, Leblond P, Pernodet JL, Challis GL. Multiple biosynthetic and uptake systems mediate siderophore-dependent iron acquisition in Streptomyces coelicolor A3(2) and Streptomyces ambofaciens ATCC 23877. Microbiology. 2006;152:3355–3366. doi: 10.1099/mic.0.29161-0. [DOI] [PubMed] [Google Scholar]
- 39.Kallifidas D, Thomas D, Doughty P, Paget MSB. The σR regulon of Streptomyces coelicolor A3(2) reveals a key role in protein quality control during disulphide stress. Microbiology. 2010;156:1661–1672. doi: 10.1099/mic.0.037804-0. [DOI] [PubMed] [Google Scholar]
- 40.Schinko E, Schad K, Eys S, Keller U, Wohlleben W. Phosphinothricin-tripeptide biosynthesis: an original version of bacterial secondary metabolism? Phytochemistry. 2009;70:1787–1800. doi: 10.1016/j.phytochem.2009.09.002. [DOI] [PubMed] [Google Scholar]
- 41.Amir A, Meshner S, Beatus T, Stavans J. Damped oscillations in the adaptive response of the iron homeostasis network of E. coli. Mol. Microbiol. 2010;76:428–436. doi: 10.1111/j.1365-2958.2010.07111.x. [DOI] [PubMed] [Google Scholar]
- 42.Hahn JS, Oh SY, Roe JH. Regulation of the furA and catC operon, encoding a ferric uptake regulator homologue and catalase-peroxidase, respectively, in Streptomyces coelicolor A3(2) J. Bacteriol. 2000;182:3767–3774. doi: 10.1128/jb.182.13.3767-3774.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Körner H, Sofia HJ, Zumft WG. Phylogeny of the bacterial superfamily of Crp-Fnr transcription regulators: exploiting the metabolic spectrum by controlling alternative gene programs. FEMS Microbiol. Rev. 2003;27:559–592. doi: 10.1016/S0168-6445(03)00066-4. [DOI] [PubMed] [Google Scholar]
- 44.Lian W, Jayapal KP, Charaniya S, Mehra S, Glod F, Kyung YS, Sherman DH, Hu WS. Genome-wide transcriptome analysis reveals that a pleiotropic antibiotic regulator, AfsS, modulates nutritional stress response in Streptomyces coelicolor A3(2) BMC Genomics. 2008;9:56. doi: 10.1186/1471-2164-9-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zwir I, Huang H, Groisman EA. Analysis of differentially-regulated genes within a regulatory network by GPS genome navigation. Bioinformatics. 2005;21:4073–4083. doi: 10.1093/bioinformatics/bti672. [DOI] [PubMed] [Google Scholar]
- 46.Okanishi M, Suzuki K, Umezawa H. Formation and reversion of streptomycetes protoplasts: cultural condition and morphological study. J. Gen. Micro. 1974;80:389–400. doi: 10.1099/00221287-80-2-389. [DOI] [PubMed] [Google Scholar]
- 47.Claessen D, Rink R, de Jong W, Siebring J, de Vreugd P, Boersma FG, Dijkhuizen L, Wosten HA. A novel class of secreted hydrophobic proteins is involved in aerial hyphae formation in Streptomyces coelicolor by forming amyloid-like fibrils. Genes Dev. 2003;17:1714–1726. doi: 10.1101/gad.264303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Thompson JD, Higgins DG, Gibson TJ. CLUSTALW: improving the sensitivity of progressive multiple sequence alignments through sequence weighting, position specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. Royal Stat. Soc. Series B (Methodological) 1995;57:289–300. [Google Scholar]
- 50.Gilks WR, Roberts GO. Strategies for improving MCMC. In: Gilks WR, Richardson S, Spiegelhalter , editors. Markov chain Monte Carlo in Practice. London: Chapman and Hall; 1996. pp. 89–114. [Google Scholar]
- 51.Geyer CJ. Markov chain Monte Carlo maximum likelihood. In: Keramidas , editor. Computing Science and Statistics: Proceedings of the 23rd Symposium on the Interface. Fairfax Station: Interface Foundation; 1991. pp. 156–163. [Google Scholar]
- 52.Altekar G, Dwarkadas S, Huelsenbeck JP, Ronquist F. Parallel Metropolis coupled Markov chain Monte Carlo for Bayesian phylogenetic inference. Bioinformatics. 2004;20:407–415. doi: 10.1093/bioinformatics/btg427. [DOI] [PubMed] [Google Scholar]
- 53.Patzer SI, Hantke K. The zinc-responsive regulator Zur and its control of the znu gene cluster encoding the ZnuABC zinc uptake system in Escherichia coli. J. Biol. Chem. 2000;275:24321–24332. doi: 10.1074/jbc.M001775200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.