


Abstract
Trf4/5p-Air1/2p-Mtr4p polyadenylation complex (TRAMP) is an essential component of nuclear RNA surveillance in yeast. It recognizes a variety of nuclear transcripts produced by all three RNA polymerases, adds short poly(A) tails to aberrant or unstable RNAs and activates the exosome for their degradation. Despite the advances in understanding the structural features of the isolated complex subunits or their fragments, the details of complex assembly, RNA recognition and exosome activation remain poorly understood. Here we provide the first understanding of the RNA binding mode of the complex. We show that Air2p is an RNA-binding subunit of TRAMP. We identify the zinc knuckles (ZnK) 2, 3 and 4 as the RNA-binding domains, and reveal the essentiality of ZnK4 for TRAMP4 polyadenylation activity. Furthermore, we identify Air2p as the key component of TRAMP4 assembly providing bridging between Mtr4p and Trf4p. The former is bound via the N-terminus of Air2p, while the latter is bound via ZnK5, the linker between ZnK4 and 5 and the C-terminus of the protein. Finally, we uncover the RNA binding part of the Mtr4p arch, the KOW domain, as the essential component for TRAMP-mediated exosome activation.
INTRODUCTION
Eukaryotic cells produce diverse types of protein-coding as well as noncoding RNAs, some of which have been recently shown to have direct regulatory role in the regulation of gene expression and chromatin structure [reviewed in (1)]. Most, if not all, RNAs are synthesized as a precursor molecule that needs to be posttranscriptionally processed and/or modified in order to form mature functional molecules. Several human genetic disorders relate to abnormalities in RNA processing and proper ribonucleoprotein assembly, such as cancer or neurodegenerative diseases (2). In yeast, nuclear RNA maturation and stability are under strict control from RNA surveillance carried out by the nuclear exosome and its cofactor the TRAMP4/5 polyadenylation complex (3–6).
The TRAMP recognizes a variety of nuclear transcripts produced by all three RNA polymerases, such as precursors of small nuclear and nucleolar RNAs (snRNAs and snoRNAs), ribosomal RNAs (rRNAs), tRNAs, as well as telomeric, and cryptic unstable transcripts (CUTs) (6–8). It adds short poly(A) tails to aberrant transcripts, creating a favorable substrate for the exosome (3–6,9), reviewed in (10). Among the complex subunits, Mtr4p provides helicase activity and Trf4p or Trf5p provide polyA-polymerase (PAP) activity, Air2p and Air1p are zinc-knuckle (ZnK) proteins with a predicted RNA-binding role within TRAMPs. The degradation presumably involves multiple polyadenylation cycles and the helicase activity of Mtr4p (5). In some instances, the RNA degradation does not require the polyadenylation activity of TRAMP (7,11,12). The mechanistic understanding of the substrate recognition and exosome activation is currently very limited.
The minimal TRAMP PAP consists of a heterodimer of one of the Trf4 or Trf5 proteins and either Air1p or Air2p (4–6). Trf4p and Trf5p are the catalytic subunits of the complex, whereas Air1p and Air2p are predicted to provide RNA-binding. Air proteins contain five tandemly arranged ZnK motifs of the CCHC-type (CX2CX4HX4C) that have previously been shown to bind nucleic acids and proteins in unrelated viral proteins (13). The ZnK regions in these proteins recognize exposed guanosines of RNA loops or in single-stranded RNA (13). The RNA helicase Mtr4p is not required for the polyadenylation activity of TRAMP; however, it regulates the length of a poly(A) tail added by Trf4p-Air2p dimer and modulates its polyadenylation rate in vitro (14).
Recently, three works reported on the structure of the full-length Mtr4p and a heterodimer of Trf4p-Air2p fragments (15–17). The structures of Mtr4p revealed a five-domain organization in which four domains constitute the DExH helicase core [RecA-1, RecA-2, winged helix (WH) and a helical bundle] (16,17). Compared to known structures of other DNA and RNA helicases, Mtr4p DExH core has an N-terminal β-hairpin extension located on the surface of the globular core. The most unique feature however is an insertion of 265 amino acids between the WH helices protruding from the globular core to form the arch domain with an arm-like structure and a fist (also termed the KOW domain) at the end. The KOW domain shares similarities with rRNA-binding proteins and contributes to RNA binding in vitro (17). The arch is dispensable for Mtr4 helicase and ATPase activities and for TRAMP integrity in vitro. A strain expressing an archless Mtr4p displays a slow growth phenotype that seems to result from an Rrp6p-dependent incomplete processing of 5.8S rRNA (16).
The study of the structure of the minimal Trf4p-Air2p heterodimer, covering the catalytic and central domains of Trf4p and only the last two ZnKs of Air2p, revealed a phylogenetically conserved surface on the central domain of Trf4p that contacts the last linker and ZnK region of Air2p (15). Nevertheless, this study still left many open questions such as what is the role of the other Air2p ZnK domains and the N- and C- terminal regions of Air2p and Trf4p. Finally, the RNA-binding mode of Trf4p-Air2p has been addressed only indirectly. The structural determination of the entire complex has proven to be difficult so far due to the instability of recombinant subunits. Moreover, it still remains a mystery as to how TRAMP interacts with and activates the downstream acting exosome. Thus despite the advances in our understanding of TRAMP function in vivo and of the individual structures, there is still very limited knowledge on the assembly of the complex and of the mechanistic details of its action.
Here, we have focused on two aspects of the TRAMP4 biochemistry. On one hand, we aimed to identify the code of recognition of structurally complicated substrates that need to be polyadenylated by TRAMP4 before exosome degradation, such as the hypomodified tRNAiMet. Second, to tackle the TRAMP4 complex assembly and molecular mechanisms of its function, we have performed detail analyses of Trf4p, Air2p and Mtr4p domain organization in vitro and in vivo. Furthermore, we succeeded to obtain the solution structure of the five zinc-knuckle region of Air2p by NMR spectroscopy. Overall, we uncovered regions required for cell viability, protein–protein interactions, RNA binding domains and regions responsible for exosome activation.
MATERIALS AND METHODS
Yeast cultures and manipulation
Yeast were cultured under standard conditions in media with selective markers corresponding to particular strains and vectors. Yeast transformations were performed by lithium acetate method. For growth test analyses, yeast were cultured overnight in minimal SD medium at 30°C, diluted with fresh SD media for OD600 0.2 and grown at 30°C to OD600 1. The cells were then serially diluted in decimal dilutions and incubated at 17°C, 25°C, 30°C and 37°C in SD media (or as indicated in each experiment). The yeast strains used in this work are listed in Supplementary Table S2.
Purification of protein complexes from yeast
The yeast culture was grown at 30°C in YPD medium to OD600 ∼2.5. Cells were harvested, frozen in liquid nitrogen and manually grinded. Lysate was melted in D100 buffer [100 mM KCl, 50 mM Tris, 10% (w/v) glycerol, 0.02% (v/v) Nonidet P–40 (NP–40), 0.2 mM EDTA, pH 7.9] with protease inhibitors, soluble fraction was recovered by centrifugation and bound to IgG-sepharose beads for 3 h at 4°C. Beads were washed thoroughly with D150 (same as D100 but 150 mM KCl) or D1000 (high-salt wash, same as D100 but 1 M KCl) buffer, and proteins were eluted by TEV protease cleavage to D150 buffer. Elutions were snap frozen and kept at −80°C. Alternatively, elutions were merged and bound o/n to Ni-NTA agarose beads. The beads were washed with D150 buffer with 10 mM imidazole and eluted by 250 mM imidazole to D50 buffer. Elutions were snap frozen and kept at −80°C.
All immunoprecipitated samples that were subsequently subjected to western blot analysis were prepared in the presence of 100 µg ml−1 of RNase A (added to the cell lysate). Samples that were used for enzymatic assays (polyadenylation, exosome degradation) were prepared in the absence of RNase in order to avoid interference of the residual RNase with the enzymatic analysis.
RNA isolation and analysis
For RNA analysis, cells were grown at 30°C o/n in liquid SD media, diluted to OD600 0.25 and grown at 25°C to OD600 1. RNA was isolated by the hot phenol extraction and stored at −80°C. Five micrograms of total RNA was denatured in 25% formamide, separated on 8% denaturing (8 M urea) polyacrylamide gel and transferred to nylon membrane using semi-dry electro-transfer. RNA was cross-linked to the membrane by UV light (120 mJ cm−2) and hybridized with a probe in Ultra-Hyb buffer (Ambion) according to manufacturer’s instructions.
Construction of yeast plasmids
Yeast genomic DNA or WT full-length TRF4, AIR2 and MTR4 inserted in pET22b (5) were used to PCR-amplify the untranslated and coding regions of TRF4, AIR2 and MTR4 genes.
TRF4 constructs
To obtain plasmids containing truncated forms of the TRF4 gene, the desired region was amplified in a standard PCR reaction using primers listed in Supplementary Table S3 and inserted into pNOPPATA1L vector via restriction sites NdeI and SalI. To obtain point mutated forms, a standard PCR mediated site directed mutagenesis with pNOPPATA1L-TRF4 (5) as a template was performed. All constructs contained N-terminal fusion proteinA tag and a TEV protease recognition sequence. With the exception of C-terminally truncated forms (that is TRF4 CΔ544, CΔ499, CΔ463 and 182–463), the constructs also contained fusion C-terminal hexahistidine anchor. The His6 tag was not used during analyses and we observed no sign of its interference with the results. To express endogenous levels of the Trf4p, the genomic region spanning 500 nt upstream of the TRF4 initiation codon was amplified using primers TRF4 506 5′-UTR XbaI For and TRF4 506 5′-UTR ATG SphI XhoI Rev and inserted into the pRS413 vector via XbaI and XhoI restriction sites. Into this construct, the TRF4 full-length gene and/or alleles D425A, DXD and/or NΔ96 from pNOPPATTA1L constructs including all fusion tags were inserted through SphI site. For Escherichia coli expression, the TRF4 coding sequence was inserted into the expression vector pET30a(+) allowing for the N-terminal fusion G-protein B1 tag (18) by using BamHI and XhoI restriction sites.
AIR2 constructs
Yeast genomic DNA or WT AIR2 gene inserted in pET22b (5) were used to PCR-amplify the untranslated and coding regions of AIR2. To prepare AIR2 constructs for phenotypic analyses in vivo, the coding sequence of AIR2 surrounded by 425-bp upstream of initiation codon and 479-bp downstream of stop codon was PCR amplified from Saccharomyces cerevisiae genomic DNA (BY4741 background) with primers AIR2 5′-UTR_For1 and AIR2 3′-UTR_Rev1. The fragment was inserted into the pRS415 vector via PstI and NotI restriction sites resulting in the plasmid V379. To prepare tagged versions of AIR2 for affinity purifications of TRAMP complexes, the full-length CDS of AIR2 was subcloned to a yeast expression vector [pNOPPATA1L, (5)] via NdeI and SalI restriction enzymes, resulting in the plasmid V53. Single point mutations of AIR2 were prepared with WT AIR2 in pNOPPATA1L as a template by site-directed mutagenesis by using oligonucleotides listed in Supplementary Table S3. AIR2 truncation mutants were constructed by PCR amplification with specific primers listed in Supplementary Table S2. The PCR product was inserted into pNOPPATA1L vector using NdeI and SalI restriction sites. To generate mutants pRS415 derived constructs, we first introduced an NdeI restriction site at the position 3-bp upstream of AIR2 start codon resulting in the construct V394 WT (with primers AIR2 NdeI ATG For and AIR2 NdeI ATG Rev). Individual mutants prepared in pNOPPATA1L were then transferred via NdeI and BamHI sites to generate constructs listed in the Supplementary Table S4.
MTR4 constructs
Full-length MTR4 and all deletion variants were cloned into pNOPPATA1L vector. C-terminal His6 tag was added via PCR amplification with an extended reverse primer (MTR4 His SalI). The full-length MTR4 was amplified from genomic DNA (primers MTR4 NdeI and MTR4 His SalI); MTR4 archless was amplified from pAV674 vector (16), which was a kind gift from Dr Van Hoof. ΔKOW MTR4 version was prepared by ligation of two PCR products into pNOPPATA1L vector (using NdeI/SalI restriction sites). N-terminal part (amplified with primers MTR4 KOW N) of MTR4 (aminoacids 1–665) was extended at the 3′-end by coding sequence for Gly-Ser-Gly-Ser-Ala-Ser and C-terminal part of MTR4 (aminoacids 819–1073, obtained with oligos MTR4 KOW C) was extended at 5′-end by coding sequence for Ala-Ser-Gly-Ser-Gly-Ser resulting in GSGSASGSGS bridge (connected via NheI restriction site in Ala-Ser coding sequence).
Preparation of unmodified tRNAiMet for enzymatic and NMR binding experiments
The plasmid ptRNAiMet for the in vitro transcription of yeast tRNAiMet (5) was linearized with BstNI. Unlabeled hypomodified tRNAiMet was produced by in vitro T7 polymerase run off transcription (19) using linearized plasmid as a template. The product was purified by anion exchange HPLC under denaturing conditions and desalted by a NAP column (Sephadex G-25). The hypomodified tRNAiMet was denatured at 95°C, pH 6.2 for 5 min and snap cooled on ice, to favor a monomeric conformation. The concentration of RNA was determined by UV-spectroscopy.
Fluorescence anisotropy
The equilibrium binding of Air2p ZnK1-5 (aminoacids 57–180) to different oligonucleotides was analyzed by fluorescence anisotropy. The 15-mer poly-A RNA and DNA oligonucleotides were 5′-labeled with N,N,N′,N′-tetramethyl-6-carboxyrhodamine (TAMRA) that was attached via a hexyl linker. The measurements were conducted on a FluoroMax-4 spectrofluorometer (Horiba Jobin-Yvon, USA). The instrument was equipped with a thermostatted cell holder with a Neslab RTE7 water bath (Thermo Scientific, USA). The system was operated using FluorEssence software (version 2.5.3.0, Horiba Jobin-Yvon, USA). The TAMRA fluorophore was excited at 561 nm, and its emission was collected at 581 nm. The widths of both excitation and emission monochromatic slits were 8 nm and integration time was set to 3 s. During the measurement 10 nM labeled oligonucleotide (volume 1.4 ml) was titrated with increasing amounts of the protein in 50 mM Tris buffer (pH 7.9), supplemented with 250 mM NaCl, 20 mM β-mercaptoethanol and 50 µM ZnSO4. The titrations were carried out at 20°C in a stirred 1.5 ml quartz cuvette. A fixed delay of 30 s was set between each aliquot addition and start of the measurement to allow the reaction to reach equilibrium. This delay was sufficient, as no further change in anisotropy was observable. Every data point is an average of three measurements. The data were analyzed in SigmaPlot 11 software (Systat Software, USA). The experimental isotherms were fit to a single-site binding model according to Heyduk and Lee (20) using nonlinear least squares regression. The data were normalized for visualization purposes.
Preparation of DNA probes for northern blot analysis
DNA probes for NEL025c and snR33 were amplified from S. cerevisiae BY4741 genomic DNA with the primers combinations (NEL025c For and Rev, and snR33 − mature + extension For and Rev, see Supplementary Table S5) and used as templates for Random Primed DNA Labeling Kit (Roche) according to manufacturer’s instructions. To detect U14 and snR13 3′-extended snoRNAs and tRNAiMet, the oligonucleotides were 5′-phosphate labeled using T4 polynucleotide kinase (NEB) and [γ-32P]-ATP.
Whole-cell protein extract preparation for western blot analysis
The yeast culture was grown at 30°C in selective medium to OD600 ∼1.0. The cells were resuspended in the D150 buffer with protease inhibitors (0.5 μg ml−1 of leupeptin, 0.8 μg ml−1 of pepstatin A, 0.6 mM PMSF and 0.6 mM DTT). The cell suspension was vortexed with glass beads at 4°C for 10 min. The beads and the cell debris were pelleted by centrifugation, a part of the cell-free extract was mixed with 1×SDS loading buffer for western blot analysis. The rest of the extract was used for protein concentration measurement by the method of Bradford.
Antibodies
The generation of specific antibodies against Trf4p, Air2p, Air1p have been described in (5,7). Mtr4p antibodies were kindly provided by Dr Patrick Linder (University of Geneva). The anti-proteinA antibodies and the anti-His6 antibodies were from Sigma. All antibodies were used at a 1:5000 dilution.
Expression and purification of recombinant proteins
Full-length Trf4p from S. cerevisiae was recombinantly expressed from pET30a vector with N-terminally fused G-proteinB1 solubility tag (18) in E. coli strain BL21-CodonPlus (DE3)-RIPL (Stratagene). The expression was induced by 0.5 mM IPTG at 37°C for 2 h in the presence of 1 mM MgCl2. Cells were lysed in Lysis Buffer (50 mM carbonate buffer, 300 mM NaCl, 5% glycerol, 5 mM imidazole, 0.1% NP-40, 1 mM MgCl2, 2 mM β-mercaptoethanol and protease inhibitors, pH 10.5) by sonication, and protein was purified in 50 mM Tris buffer, pH 8.0 containing 300 mM NaCl, 5% glycerol, 0.02% NP-40, 1 mM MgCl2, 1 mM β-mercaptoethanol using affinity Ni-NTA chromatography. For NMR studies, the GB1-Trf4 fusion protein was dialyzed into a buffer containing 50 mM Tris, pH 8.0, 300 mM NaCl, 3% glycerol and 1 mM MgCl2 and subsequently concentrated up to 230 μM by VIVASPIN column (Sartorius).
Wild-type and mutant forms of AIR2 were recombinantly expressed from the pET22b vector in E. coli strain BL21-CodonPlus (DE3)-RIPL (Stratagene). The expression was induced by 0.4 mM IPTG at 37°C for 4 h in the presence of 0.5 mM ZnSO4. Cells were lysed in the lysis buffer (50 mM carbonate buffer, 400 mM NaCl, 25% sacharose, 2 mM MgCl2 10% glycerol, 400 μM ZnSO4, pH 10.5), and all proteins were purified in buffer containing 50 mM carbonate buffer, pH 10.5, 400 mM NaCl, 10% glycerol, 400 μM ZnSO4 using affinity Ni-NTA chromatography followed by gel filtration.
Expression and preparation of Air2 ZnK protein for NMR studies
The coding sequence of Air2 protein comprising residues 57–180 from S. cerevisiae was expressed with the N-terminal fusion Smt3 protein (21). The expression was induced by 100 µM IPTG in E. coli BL21-Codon Plus (DE3)-RIPL (Stratagene) strain overnight at 16°C in M9 minimal medium, supplemented with 50 µM ZnSO4. For isotope labeling, the medium was supplemented with 15NH4Cl and [U-13C6] glucose. Cells were harvested, resuspended in denaturing buffer (50 mM Tris, 500 mM NaCl, 8 M Urea, pH 8.0) and lysed by sonication, and proteins were purified using affinity Ni-NTA chromatography. The protein immobilized on beads was extensively washed and subsequently refolded in buffer containing 50 mM Tris pH 7.9, 500 mM NaCl, 20 mM β-mercaptoethanol, 5 mM imidazole, 50 µM ZnSO4, and protease inhibitors. Refolded protein was eluted and dialyzed against buffer containing 50 mM Tris pH 7.9, 500 mM NaCl, 20 mM β-mercaptoethanol and 50 µM ZnSO4. The Smt3 fusion protein was cleaved by Ulp1p protease and removed on Ni-NTA column. For NMR measurements the protein was concentrated using a Vivaspin 20 concentrator (Sartorius).
NMR spectroscopy and structure determination
The protein used in NMR spectroscopy was concentrated to ∼150 μM. Due to a high salt buffer [50 mM sodium phosphate buffer, 500 mM NaCl, 10 mM β-mercaptoethanol pH 8.0 (90% H2O/10% D2O)], 3 mm or shaped NMR tubes were used. All NMR experiments were recorded at 20°C on a Bruker AVANCE 600 MHz and 900 MHz spectrometers equipped with a cryogenic triple resonance probe (Bruker BioSpin). The chemical shifts of backbone and side-chains were assigned using standard triple resonance experiments (22,23). All distance restraints were derived from the 3D 15N- and 13C-edited NOESYs (with mixing time of 80 ms) collected at 900 MHz spectrometer. The structure calculations were performed with the automated NOE assignment module implemented in the CYANA program (24). Initial structures were further refined with the CYANA-generated restraints along with the restraints for the tetrahedral zinc coordination with AMBER 10.0 software (25) as described previously (26,27). From 40 refined structures, the 20 conformers with the lowest AMBER energy were selected to form the final ensemble of structures.
Enzymatic assays
Polyadenylation assays were carried out in 15 μl reaction mixtures containing 5–50 ng of affinity purified complex, 50 fmol 5′-end labeled RNA, 0.5 mM ATP, 5 mM MgCl2, 25 mM Tris, pH 7.9, 20 mM KCl, 10% glycerol, 0.1 mg ml−1 BSA, 1 mM DTT, 0.02% Nonidet P-40, 5U of RNasin Plus (Promega) with or without addition of 50 ng of affinity purified Rrp6-TAP eluate (5). Reaction mixture was incubated at 30°C, and 5 μl aliquotes were collected at times indicated. The reaction was stopped by the addition of 25 mM EDTA and proteins removed by incubation with ProteinaseK (Promega) for 10 min at 37°C. Sample was mixed with one volume of formamide loading buffer and resolved on denaturing polyacrylamide gel.
Database searches and sequence analysis
BLAST searches were conducted by using the NCBI genome databases (http://www.ncbi.nlm.nih.gov/BLAST/) or other nonredundant or EST databases at the Swiss EMBnet node (28). The multiple sequence alignment was performed by using ClustalW2 (29) at the http://www.ebi.ac.uk/Tools/services/web_clustalw2 interface. The Boxshade server (http://www.ch.embnet.org/software/BOX_form.html) was used to color the similarity in the alignment.
RESULTS
Identification of Air2p regions required for intermolecular contacts with Trf4p and Mtr4p
Trf4/5p, unlike canonical PAPs, possess no identifiable RNA-binding domain and thus are inactive on their own. We have previously demonstrated that Air2p activates Trf4p, likely through its RNA-binding properties (5). To identify Air2p regions that contact RNA, interact with Trf4p or Mtr4p, and are required for yeast viability, we have prepared Air2p mutants carrying point mutations in the second zinc-coordinating cysteine in each of the five ZnKs (ZnK1 to ZnK5) to disrupt the ZnK fold (Figure 1A and Supplementary Figure S1A). In order to tackle the importance of the surrounding N- and C-termini, we generated deletion mutants lacking either the N- or C- terminus or both (Figure 1A). Mutants ZnK4, ZnK5, CΔ180 and ZnK1-5L were not able to rescue growth defect of the air1Δair2Δ strain at higher temperature and mutants in the first three ZnKs and NΔ57 showed reduced fitness (Figure 1B). The growth defects observed at 30°C were not caused by reduced expression nor stability of Air2p variants nor other TRAMP subunits, as the western blot analysis of protein extracts prepared from cells grown at 30°C revealed comparable protein levels for the mutants tested (Supplementary Figure S1B and S1C). This pointed to the importance of the last two zinc knuckles and the Air2p C-terminus. Our previous yeast two-hybrid results indicated the minimal region in Air1 and Air2 proteins required for Trf4p interactions positioned across the zinc knuckles ZnK3 to ZnK5 (5). Accordingly, in NMR titration experiments, we observed that the fourth and the fifth ZnKs of Air2p associated with recombinant Trf4p (data not shown). Furthermore, we observed chemical shift perturbation in the third (L3) and fourth linker (L4) regions upon Trf4p binding. It concerned mainly residues 117–120 (L3) and residues S138, I139, W140, R141 and Y143 (L4), most of them being highly conserved between Air homologs from distant species (Supplementary Figure S1A). The analysis of additional mutants in those regions revealed that double mutants in the last two ZnKs ZnK4+5 and in the fourth linker (W140G+R141E), respectively, exhibited poor fitness comparable to the WT strain at all temperatures tested (Figure 1B). The growth defects observed at 24°C correlated with deficiencies in TRAMP function in vivo as northern blot analyses revealed a strong accumulation of NEL025c CUT and snR33 pre-snoRNA in ZnK4+5, L4, CΔ180 and ZnK1-5L mutants cultivated at 24°C (Figure 1C). Cold sensitivity and to a certain extent RNA degradation defects seen at 24°C were observed also in all the other ZnK point mutants indicating that all ZnKs contribute to some degree to TRAMP activity in vivo (Figure 1B and C).
Figure 1.

Air2p N- and C-termini and ZnK4 and ZnK5 domains are important for TRAMP functions and integrity. (A) Overview of mutant forms of AIR2 used in this study: ZnK, zinc knuckle. The aminoacid substitutions of individual point mutants are designated in brackets. (B) Growth test analysis of WT and mutant forms of Air2p episomally expressed in air1Δair2Δ strain, serially diluted on SD-Leu media and incubated for 3 days at temperatures indicated. (C) Northern blot analysis of total RNA isolated from AIR2 WT and mutant yeast strains grown at 25°C, with specific probes for RNAs indicated. The number below each lane represents percentage of unprocessed snoRNA precursor. (D) Western blot analysis of the composition of purified TRAMPs containing WT and mutant Air2p variants. TRAMP subunits were detected with antibodies indicated: HSW, high-salt wash (1M KCl).
To address whether the in vivo phenotypes reflected defects in TRAMP assembly or catalytic activity, we isolated and analyzed yeast complexes containing the individual mutated Air2p versions. In agreement with our NMR studies, L4 and ZnK5 mutants failed to efficiently copurify Trf4p and Mtr4p, whereas the other point mutations did not affect TRAMP4 assembly (Figure 1D). Western blot analysis of the input fractions of individual Air2p mutants revealed comparable levels of all three TRAMP subunits in WT, ZnK and L4 mutant strains. Thus the lack of Trf4p and Mtr4p copurification with L4 and ZnK5 was not caused by their decreased stability or expression in these mutant strains. As the absence of Mtr4p does not disrupt the interaction with Trf4p (Figure 1D, HSW) (4,5), we conclude that the fourth linker and last zinc knuckle regions of Air2p are crucial for the binding to Trf4p. The lack of Trf4p and Mtr4p copurification in ZnK5 samples was reflected in the lack of PAP activity of the purified protein sample (Figure 2A). Interestingly, although the ZnK4 mutant was able to maintain the TRAMP integrity (Figure 1D), it was strongly defective in polyadenylation in vitro (Figure 2A). Accordingly, recombinant Air2 proteins mutated in ZnK4 or ZnK5 failed to activate yeast Trf4p in vitro (Figure 2B).
Figure 2.

Air2p ZnK4 and ZnK5 are essential for TRAMP polyadenylation activity in vitro. (A) In vitro polyadenylation assay with affinity purified WT and mutant Air2p TRAMPs was performed in the presence of 0.5 mM ATP and 50 fmol of radioactively labeled hypomodified tRNAiMet (migration indicated by an arrow). Reactions were stopped after 15, 30 and 60 min, respectively. (B) Reconstitution of Trf4p PAP activity with recombinant Air2p. In vitro polyadenylation assay was performed with 60 ng of WT and mutant recombinant Air2p and 20 ng of WT Trf4p affinity purified from air1Δair2Δ yeast strain in the presence of [α-32P]-ATP and 50 fmol unlabeled hypomodified tRNAiMet (migration indicated by an arrow). The presence of a radioactive signal in a lane is the result of polyadenylation activity. Reactions where Trf4p was omitted (np) serves as a negative control for bacterial PAP contamination. Trf4p purified from air1Δair2Δ strain has no detectable activity alone (lane 4, yTrf4p). Trf4p purified from WT yeast strain (TRAMP4) was used as a positive control (lane 3, TRAMP4). Reactions were stopped after 60 min in the case of recombinant proteins or 10 min in the case of TRAMP4. The purity of recombinant Air2 proteins used in the assay separated on 12% polyacrylamide gel and stained with Coomassie blue are shown on the right. (C) Rrp6p-TAP purified exosome fraction used in all degradation assays of this work is compared to profile of an exosome purified via the core subunit Rrp4p. The purified complexes were resolved on 10% SDS-PAGE and silverstained. The migration position of Rrp6p is marked by an arrow. M is molecular weight marker. (D) ZnK1, 2 and 3 are dispensable for exosome activation. In vitro coupled polyadenylation/exosome assays with 30 ng TRAMP4 obtained via affinity purification of WT and mutant Air2p. The reactions were performed in the presence of 0.5 mM ATP and 50 fmol of radioactively labeled hypomodified tRNAiMet (migration indicated by an arrow). Reactions were stopped after 15, 30 and 60 min, respectively. RNAs were resolved on 20% PAGE.
We have previously reported that purified TRAMP4 complex activates exosomes in vitro (5). In order to test whether some of the mutants may affect exosome activation, we performed coupled polyadenylation/exosome assays. In this assay, the 5′-end labeled hypomodified tRNAiMet (a well-defined in vivo target of this pathway) is incubated in the presence of WT or mutant TRAMPs and yeast nuclear exosome purified via the Rrp6p subunit (5). Figure 2C shows Rrp6p-TAP purified fractions that were used in all subsequent degradations assays in this work compared to exosome obtained via the core subunit Rrp4p. The coupled polyadenylation/degradation assays revealed that none of the polyadenylation-competent Air2p point mutants further affected exosome activation in vitro (Figure 2D).
The analysis of deletion mutants indicated the importance of the first 25 N-terminal aminoacids for the interaction with Mtr4p. We observed that both the NΔ57 (residues 57-344) and ZnK1-5L (residues 26-205) proteins did not co-purify Mtr4p (Figure 1D, lanes NΔ57 and ZnK1-5L). Interestingly, the NΔ57 protein on contrary pulled down significantly higher amounts of Trf4p than WT even in the presence of 1M KCl (HSW). At present, we do not know the reason for higher yields of Trf4p in this mutant. These results were not due to significantly altered levels of Mtr4p nor Trf4p in these mutants (Supplementary Figure S1C). In addition, the deletion of the last 139 amino acids weakened the interaction with both Mtr4p and Trf4p (Figure 1D, samples CΔ180 and ZnK1-5L).
Air2p is a genuine RNA-binding protein
To further investigate why TRAMP4 mutant in Air2p ZnK4 lacks PAP activity, we studied the central region of Air2p (ZnK1-5, residues 57-180) by NMR spectroscopy. The Air2p was purified under denaturing conditions and refolded on Ni-NTA column in the presence of N-terminal Smt3 fusion protein. We first confirmed, that this fragment is sufficient to activate the PAP activity of Trf4p (Figure 2B, first lane). For NMR experiments, a high-salt concentration of 500 mM NaCl was used to prevent the protein aggregation. Although this protein construct suffered from a very low solubility limit (150 µM), we were able to achieve 1H, 13C and 15N resonance assignments of ZnK1-5 and determine its three-dimensional structure (Figure 3A). The chemical shift deviations of Cα and Hα of the assigned backbone resonances of ZnK1-5 from the sequence-dependent random coil values show no pattern for the ZnK1-5 region. This agrees well with the absence of secondary structure elements in the topology of zinc knuckles (13,22). The histidines of the CCHC motif of ZnKs, namely H71, H90, H109, H131 and H172, are involved in coordination of zinc ions as the N-H correlations of these residues span the same region in the 2D 1H-15N HSQC (Figure 3B) that is characteristic for zinc knuckles (22). Furthermore, the presence of folded zinc knuckles was confirmed by the addition of EDTA in excess of zinc ions that resulted in a loss of resonance dispersion in the 2D 1H-15N HSQC (data not shown). The NOESY spectra suffered from a low signal-to-noise ratio as a result of low solubility of the ZnK1-5 protein and the presence of high-salt buffer. Therefore, the resulting NMR structure is of a low resolution (Supplementary Table S1, Supplementary Figure S2). The structure of this Air2p region consists of five independent zinc knuckles that all adopt a canonical zinc knuckle fold of the CCHC type. The domains are connected by flexible linkers, and no inter-domain contacts have been found (Figure 3A). The conserved IWRxY motif of linker 4 is also unstructured in the free form, whereas it forms a short α-helix upon binding to Trf4p (15).
Figure 3.

Structure and RNA-binding of ZnK1-5 Air2p. (A) The lowest energy structure of the Air2p ZnK1-5. The protein is shown as a ribbon model (in red) with the zinc-coordinating residues (CCHC) shown in yellow. The zinc ions are shown in blue. (B) Close-up view of the 1H-15N TROSY spectrum, showing the N-H correlations of histidines that are involved in the coordination of zinc ions. (C) Air2p ZnK1-5 binds A15 RNA. Binding isotherms for equilibrium binding of ZnK1-5 Air2p to fluorescently labeled A15 RNA, monitored by fluorescence anisotropy. (D) Air2p ZnK1-5 binding to dA15 DNA. The ionic strength and pH of the binding buffer was the same for both measurements. The dissociation constant (Kd) was calculated from the best fit of data using a single-site binding isotherm. (E) 1H-15N TROSY spectra of Air2p ZnK1-5 alone (in blue) and in the presence of one equivalent of hypomodified tRNAiMet (in red; 1H-15N HSQC) at 20°C. (F) Summary of chemical shift perturbations and line broadening of Air2p ZnK1-5 upon binding to hypomodified tRNAiMet. Affected residues (qualitatively described by perturbation index; yes=1, no=0) are plotted against the amino-acid residue number. The assignments of residues indicated by asterisks could not be obtained. (G) Solvent-accessible surface representation of the RNA-binding ZnK of Air2p (ZnK2, ZnK3 and ZnK4) colored by electrostatic potential (blue, positive; red, negative) of the representative structure.
To investigate whether Air2p is a genuine RNA-binding protein, we performed a quantitative solution-binding assay using fluorescence anisotropy (FA) experiments. We found that Air2p ZnK1-5 binds unstructured A15 RNA with a KD in a low micromolar range (Figure 3C), whereas the binding affinity to dA15 DNA could not be detected in our experimental setup (KD >> 100µM) (Figure 3D). Next, we used NMR spectroscopy to reveal which ZnK domains of Air2p are used to bind to hypomodified tRNAiMet, a substrate that is used for in vitro assays in this work. The chemical shift changes and resonance broadening observed in the 1H-15N HSQC spectrum of Air2p ZnK1-5 upon the addition of tRNAiMet established that tRNAiMet interacts with the second, the third, and the fourth ZnKs (Figure 3E and F). In addition, the chemical shifts of the linkers connecting these ZnKs were perturbed as well. This suggested that these highly conserved linkers that are unstructured in the free form, become structured upon RNA-binding akin to the linkers of viral nucleocapsid (NC) proteins (13). Our attempts to compare binding of Air2p to hypomodified tRNAiMet and native tRNAiMet (the latter being a poor substrate for TRAMPs) failed, as we were not able to prepare sufficient quantities of native tRNAiMet from yeast for NMR experiments.
The minimal functional Trf4p
The recent crystal structure of truncated Trf4p bound to a fragment of Air2p encompasses only the conserved catalytic (CAT) and central domains (CD) of Trf4p (residues 161–481) (15). We observed that these regions alone (fragments 182–463 or 161–481) are not functional in vivo as they were not able to rescue viability of the double deletion strain trf4Δtrf5Δ (Figure 4A and C and Supplementary Figure S3A and S3B). To search for the minimal functional Trf4p and to address the role of individual Trf4p domains, we prepared a set of deletion and point mutant constructs of TRF4 (Figure 4B). The deletions were designed to remove poorly conserved, potentially unstructured parts of the protein. In addition to the catalytically inactive DXD mutant (D236A and D238A) (5), we introduced two point mutations: (i) D425A residing in a highly conserved aspartate residue in the central domain (30), which is not present in canonical PAPs, and (ii) K552A, which has been proposed to reside in the catalytic center for Trf4p dRP lyase activity (31).
Figure 4.

Mutational analysis of Trf4p reveals minimal functional Trf4p and indicates the importance of D425 in PAP activity. (A) Fragment of Trf4p (aminoacids 161–481) used for previous structural studies does not support growth in trf4Δtrf5Δ background. Growth test analysis of wt and mutants of TRF4 episomally expressed in trf4Δtrf5Δ strain complemented with WT TRF4 on URA3 plasmid and serially diluted and spotted on media with 5-FOA. (B) Overview of mutant forms of TRF4 used in this study: CAT, catalytical domain; CD, central domain. (C) Minimal viable allele of TRF4 spans the amino acids 96–499. Drop tests were performed analogously to (A). (D) Northern blot analysis of total RNA isolated from WT and mutant TRF4 yeast strains with DNA probes specific for indicated RNAs. (E) In vitro polyadenylation assays with 20 ng affinity purified WT and mutant TRAMP4 were performed in the presence of 0.5 mM ATP, 10 ng radioactively labeled hypomodified tRNAiMet (migration indicated by an arrow). Reactions were stopped after 15, 30 and 60 min, respectively. The first lane (np) represents control sample with no protein added. RNAs were separated on 20% PAGE. (F) Coupled polyadenylation and exosome assay. The 5′-end-labeled unmodified tRNAiMet incubated with 40 ng of affinity-purified TRAMP4 (ProtA-Trf4p) and 40 ng of nuclear exosome (Rrp6-TAP) for 15, 30 and 60 min. The first lane (np) represents control sample with no protein added. (G) D425A mutation lowers Trf4p affinity to ATP. Polyadenylation assay was performed with 25 ng of affinity purified D425A Trf4p, 50 fmol 5′-end-labeled unmodified tRNAiMet in the presence of ATP concentrations indicated. Reactions were stopped after 60 min and resolved on 10% denaturing gel.
Deletions of the entire N- (NΔ181) or C- (CΔ463) termini surrounding the CAT and CD parts of Trf4p did not support viability of the double trf4Δtrf5Δ strain (Figure 4C, Supplementary Figure S3A and S3B) and exhibited an accumulation of three well-characterized TRAMP4 substrates, NEL025c CUT and precursors of SnR13 and U14 snoRNAs when expressed in trf4Δ strain (Figure 4D). Although we were able to detect the expression of these fragments by western blot (Supplementary Figure S3C), our attempts to purify these mutants to assess the interaction with other TRAMP components and their activities failed. Thus, we hypothesize that the deletion resulted in protein misfolding and instability. The analyses of the rest of the deletion mutants indicated the minimal functional Trf4p region (spanning residues 97–499) containing CAT and CD of Trf4p surrounded by short extensions as NΔ96 and CΔ499 were sufficient for TRAMP4 assembly (Supplementary Figure S3D). In vivo, CΔ499 displayed cold sensitivity and impaired growth at 30°C when expressed in trf4Δtrf5Δ deletion background (Figure 4C, Supplementary Figure S3A and S3B). When expressed in the single trf4Δ strain, CΔ499 along with NΔ96 were able to suppress the RNA degradation/processing defects of the trf4Δ in vivo (Figure 4D). TRAMPs purified via proteinA-tagged NΔ96 and CΔ499 showed WT-like PAP activities (Figure 4E) and were able to activate tRNAiMet degradation in the presence of Rrp6p-TAP exosomes in vitro (Figure 4F).
In agreement with previously published data, the catalytically inactive DXD TRF4 mutant suppressed only partially the NEL025C accumulation (6,7). This implies that TRAMP can also act in polyadenylation independent manner in vivo. In addition to DXD, we have identified D425A as the second point mutant with impaired PAP activity (Figure 4F). Compared to DXD, the D425A exhibited very weak, only above the background accumulation of TRAMP targets seen by northern blot analysis (Figure 4D). Moreover, it was able to support growth of trf4Δtrf5Δ strain (Figure 4C). The purified TRAMP4 containing the point mutation in D425 failed to polyadenylate hypomodified tRNAiMet in vitro under standard assay conditions and accordingly to activate the exosome (Figure 4E and F), although the complex integrity was not affected (Supplementary Figure S3D). We found that increasing ATP concentration resulted in a gradual increase of the PAP activity in the D425A mutant up to the 4 mM concentration tested, whereas it did not change any further activity of the saturated WT protein (Figure 4G). This result indicated that the mutation reduced Trf4p affinity for ATP. We propose that lowered ATP substrate affinity of D425A slows down PAP activity of the complex, which is however sufficient for TRAMP4 function in vivo.
The Mtr4p-arch domain is required for TRAMP4 complex-mediated exosome activation in vitro
We have observed that in the absence of Air proteins the stability of the Trf4p-Mtr4p heterodimer is compromised even under low salt conditions (Figure 5A) implying that Air proteins mediate interaction between Trf4p and Mtr4p. This is in agreement with similar recent findings in Schizosaccharomyces pombe (32), suggesting a conserved mode of Mtr4p interaction within TRAMP in two distant yeast species. Based on sequence conservation and molecular surface scanning, several patches were proposed as possible binders for Trf4p-Air2p (17). To identify the interaction surface of Mtr4p with the rest of the TRAMP complex, we prepared a set of terminal and internal deletion variants N-terminally fused with cleavable 2×proteinA tag (Figure 5B). As expected, the mutants in the conserved core were not able to support growth in the absence of WT Mtr4p, while the removal of the first 143 amino acids (NΔ143) resulted in strong growth defect (Figure 5C). The archless mutant shows reduced growth as reported previously (16). Interestingly, the deletion of the KOW domain was sufficient to reproduce this phenotype (Figure 5D).
Figure 5.

The RNA-binding KOW domain of Mtr4p is required for activation of exosome by the TRAMP complex. (A) Air2p is required for Mtr4p association with Trf4p. Western blot analysis of affinity purified ProteinA-Trf4p from WT or air1Δair2Δ strains. TRAMP subunits were detected with antibodies indicated. HSW, 1 M KCl high-salt wash used during purification. (B) Overview of mutant forms of MTR4 used in this study: N-term, N-terminal β-hairpin extension, WH, winged-helix domain. (C and D) Growth test analysis of WT and mutants of MTR4 episomally expressed in mtr4Δ strain complemented with WT MTR4 on URA3 plasmid and serially diluted and spotted on media with 5-FOA. (E) Western blot analysis of the composition of purified TRAMPs containing different Mtr4p variants. TRAMP subunits were detected with antibodies indicated. (F and G) Coupled in vitro polyadenylation and exosome activation assay with 40 ng affinity purified Mtr4p variants were performed with 0.5 mM ATP and 50 fmol radioactively labeled hypomodified tRNAiMet (migration position indicated by an arrow). Reactions were stopped after 15, 30 and 60 min, respectively. Samples np stand for controls with no protein included. The entire gel is shown in Supplementary Figure S4A.
Using affinity purification from S. cerevisiae and subsequent western blotting, we confirmed the previous results from in vitro (17) showing that the arch domain is also dispensable for TRAMP formation in vivo (Figure 5E). Furthermore, we observed the full complex formation after deletion of the unusual RecA-1N-terminal β-hairpin extension (NΔ143) found in the structure (17), which is not conserved outside of the Ski2 protein family (Figure 5E). Deletion of any of the domains that form the helicase core, led to the loss of co-purification of TRAMP components (Figure 5E). Nevertheless, we cannot rule out the possibility of misfolding or cellular mislocalization. Since the base area of the helical core around the RNA exit tunnel was proposed to serve for exosome interaction (16), we narrowed down the interaction site with the Trf4p/Air2p heterodimer to the side of the barrel formed by RecA-2, winged helix and helical bundle domains.
Since Mtr4p is dispensable for TRAMP polyadenylation activity but is required for the activation of the exosome (4,5), we tested whether the loss of N-terminal extension or arch domains had any effect on the ability of the TRAMP complex to activate degradation in coupled in vitro polyadenylation and exosome assay. While the NΔ143 mutant did not show any deviation from WT, we observed almost complete loss of exosome activation in the presence of the archless mutant on two different structured substrates (Figure 5F, Supplementary Figure S4A and S4B). To investigate the importance of the KOW domain, the RNA binding element of the arch (17), we conducted the assay in the presence of Mtr4p variant lacking this domain. As shown in Figure 5G, removal of the KOW domain recapitulated the inability of the archless mutant to efficiently activate exosome in vitro. Since we observed apparently lowered polyadenylation in KOWless TRAMPs, we next compared the exosome activation in the presence of lowered amounts of WT Mtr4p. Despite the resulting reduction in observed polyadenylation activity, the WT Mtr4p TRAMP still activated Rrp6p-TAP, unlike KOWless mutant (Supplementary Figure S4C). This together with the slow growth phenotypes (Figure 5D) suggested that arch and particularly the KOW domains are crucial for exosome activation.
DISCUSSION
We have identified several key features of the assembly, RNA binding and exosome activation of the yeast TRAMP RNA surveillance complex. While we focused on the analysis of the TRAMP4 complex, due to the similar composition of TRAMP5 (33) and high level of sequence conservation between Trf5p and Trf4p, and between Air1p and Air2p (5), data presented here are likely valid also to TRAMP5.
Dual function of Air1/2p in the TRAMP complex
Here, we demonstrate that Air2p is a bona fide RNA-binding protein. The Air2p ZnKs 2, 3 and 4 bind aberrant tRNA (Figure 3E and F); however, only the fourth ZnK is fully essential for TRAMP PAP activity in vitro (Figure 2A and B). Although other study by Hamill et al. (15) proposed its involvement in RNA binding, our analyses have not revealed any specific role for ZnK1. It is possible that ZnK1 binds other RNAs, not used in our tests. Alternatively, it may be involved in the interaction with other TRAMP-associated proteins. Structural and sequence alignment analyses indicate that the RNA-binding ZnKs of Air2p use a different binding mode compared to the NC proteins. They lack the hydrophobic pocket, which accommodates guanine base in the NC proteins. Instead, they have large electropositive patches (Figure 3G) created out of lysines, arginines and serines. We speculate that these residues could be responsible for the recognition of a broad spectrum of RNA substrates based on a low-sequence specificity interactions with the sugar-phosphate backbone.
The analysis of deletion mutants indicated the importance of the first 25 N-terminal aminoacids for the interaction with Mtr4p, as both the NΔ57 (residues 57–344) and ZnK1-5L (residues 26–205) proteins did not co-purify Mtr4p (Figure 1D, lanes NΔ57 and ZnK1-5L). The N-terminus is poorly conserved among Air1/2p homologs. In fact, a metazoan Air1-like protein (also designated ZCCHC7) contains a long N-terminal extension (data not shown). Given the observation that the human Mtr4p (SKIV2L2) forms the NEXT (Nuclear EXosome Targeting) complex independently of PAPD5/ZCCHC7 (the human homologs of Trf4p/Air1/2p) (34), it is possible that the interaction between noncanonical PAP (Trf4p/Air2p) and Mtr4p helicase has diverged during evolution depending on the organism complexity.
On the other hand, the Trf4p interacting residues in Air2p identified by NMR titration experiments at positions 139–143, W140G and R141E reside in a stretch of aminoacids forming the (P/T)xIWRxYxL motif conserved among Air-like proteins from distant species including humans (Supplementary Figure S1A). The protein pulldown experiments revealed strong defects in Trf4p and Mtr4p copurification with mutants in this conserved motif (L4 mutant) as well as in the mutant with the disrupted last fifth zinc knuckle fold (ZnK5) (Figure 1D). Mtr4p is not required for Trf4p–Air2p interaction. It is likely that dimerization of Air2p and Trf4p is needed for efficient assembly with Mtr4p, as the absence of either one results in loss of Mtr4p copurification (Figures 1D and 5A). Therefore, we believe that linker 4 and ZnK5 of Air2p are crucial regions responsible for interaction with Trf4p. The last ZnK also displays the highest degree of conservation among the five Air ZnKs (Supplementary Figure S1A). Likewise, the Trf4p surface interacting with the ZnK4–ZnK5 region is conserved among other Trf4-like proteins (15). This implies a phylogenetically conserved mode of interaction between Air1/2p homologs and Trf4p-like noncanonical PAPs. Although the human PAPD5 does not apparently require a cofactor for PAP activity in vitro (35), it does form a complex with ZCCHC7 and other putative RNA-binding proteins (34), suggesting that Air-like proteins mainly serve as RNA substrate specificity factors. During the preparation of this manuscript, Fasken and co-workers (36) reported similar results on Air1p as regard to the importance of the last two zinc knuckles for cell viability and IWRxY motif conservation, NEL025C degradation and Trf4p interaction (36). Our analysis goes beyond this work by distinguishing the different key roles of ZnK4 in RNA binding and Trf4p activation (see below) and ZnK5 in Trf4p interaction. Fasken and colleagues found that Air1p and Air2p mutants in ZnK5 are unstable at 37°C and to a lesser extend at 25°C, which is likely the reason of strong growth defects at high temperatures. Our western blot analysis of expression levels of the different Air2p variants shows only slight decrease in ZnK5 Air2p mutant at 25°C (Supplementary Figure S1B). Moreover, our in vitro reconstitution assays (Figure 2B) suggest that this mutant is unable to form functional Trf4p-Air2p PAP even in amounts comparable to WT. Thus we conclude that the absence of Trf4p in ZnK5 immunoprecipitates is not caused by lowered in vivo expression of Trf4p, but due to weakened interactions between the two proteins as a response to the disruption of the fifth Air2p zinc knuckle. Furthermore, we show that Air2p mediates the interaction between Trf4 and Mtr4 proteins (Figure 5A). Our work extends to the analysis of the N- and C- termini of Air2p revealing crucial interactions with Mtr4p. The Air2p termini enhance Mtr4p binding, while the main surface for interaction with Trf4p is located in the fifth ZnK and the adjacent linker 4 regions (Figure 1D). Based on these mapping analyses and the prediction that the bottom of the Mtr4p helicase barrel serves for interaction with exosome (16), we propose a model of the quarternary interactions within the TRAMP complex. In this model, Trf4p/Air2p binds to the Mtr4p DExH globular core, at the side of barrel formed by RecA-2, winged helix and helical bundle domains. We suggest that the RNA substrates are first screened by the Air2p ZnK2 – ZnK4 region that protrudes from Mtr4 surface and are consequently loaded to the Trf4p active site. We speculate that the RNA 3′-oligo(A) terminus leaves Trf4p in the direction toward the KOW-RecA-2 interface, where the RNA 3′-end is expected to enter the helicase core (17) (Figure 6).
Figure 6.
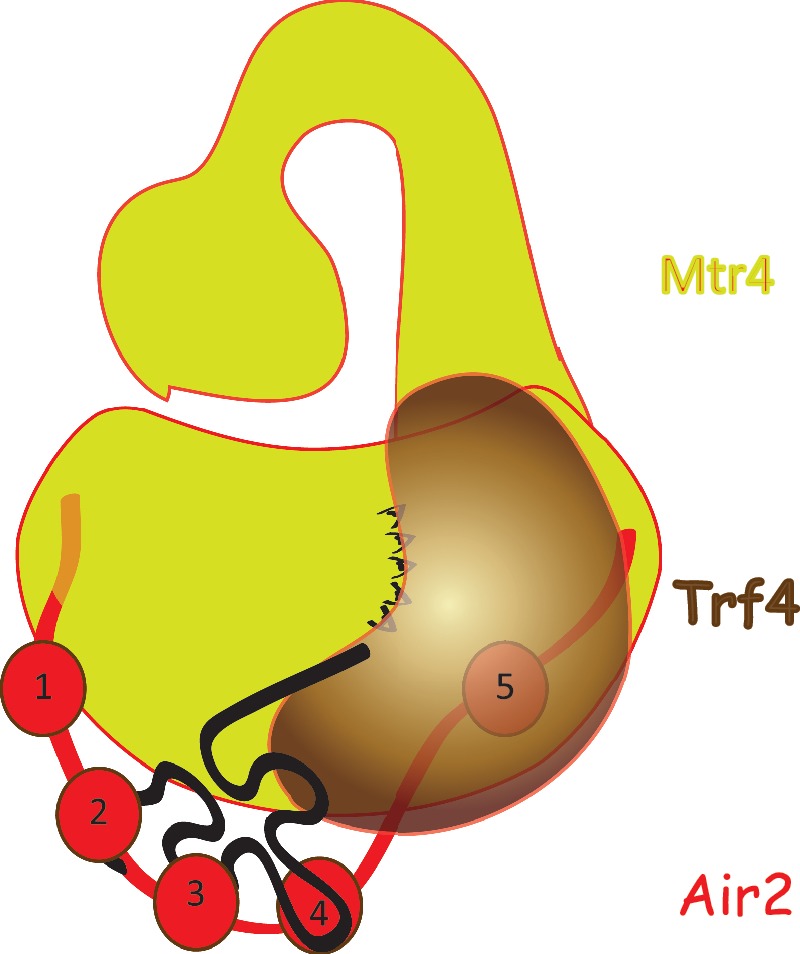
Model of TRAMP assembly and RNA binding. The Air2 protein is bridging the Mtr4p (interaction via N- and C-terminus) and Trf4p (interaction via L4, ZnK5 and C-terminus). The ZnKs 2, 3 and 4 are involved in RNA binding. The KOW domain of Mtr4p is required for exosome activation.
TRF4 catalytical mechanism
We found that increasing ATP concentration resulted in a gradual increase of the PAP activity in the D425A mutant up to the 4 mM concentration tested, whereas it did not change any further activity of the saturated WT protein (Figure 4G). This result indicated that the mutation reduced Trf4p affinity for ATP.
By examining the available crystal structure (15), we observed that the D425 residue is situated approximately 20 Å from the active site. Close inspection of the mechanism of the homologous yeast canonical poly(A) polymerase Pap1p catalysis revealed that hydrogen bonding in this region is required for stabilization of conformational movements leading from an open to closed conformation of the protein (37). When the N189A (CAT domain) mutation is introduced in Pap1p, the equilibrium between the open and closed state is shifted, resulting in increased KM for ATP (37). N189 forms a hydrogen bond with the main chain carbonyl of the Y307 (CD) in the closed conformation (Supplementary Figure S3E and S3F). Interestingly, the Y307 of Pap1p resides in both, sequentially (30) and spatially, identical position to D425 of Trf4p (Supplementary Figure S3G). Therefore, we suggest that the side chain of D425 may serve as a hydrogen bond acceptor analogous to Y307 main chain carbonyl. We have not identified any hydrogen bond formed by D425 in the published structure [(15) and Supplementary Figure S3G]. Notably, the structure was solved for the Trf4p apoenzyme; hence it is likely to represent the open state status. We, therefore, suggest that a conformational change analogous to Pap1p may occur in Trf4p upon RNA and ATP binding, thus indicating the conservation of the catalytic mechanism of both related proteins.
RNA binding KOW domain is involved in exosome activation
The role of the arch domain in exosome function in vivo has been proposed previously by Jackson et al. (16) who observed the accumulation of 5.8S+30 rRNA and stabilization of 5′-ETS, both being specific substrates of Rrp6p activity (38). Here we report that the arch, particularly the KOW domain, of Mtr4p is necessary for the TRAMP-mediated exosome activation in vitro. We show that TRAMPs lacking arch or KOW are able to add poly(A) tails, but they almost completely fail to activate exosomes to degrade structured RNAs in vitro (Figure 1F and G; Supplementary Figure S4). Recently Jia et al. (14) showed that Mtr4p regulates the individual steps of TRAMP polyadenylation as well as the length of the poly(A) tails added. We do not exclude the possibility that arch/KOW is also involved in this regulation. However, in our assays, NΔ143 and ΔKOW TRAMPs showed comparable polyadenylation, but only NΔ143 was fully able to initiate exosomal degradation, whereas ΔKOW was highly defective. Therefore, we conclude that the observed differences in polyadenylation seen in KOW-less TRAMP cannot explain the lack of exosome activation, and we propose that the KOW domain is crucial for TRAMP-mediated exosome activation.
As neither the helicase nor ATPase activities of Mtr4p are affected by arch removal (16,17), we speculate that the RNA-binding properties of the arch might be necessary to handle the substrate between different activities of the TRAMP-exosome pathway. Both the close proximity of the KOW to the helicase RNA entry site and the possible flexibility in the elbow of the arm (16) suggest that arch transports RNA between Trf4p-Air2p, the helicase core and potentially the Rrp6p. Alternatively, the arch is required for the displacement of other RNA-binding factors and shuttling the substrate to the helicase core. We also cannot exclude the possibility that the arch serves for protein–protein interactions with Rrp6p or other core exosome components. This scenario is supported by the presence of a similar structure recently uncovered in the cytoplasmic exosome cofactor Ski2p (16,17,39).
The nuclear exosomes contain two confirmed exoribonucleases—the Rrp6p and Rrp44p. Currently, it is still not fully understood how these two subunits interplay on divergent exosome substrates in vivo. However, it was shown that in the conditions used in our in vitro assays (5 mM Mg2+) the nuclease Rrp44p is inactive (12). Therefore, we propose that nuclease activity observed in our assays can be fully attributed to Rrp6p. Therefore, it will be interesting to address whether RNAs bound by KOW can also undergo processing by the core exosome or whether they are specifically triggered for the degradation by Rrp6p. The exact mechanistic details remain obscure and will be the subject of following studies.
ACCESSION NUMBERS
The atomic coordinates and restraints (code 2lli) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Tables 1–5, Supplementary Figures 1–4 and Supplementary Reference [40].
FUNDING
Wellcome Trust (084316/Z/07/Z to S.V.); EMBO Installation Grant (1642 to S.V.); Czech Science Foundation (305/11/1095 to S.V, 305/10/1490 to R.S., P305/12/G034 to S.V. and R.S.); CEITEC–Central European Institute of Technology (CZ.1.05/1.1.00/02.0068) from European Regional Development Fund; the NOESY spectra were obtained at the BMRZ NMR facility supported by the EU-NMR program (RII3-026145); the EC FP-7 (grant no 205872 to M.S.A.); the Brno City Municipality Scholarship for Talented Ph.D. Students (to P.H. and F.H.). Funding for open access charge: Senior International Wellcome Trust Fellowship.
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Leona Svajdova for excellent technical assistance; Walter Keller and Georges Martin for helpful advice; and John LaCava, Jonathan Houseley and Andrzej Dziembowski for critical reading and comments on the manuscript. We also thank David Tollervey, Ambro Van Hoof and Domenico Libri for yeast strains and constructs, Patrick Lindner for Mtr4p antibodies, Gerhard Wagner for G-proteinB1 vector and Christopher Lima for Smt3p and Ulp1p expression vectors.
REFERENCES
- 1.Brosnan CA, Voinnet O. The long and the short of noncoding RNAs. Curr. Opin. Cell. Biol. 2009;21:416–425. doi: 10.1016/j.ceb.2009.04.001. [DOI] [PubMed] [Google Scholar]
- 2.Philips AV, Cooper TA. RNA processing and human disease. Cell. Mol. Life Sci. 2000;57:235–249. doi: 10.1007/PL00000687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kadaba S, Krueger A, Trice T, Krecic AM, Hinnebusch AG, Anderson J. Nuclear surveillance and degradation of hypomodified initiator tRNAMet in S. cerevisiae. Genes Dev. 2004;18:1227–1240. doi: 10.1101/gad.1183804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.LaCava J, Houseley J, Saveanu C, Petfalski E, Thompson E, Jacquier A, Tollervey D. RNA degradation by the exosome is promoted by a nuclear polyadenylation complex. Cell. 2005;121:713–724. doi: 10.1016/j.cell.2005.04.029. [DOI] [PubMed] [Google Scholar]
- 5.Vanacova S, Wolf J, Martin G, Blank D, Dettwiler S, Friedlein A, Langen H, Keith G, Keller W. A new yeast poly(A) polymerase complex involved in RNA quality control. PLoS Biol. 2005;3:e189. doi: 10.1371/journal.pbio.0030189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wyers F, Rougemaille M, Badis G, Rousselle JC, Dufour ME, Boulay J, Regnault B, Devaux F, Namane A, Seraphin B, et al. Cryptic pol II transcripts are degraded by a nuclear quality control pathway involving a new poly(A) polymerase. Cell. 2005;121:725–737. doi: 10.1016/j.cell.2005.04.030. [DOI] [PubMed] [Google Scholar]
- 7.San Paolo S, Vanacova S, Schenk L, Scherrer T, Blank D, Keller W, Gerber AP. Distinct roles of non-canonical poly(A) polymerases in RNA metabolism. PLoS Genet. 2009;5:e1000555. doi: 10.1371/journal.pgen.1000555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wlotzka W, Kudla G, Granneman S, Tollervey D. The nuclear RNA polymerase II surveillance system targets polymerase III transcripts. EMBO J. 2011;30:1790–1803. doi: 10.1038/emboj.2011.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Houseley J, Kotovic K, El Hage A, Tollervey D. Trf4 targets ncRNAs from telomeric and rDNA spacer regions and functions in rDNA copy number control. EMBO J. 2007;26:4996–5006. doi: 10.1038/sj.emboj.7601921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Houseley J, Tollervey D. The nuclear RNA surveillance machinery: the link between ncRNAs and genome structure in budding yeast. Biochim. Biophys. Acta. 2008;1779:239–246. doi: 10.1016/j.bbagrm.2007.12.008. [DOI] [PubMed] [Google Scholar]
- 11.Thiebaut M, Kisseleva-Romanova E, Rougemaille M, Boulay J, Libri D. Transcription termination and nuclear degradation of cryptic unstable transcripts: a role for the nrd1-nab3 pathway in genome surveillance. Mol. Cell. 2006;23:853–864. doi: 10.1016/j.molcel.2006.07.029. [DOI] [PubMed] [Google Scholar]
- 12.Callahan KP, Butler JS. TRAMP complex enhances RNA degradation by the nuclear exosome component Rrp6. J. Biol. Chem. 2010;285:3540–3547. doi: 10.1074/jbc.M109.058396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.D'Souza V, Summers MF. How retroviruses select their genomes. Nat. Rev. Microbiol. 2005;3:643–655. doi: 10.1038/nrmicro1210. [DOI] [PubMed] [Google Scholar]
- 14.Jia H, Wang X, Liu F, Guenther UP, Srinivasan S, Anderson JT, Jankowsky E. The RNA helicase Mtr4p modulates polyadenylation in the TRAMP complex. Cell. 2011;145:890–901. doi: 10.1016/j.cell.2011.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hamill S, Wolin SL, Reinisch KM. Structure and function of the polymerase core of TRAMP, a RNA surveillance complex. Proc. Natl Acad. Sci. USA. 2010;107:15045–15050. doi: 10.1073/pnas.1003505107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jackson RN, Klauer AA, Hintze BJ, Robinson H, van Hoof A, Johnson SJ. The crystal structure of Mtr4 reveals a novel arch domain required for rRNA processing. EMBO J. 2010;29:2205–2216. doi: 10.1038/emboj.2010.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Weir JR, Bonneau F, Hentschel J, Conti E. Structural analysis reveals the characteristic features of Mtr4, a DExH helicase involved in nuclear RNA processing and surveillance. Proc. Natl Acad. Sci. USA. 2010;107:12139–12144. doi: 10.1073/pnas.1004953107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhou P, Lugovskoy AA, Wagner G. A solubility-enhancement tag (SET) for NMR studies of poorly behaving proteins. J. Biomol. NMR. 2001;20:11–14. doi: 10.1023/a:1011258906244. [DOI] [PubMed] [Google Scholar]
- 19.Price S, Oubridge C, Varani G, Nagai K. In: RNA–Protein Interaction: Practical Approach. Smith C, editor. Oxford: Oxford University Press; 1998. pp. 37–74. [Google Scholar]
- 20.Heyduk T, Lee JC. Application of fluorescence energy transfer and polarization to monitor Escherichia coli cAMP receptor protein and lac promoter interaction. Proc. Natl Acad. Sci. USA. 1990;87:1744–1748. doi: 10.1073/pnas.87.5.1744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mossessova E, Lima CD. Ulp1-SUMO crystal structure and genetic analysis reveal conserved interactions and a regulatory element essential for cell growth in yeast. Mol. Cell. 2000;5:865–876. doi: 10.1016/s1097-2765(00)80326-3. [DOI] [PubMed] [Google Scholar]
- 22.Sanudo M, Jacko M, Rammelt C, Vanacova S, Stefl R. 1H, 13C, and 15N chemical shift assignments of ZCCHC9. Biomol. NMR Assign. 2011;5:19–21. doi: 10.1007/s12104-010-9257-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sattler M, Schleucher J, Griesinger C. Heteronuclear multidimensional NMR experiments for the structure determination of proteins in solution employing pulsed field gradients. Prog. Nucl. Magn. Reson. Spectrosc. 1999;34:93–158. [Google Scholar]
- 24.Herrmann T, Guntert P, Wuthrich K. Protein NMR structure determination with automated NOE-identification in the NOESY spectra using the new software ATNOS. J. Biomol. NMR. 2002;24:171–189. doi: 10.1023/a:1021614115432. [DOI] [PubMed] [Google Scholar]
- 25.Case DA, Darden TA, Cheatham TE, III, Simmerling CL, Wang J, Duke RE, Luo R, Crowley M, Walker RC, Zhang W, et al. AMBER 10, Users' Manual. San Francisco, CA, USA: University of California; 2008. [Google Scholar]
- 26.Stefl R, Oberstrass FC, Hood JL, Jourdan M, Zimmermann M, Skrisovska L, Maris C, Peng L, Hofr C, Emeson RB, et al. The solution structure of the ADAR2 dsRBM-RNA complex reveals a sequence-specific readout of the minor groove. Cell. 2010;143:225–237. doi: 10.1016/j.cell.2010.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hobor F, Pergoli R, Kubicek K, Hrossova D, Bacikova V, Zimmermann M, Pasulka J, Hofr C, Vanacova S, Stefl R. Recognition of transcription termination signal by the nuclear polyadenylated RNA-binding (NAB) 3 protein. J. Biol. Chem. 2011;286:3645–3657. doi: 10.1074/jbc.M110.158774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Falquet L, Bordoli L, Ioannidis V, Pagni M, Jongeneel CV. Swiss EMBnet node web server. Nucleic Acids Res. 2003;31:3782–3783. doi: 10.1093/nar/gkg547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rogozin IB, Aravind L, Koonin EV. Differential action of natural selection on the N and C-terminal domains of 2′-5′ oligoadenylate synthetases and the potential nuclease function of the C-terminal domain. J. Mol. Biol. 2003;326:1449–1461. doi: 10.1016/s0022-2836(03)00055-x. [DOI] [PubMed] [Google Scholar]
- 31.Gellon L, Carson DR, Carson JP, Demple B. Intrinsic 5′-deoxyribose-5-phosphate lyase activity in Saccharomyces cerevisiae Trf4 protein with a possible role in base excision DNA repair. DNA Repair (Amst) 2008;7:187–198. doi: 10.1016/j.dnarep.2007.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Keller C, Woolcock K, Hess D, Buhler M. Proteomic and functional analysis of the noncanonical poly(A) polymerase Cid14. RNA. 2010;16:1124–1129. doi: 10.1261/rna.2053710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Houseley J, Tollervey D. Yeast Trf5p is a nuclear poly(A) polymerase. EMBO Rep. 2006;7:205–211. doi: 10.1038/sj.embor.7400612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lubas M, Christensen MS, Kristiansen MS, Domanski M, Falkenby LG, Lykke-Andersen S, Andersen JS, Dziembowski A, Jensen TH. Interaction profiling identifies the human nuclear exosome targeting complex. Mol. Cell. 2011;43:624–637. doi: 10.1016/j.molcel.2011.06.028. [DOI] [PubMed] [Google Scholar]
- 35.Rammelt C, Bilen B, Zavolan M, Keller W. PAPD5, a noncanonical poly(A) polymerase with an unusual RNA-binding motif. RNA. 2011;17:1737–1746. doi: 10.1261/rna.2787011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fasken MB, Leung SW, Banerjee A, Kodani MO, Chavez R, Bowman EA, Purohit MK, Rubinson ME, Rubinson EH, Corbett AH. Air1 zinc knuckles 4 and 5 and a conserved IWRXY motif are critical for the function and integrity of the Trf4/5-Air1/2-Mtr4 polyadenylation (TRAMP) RNA quality control complex. J. Biol. Chem. 2011;286:37429–37445. doi: 10.1074/jbc.M111.271494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Balbo PB, Bohm A. Mechanism of poly(A) polymerase: structure of the enzyme-MgATP-RNA ternary complex and kinetic analysis. Structure. 2007;15:1117–1131. doi: 10.1016/j.str.2007.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Allmang C, Kufel J, Chanfreau G, Mitchell P, Petfalski E, Tollervey D. Functions of the exosome in rRNA, snoRNA and snRNA synthesis. EMBO J. 1999;18:5399–5410. doi: 10.1093/emboj/18.19.5399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Halbach F, Rode M, Conti E. The crystal structure of S. cerevisiae Ski2, a DExH helicase associated with the cytoplasmic functions of the exosome. RNA. 2012;18:124–134. doi: 10.1261/rna.029553.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bard J, Zhelkovsky AM, Helmling S, Earnest TN, Moore MJ, Bohm A. Structure of yeast poly(A) polymerase alone and in complex with 3′-dATP. Science. 2000;289:1346–1349. doi: 10.1126/science.289.5483.1346. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.