


Abstract
DNA polymerase III (DNA pol III) efficiently replicates the Escherichia coli genome, but it cannot bypass DNA damage. Instead, translesion synthesis (TLS) DNA polymerases are employed to replicate past damaged DNA; however, the exchange of replicative for TLS polymerases is not understood. The umuD gene products, which are up-regulated during the SOS response, were previously shown to bind to the α, β and ε subunits of DNA pol III. Full-length UmuD inhibits DNA replication and prevents mutagenic TLS, while the cleaved form UmuD′ facilitates mutagenesis. We show that α possesses two UmuD binding sites: at the N-terminus (residues 1–280) and the C-terminus (residues 956–975). The C-terminal site favors UmuD over UmuD′. We also find that UmuD, but not UmuD′, disrupts the α–β complex. We propose that the interaction between α and UmuD contributes to the transition between replicative and TLS polymerases by removing α from the β clamp.
INTRODUCTION
DNA polymerase III (DNA pol III) is the main DNA polymerase in Escherichia coli and is responsible for replicating the entire genome. It contains ten subunits that allow for accurate and processive replication to occur (1–4). These 10 subunits are organized into three subassemblies: the polymerase core, the β processivity clamp, and the clamp loader complex (5). The polymerase core consists of the α polymerase subunit, the ε proofreading subunit and the θ subunit whereas the clamp loader complex loads the processivity clamp onto primer:template DNA (6) and coordinates continuous replication of both leading and lagging strands of DNA (7–9). Once the β clamp is positioned, the clamp loader complex regulates the loading of the polymerase core onto the β clamp (7). This ensemble of proteins allows for continuous replication to occur on the leading strand and discontinuous replication to occur on the lagging strand (10), allowing for efficient replication of DNA.
A crystal structure has been solved of a truncated form of the E. coli α subunit (residues 1–917) (Figure 1A) (11). With a resolution of 2.3 Å, it is possible to distinguish the characteristic domains of DNA polymerases including the palm domain containing the conserved aspartic acid residues of the active site and the fingers and thumb domains (12,13). The N-terminal domain, which was thought to possess pyrophosphatase activity (14), is also present in the crystal structure. This domain contains the binding site of the ε proofreading subunit (15). The C-terminal domain, not included in the crystal structure, contains the oligonucleotide/oligosaccharide binding (OB) fold (11,16) responsible for binding single-stranded DNA (17), the binding site for the τ subunit of the clamp loader complex (7,18–20) and the binding site for the β processivity clamp (Figure 1A) (21,22). It was originally suggested through the use of truncations of α that the C-terminus is responsible for binding to the β clamp (21); however, site-directed mutagenesis experiments revealed that residues 920–924 are primarily responsible for binding to the β clamp and that the C-terminus plays a minor role (22). Because of these findings, we refer to residues 920–924 as the β binding site on α.
Figure 1.

Models of α and UmuD. (A) The X-ray crystal structure of α 1–917 [PDB code 2hnh (11)] (left) shows the major domains of the polymerase, with the active site and the binding site for ε indicated (23). A homology model of full-length α (24) (right) depicts the C-terminal domain that is not present in the crystal structure containing the β (25) and τ (26) binding sites as well as the OB domain (11,17). (B) The homology model of UmuD (27) and the X-ray crystal structure of UmuD′ (28) showing one monomer in green and the other in purple. The first 24 N-terminal residues missing from the UmuD′ structure (yellow) are cleaved as part of a primitive DNA damage checkpoint by the active site (orange). The single cysteine residues at C24 are shown (red) in full-length UmuD.
The α subunit, together with the other subunits of DNA pol III, is capable of efficiently replicating undamaged DNA. But when it encounters DNA damage, replication by the polymerase is disrupted (29). DNA pol III is capable of inserting nucleotides opposite some DNA lesions, but usually cannot extend beyond the unusual primer terminus that it generates. Thus, it can become trapped in a futile cycle of insertion and exonucleolytic proofreading at sites of DNA damage (30). On the leading strand, the blockage of DNA synthesis is disruptive, causing the inhibition of replication fork progression (31,32). On the lagging strand however, progression of the replication fork is less affected because the polymerase can reassemble on the next primer downstream (31). In order to withstand DNA damage, the SOS response is employed by the cell (33).
The initial molecular trigger for the SOS response is an abundance of ssDNA caused by the stalled DNA polymerase (34). RecA coats the ssDNA forming a nucleoprotein filament that facilitates the cleavage of the LexA repressor and up-regulates the expression of at least 57 SOS genes (35). The products of these genes are involved in DNA damage tolerance mechanisms, such as recombination, DNA repair and translesion synthesis (TLS), which is the process involved in replicating damaged DNA. TLS is carried out by potentially mutagenic Y family DNA polymerases that can replace a stalled replicative DNA polymerase (36–40).
Y family DNA polymerases have the specialized ability to bypass DNA damage by inserting nucleotides opposite the lesion (36–43). Of the five DNA polymerases present in E. coli, DNA pol IV (DinB) and DNA pol V (UmuD′2C) are members of this family (44–46). DNA pol V is composed of the UmuD′2 dimer, which is the RecA/ssDNA-facilitated cleavage product of UmuD2, and UmuC, which possesses polymerase activity (44,45). During the first 20–40 min after the activation of the SOS response, UmuD2 is the predominant species (47,50). UmuD2 together with UmuC serves to affect a primitive DNA damage checkpoint, inhibiting replication to allow time for accurate DNA repair processes to act (48,49,50). After ∼40 min, UmuD′2 becomes the predominant species, releasing the primitive DNA damage checkpoint (50–52). UmuD′2 and UmuC form DNA pol V (44,45,53), which is the form active in TLS. UmuD2 is a very tight dimer (Kd < 10 pM) (54) and is expected to be dimeric in all experiments reported here; therefore we use UmuD to refer to homodimeric UmuD and variants. Each UmuD monomer consists of two domains: the N-terminal arms and a C-terminal globular domain. One model of UmuD places the N-terminal arms folded down onto the globular domain, positioning the cleavage site (between C24 and G25) at the active site (S60 and K97) (an ‘arms down’ conformation) (Figure 1B) (27,55). But because of the dynamic nature of the arms, UmuD can exist in a conformation where the arms are unbound (an ‘arms up’ conformation) exposing the globular domain (27,54,56).
The umuD gene products have also been shown to interact with other proteins such as DNA pol IV (Kd 0.62 µM) (57), and components of DNA pol III including the α polymerase, the ε proofreading subunit and the β processivity clamp (58). UmuD and UmuD′ differentially interact with components of DNA pol III (58). Specifically, affinity chromatography experiments suggested that UmuD binds more strongly to the β processivity clamp than to α (58), supporting the role of UmuD in inhibiting DNA synthesis as part of a primitive DNA damage checkpoint (51); however, the α-UmuD interaction has not been characterized at the molecular level. It has also been suggested that upon binding to the β clamp or to DinB, the structure of UmuD becomes less disordered (54). These interactions suggest that both UmuD and UmuD′ play a role in replication fork management.
The process by which exchange of the α polymerase subunit and a TLS polymerase occurs is not fully understood. There are likely a number of factors that facilitate such exchange; for example, it has been shown that the presence of DinB inhibits DNA pol III replication (59). In this work, we characterized the interactions of α with the umuD gene products. We determined the binding affinity between UmuD and α. We also determined that there are two UmuD binding sites on α, one of which favors UmuD over UmuD′. The UmuD-specific site overlaps with the β binding site on α and consequently, we found that UmuD, but not UmuD′, specifically disrupts the α–β interaction. Therefore, we find that UmuD plays a key role in regulating polymerase access to the β clamp.
MATERIALS AND METHODS
Proteins and plasmids
UmuD and UmuD′ were expressed from pSG5 and pSG4 plasmids, respectively, as previously described (60). UmuD3A (UmuD containing the triple mutation T14A, L17A and F18A), UmuD-S60A and UmuD′-S60A variants were previously described (27). Plasmids that express His-tagged wild-type α and the truncations α1–280, α1–917, α1–956 and α1–975 were provided by Dr Meindert Lamers and Prof. John Kuriyan, UC Berkeley (11). The plasmids encoding the truncations α1–835 and α917–1160 were described previously (17). Wild-type α and truncations were expressed in Tuner (DE3) (Novagen) competent cells and purified using the established protocol and stored at −20°C in a buffer containing 50 mM Hepes (pH 7.5), 100 mM NaCl and 50% glycerol (protein storage buffer) (11). The β clamp was purified according to published protocols (61). LexA was purified from pJWL228 (provided by Dr Ronaldo Mohana-Borges, Instituto de Biofísica Carlos Chagas Filho-UFRJ, Brazil) as described (62).
To improve the stability of the α protein, various conditions were tested including different buffer conditions and varying temperatures. The most stable proteins were those stored at −20°C in a buffer containing 50% glycerol, at 25 µM or lower concentrations. The high concentration of glycerol was necessary to keep the proteins from freezing. It was noted that the stability of α was reduced when the protein was frozen and thawed before use. In solutions with lower glycerol concentrations, aggregation was observed. We found that the high concentration of glycerol interfered with many of the experiments described here; therefore, before each experiment, the buffer was exchanged to the relevant buffer for each assay. This was accomplished by using Zeba Desalt Spin Columns (Thermo Scientific) with the buffer exchange protocol provided.
Tryptophan fluorescence assay
The intrinsic tryptophan fluorescence of α in the presence of increasing amounts of UmuD was used to determine the equilibrium constant for α binding to UmuD. UmuD has no tryptophan residues and α has eight tryptophans. To determine the fluorescence of α alone, 60 µL of 5 µM α in 50 mM Hepes (pH 7.5), 100 mM NaCl was excited at 278 nm and emission was monitored from 300 to 500 nm. To determine the binding constant between α and UmuD, the sample of α was then titrated with UmuD. After each addition, the sample was excited at 278 nm and emission was monitored from 300 to 500 nm, with both the excitation and emission slits set to 5 nm. To analyze the change in fluorescence of α, the maximum of each peak was determined and corrected for the dilution caused by adding UmuD. To correct for any fluorescence contribution from UmuD alone, fluorescence emission was monitored from UmuD in reaction buffer at the same concentrations used to titrate α. To produce a binding curve, the fraction of fluorescence from α quenched by the addition of UmuD (Q) was plotted versus the ratio of UmuD to α. The curve was fit to the following equation using GraphPad Prism (La Jolla, CA):
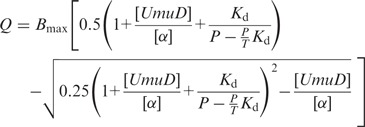 |
(1) |
where P is the initial concentration of α and T is the concentration of the UmuD stock used in the titration. The binding constant at pH 10 for α and UmuD was obtained in alkaline cleavage buffer (see below).
Thermal-shift assays
Reactions were assembled in 96-well PCR plates (Applied Biosystems) in which each sample (16 μl total volume) consisted of thermal-shift assay buffer (50 mM Hepes (pH 7.5), 100 mM NaCl), and 25 × Sypro Orange (Invitrogen) excited at 490 nm. In experiments in which the melting transitions of UmuD were observed in the presence of α, each sample contained 45 µM UmuD (monomer concentration) and 1 μM α. Samples were incubated for 2 h at room temperature before detection. In experiments in which the stability of the truncations of α was compared with that of wild-type α, each construct of α was added to a final concentration of 5 µM without incubation before detection. In order to increase the thermal stability of the α proteins for ease of detection, 30% glycerol was added to each sample. Once the plate was sealed with optical adhesive film (Applied Biosystems), an iCycler iQ5 Real-Time PCR (Bio-Rad) was used to increase the temperature from 25°C to 75°C with an increment of 0.1°C and a 10 s dwell time per temperature increment. The fluorescence intensities emitted at 575 nm and detected by the built-in CCD camera were plotted versus temperature. The melting temperature (Tm) was calculated as described (63,64). The assays were repeated several times with similar observations and consistent melting temperatures.
UmuD in vitro cleavage assays
RecA/ssDNA nucleoprotein filament-facilitated UmuD cleavage reactions were carried out as described in LG Buffer (60). Samples containing α, UmuD and buffer were incubated for 2 h at room temperature before the addition of RecA/ssDNA, after which reactions were carried out at either 37°C or 30°C for 45 min. The reaction temperature was adjusted to 30°C wherever possible because of the relatively low melting temperature of α (∼38°C). To rule out any non-specific interactions, BSA was used instead of α, or, alternatively, LexA cleavage was assayed in the presence of α. Alkaline cleavage of UmuD was also carried out as previously described (60). The alkaline cleavage buffer [100 mM Glycine (pH 10), 10 mM CaCl2, 50 mM NaCl, 10 mM dithiothreitol (DTT) and 0.25 mg/mL BSA] was added after a 30 min incubation at room temperature, and the reactions were carried out at either 37°C or 30°C for 48 h. Reactions of cleavage assays were analyzed by using 18% SDS–PAGE.
Cross-linking of UmuD using bis-maleimidohexane
Bis-maleimidohexane (BMH, Pierce) cross-linking (27,65) was carried out as described by incubating 10 µM UmuD-S60A in the presence of 10 µM wild-type α, α917–1160 or α1–280 in 10 mM sodium phosphate (pH 6.8), 100 mM NaCl and 1 mM BMH. The reactions were incubated for 5 min at room temperature, after which the reactions were quenched with 50 mM DTT. Cross-linked dimers of UmuD-S60A were resolved from monomers by 4–20% SDS–PAGE (Bio-Rad). An immunoblot was used to identify those bands containing UmuD-S60A using rabbit anti-UmuD as the primary antibody as described (27,60).
Fluorescence resonance energy transfer
Protein labeling was carried out using Alexa Fluor 488 C5-maleimide (Invitrogen) for the β clamp and Alexa Fluor 647 C2-maleimide (Invitrogen) for the α subunit, in a reaction buffer containing 50 mM Hepes (pH 7.5) and 200 mM NaCl. The β clamp (100–150 μM) and α (20–50 μM) were each labeled by adding 5–10 molar excess of the respective Alexa Fluor maleimide at room temperature, followed by overnight incubation at 4°C in the dark. Unreacted reagent was separated from labeled protein on a Sephadex G-50 (GE Healthcare) column at 4°C using a buffer containing 25 mM Hepes (pH 7.5) and 200 mM NaCl. Fractions were analyzed by SDS–PAGE and the gels were analyzed on a Storm 860 phosphorimager (Molecular Dynamics) by monitoring fluorescence after irradiating the gels at 450 nm. The degree of labeling was determined as described by the manufacturer and was on average 3–4 fluorophores per α subunit and 1–2 fluorophores per β monomer.
The interaction between the α subunit and the β clamp was monitored by fluorescence resonance energy transfer (FRET). A reaction mixture containing 350 nM donor-labeled β subunit (βA488) and 1 μM acceptor-labeled α subunit (αA647) in 70 μL FRET buffer [25 mM Hepes (pH 7.5), 25 mM NaCl, 1 mM DTT and 0.5 mM EDTA] was incubated for 15 min on ice, followed by analysis at room temperature with a Cary Eclipse spectrofluorimeter (Varian). Samples were excited at 495 nm and emission spectra were recorded from 500 to 700 nm, with slits for both excitation and emission set to 5 nm. The effects of UmuD, UmuD′ and UmuD-S60A were investigated by adding up to 40 μM UmuD or variants to each sample prior to analysis. FRET efficiency (E) was calculated by finding the ratio between the emission intensity of the donor in the presence of the acceptor (Fd′) to the emission intensity of the donor alone (Fd), which is then subtracted from one [Equation (2)].
| (2) |
RESULTS
α binds UmuD via two UmuD binding sites on α
In order to develop a more complete picture of the interactions of UmuD with subunits of the replisome, we probed the interaction between α and the umuD gene products. Because α has eight tryptophans and UmuD has none, we were able to use the intrinsic tryptophan fluorescence of α to determine the equilibrium binding constant for the interaction between α and UmuD. By analyzing the extent of α fluorescence quenching observed with the addition of UmuD, the Kd was determined to be 1.1 ± 0.6 µM (Figure 2). This binding constant is similar to that for other protein interactions with UmuD [UmuD and the β clamp: 5.5 ± 0.8 μM (27); UmuD and DinB: 0.62 μM (57)]. To localize the binding site of UmuD on α, a number of truncations of α were constructed and dissociation constants for their interactions with UmuD were determined (Figure 3A). The truncation α1–975 has a binding constant (3.1 ± 1.0 µM) similar to that of full-length α indicating that the binding site is located within this region. When the truncation α1–956 was analyzed, a significant decrease in affinity was observed (Kd = 14.7 ± 1.9 µM), suggesting that there is a UmuD binding site between residues 956–975, which is located in the C-terminal domain (Figure 1A). This region is also near the β-binding site (residues 920–924) (22).
Figure 2.

UmuD binds wild-type α with a Kd = 1.1 μM. The curve representing the fraction of α tryptophan fluorescence quenched by various concentrations of UmuD was fit to Equation (1), which produced a Kd value of 1.1 ± 0.6 µM at pH 7.5. The error bars represent the standard deviation from five independent experiments.
Figure 3.

UmuD binds α at two distinct binding sites. (A) Kd values for UmuD binding to various α truncations. The large difference between the Kd values for the truncations α1–975 and α1–956 indicates that one binding site is between residues 956–975. The reduced but still significant binding constants for the truncations α1–956, α1–917, α1–835 and α1–280 suggests another UmuD binding site is present in the N-terminal domain α1–280. The domains of α are labeled above the schematic of the protein; ε, β and τ indicate the location of the respective binding site for each protein, HhH indicates the helix-hairpin-helix domain involved in double-stranded DNA binding, OB indicates the OB-fold domain involved in single-stranded DNA binding. (B) Melting curves for the various α truncations used show comparable stability to that of full-length α.
The Kd values for truncations α1–917, α1–835, α1–280 are all similar to that of truncation α1–956. That binding was still detected with these truncations suggests that there is a second UmuD binding site within α. N-terminal truncations smaller than 280 residues were expressed poorly and so could not be analyzed. As a result, this second binding site could not be localized any further than to residues 1–280, which is the PHP domain (Figure 1A). The Kd value for UmuD binding to the truncation α917–1160, which contains the proposed C-terminal UmuD binding site (α residues 956–975), was determined to be 13.9 ± 5.1 µM which is somewhat similar to that for truncation α1–280 (7.3 µM) but greater than that of full-length α (1.1 µM). The increased affinity for UmuD seen with full-length α as opposed to that of the truncations containing only one binding site may be due to the simultaneous binding of two UmuD dimers to α, as the two UmuD binding regions are spatially somewhat distant (Figure 1A).
A thermal-shift assay was used to determine whether any of the truncations assayed here were unstable. This assay involves the use of an environmentally-sensitive fluorescent dye where the fluorescence intensity is proportional to the hydrophobicity of its environment. When a folded protein is present, the dye is in an aqueous environment and so exhibits low fluorescence. As the temperature reaches the melting point of the protein, the dye binds to the exposed hydrophobic residues of the unfolding protein, increasing the fluorescence intensity of the dye. The temperature at the midpoint of the transition between the folded and unfolded states is known as the melting temperature, or Tm. Because the Tm is related to the stability of the protein, a protein with a higher Tm is more stable than a protein with a lower Tm. All α truncations were found to be about as stable as full-length α (Figure 3B); while the truncations α1–280 and α917–1160 were even more stable than full-length α.
α inhibits RecA/ssDNA-facilitated cleavage in vitro
It was previously shown that overexpression of α inhibited UmuD cleavage in vivo (58). We wanted to determine whether this phenomenon could be observed in vitro and therefore could be used to further characterize the UmuD binding site on α. Assays of RecA/ssDNA-facilitated UmuD cleavage were conducted in the presence of 10–25 µM α, which resulted in ∼50% less cleavage of UmuD at 37°C than in assays without α (Figure 4A). Because the melting point of α is ∼38°C at pH 7.5 (Figure 4B), the assay was also conducted at 30°C to minimize denaturation of α present in the reaction. This resulted in even less UmuD cleavage, ∼60% less cleavage than with UmuD alone. In order to determine whether the inhibition observed was specific for both UmuD and α, two controls were employed: (i) UmuD cleavage in the presence of BSA and (ii) LexA cleavage in the presence of α, as LexA also undergoes RecA/ssDNA-facilitated self-cleavage (62). In both cases, there was minimal change in cleavage efficiency (Figure 4D), indicating that the inhibition of UmuD cleavage by α is specific.
Figure 4.

The presence of α inhibits UmuD cleavage. After each cleavage reaction, UmuD and UmuD′ were resolved by 18% SDS–PAGE. Representative data are shown in A and C. (A) RecA/ssDNA nucleoprotein filament-facilitated UmuD cleavage assays were performed in the presence of 10 and 25 µM α wild type and at 37°C and 30°C. (B) To determine stability, the melting temperature was obtained for full-length α at pH 7.5 [25 µM α, 50 mM Hepes (pH 7.5)] and at pH 10 (25 µM α in alkaline cleavage buffer). (C) Cleavage assays were conducted under alkaline conditions in the absence of RecA/ssDNA. (D) A comparison of the cleavage assay performed under different conditions: RecA/ssDNA facilitated UmuD cleavage in the presence of α (red); UmuD cleavage under alkaline conditions in the presence of α (purple); RecA/ssDNA facilitated UmuD cleavage in the presence of BSA (blue); RecA/ssDNA facilitated LexA cleavage in the presence of α (green). Percentage of cleavage was calculated by comparing the density of cleaved product to the total amount of protein present. Each point represents the average of at least three experiments and the error bars represent the standard deviation from three independent experiments. (E) The Kd at pH 10 was determined to be 21.7 ± 11.7 µM.
Inhibition of UmuD cleavage in the RecA/ssDNA-facilitated cleavage assay may be due to the competitive binding of RecA/ssDNA and α for UmuD. When bound to α, UmuD cannot bind to the RecA/ssDNA filament and so cleavage cannot occur under these conditions. It is also possible that in addition to competitive binding, α may directly affect the ability of UmuD to cleave itself. In order to assess the possibility that α inhibited UmuD cleavage directly, rather than via competition with the RecA/ssDNA filament, cleavage assays were conducted under alkaline conditions (pH 10) (60) without the RecA/ssDNA nucleoprotein filament (Figure 4C). It was previously shown that UmuD can undergo autocleavage, albeit slowly, in the absence of RecA/ssDNA under alkaline conditions, as the elevated pH apparently facilitates deprotonation of the S60 nucleophile on UmuD (60,66). Under these conditions, no change in cleavage efficiency was observed, except at the highest concentrations of α and at 30°C. At pH 10, α was determined to be stable with a melting temperature of ∼36°C (Figure 4B). However, we found that at pH 10, the Kd was higher than at pH 7.5, indicating a decrease in affinity of α for UmuD (Figure 4E). Consequently, the observation that α did not substantially change the cleavage efficiency of UmuD under alkaline conditions may be attributed to the decreased affinity at pH 10. As a result, we conclude that in the case of RecA/ssDNA-facilitated cleavage, cleavage is inhibited likely due to competition for binding to UmuD between α and the RecA/ssDNA filament.
To confirm the binding sites between α and UmuD, the RecA/ssDNA-facilitated cleavage assay was conducted in the presence of two α truncations: the α1–280 truncation containing the N-terminal binding site (Figure 5A) and the α917–1160 truncation containing the C-terminal binding site (Figure 5B). As the concentration of α1–280 was increased from 0 µM to 40 µM, inhibition of cleavage was observed (Figure 5A). Approximately four times more α1–280 than wild-type α is required to observe the same extent of cleavage inhibition, consistent with the decreased affinity of α1–280 for UmuD. In contrast, the presence of 0–5 µM α917–1160 modestly increased UmuD cleavage efficiency (Figure 5B), which suggests that UmuD binds the C-terminal binding site in a conformation that may facilitate cleavage.
Figure 5.

The α truncation 1–280 inhibits UmuD cleavage. RecA/ssDNA nucleoprotein filament-facilitated UmuD cleavage was carried out at 30°C in the presence of (A) the α1–280 truncation and (B) the α917–1160 truncation. SDS–PAGE analysis showed that approximately four times as much α1–280 is needed to produce the same extent of cleavage inhibition as wild-type α. On the other hand, the presence of α917–1160 increased cleavage efficiency. Percentage of cleavage, shown below each lane, was calculated by comparing the density of cleaved product to the total amount of UmuD present.
The interaction between α and UmuD is conformation dependent
A thermal-shift assay was used to determine whether the presence of α had an effect on the conformational dynamics of UmuD. Previously, it had been shown that UmuD exhibits two distinct melting transitions: one at ∼28°C and another at ∼60°C (64). The absence of the first transition in the case of UmuD′ suggests that the lower-temperature transition represents the dissociation of the N-terminal arms of UmuD from the globular domain (56,64). In order to observe the effect of α on these two melting transitions, the thermal-shift assay was conducted in the presence of α (Figure 6A). By comparing the melting curve of UmuD alone with the melting curve of UmuD with α, it is clear that upon addition of α, the first melting transition is diminished. This suggests that α binds UmuD in a conformation-dependent manner. Two scenarios are possible (Figure 6C) when α binds to UmuD: (i) the arms are confined in such a way that they cannot dissociate from the body (‘arms down’) or (ii) the arms are not bound to the body and therefore do not undergo the transition to the unbound conformation (‘arms up’). The second melting transition is essentially unchanged in the presence of α.
Figure 6.

Binding to α alters the conformation of UmuD. (A) Melting curves of 45 μM UmuD (solid line), 45 μM UmuD with 1 μM α (dotted line) and 45 μM UmuD′ (dashed line). The solid line shows the melting transitions for UmuD at 32°C and 60°C. The disappearance of the first melting transition suggests that the interaction between α and UmuD influences the conformation of the arms. (B) Percent UmuD-S60A cross-linked by using BMH in the presence of either full-length α, α1–280 or α917–1160. (C) Cartoon showing two possible conformations of the UmuD N-terminal arms. The star indicates the position of the C24 residues, which are cross-linked by BMH.
To determine whether the C-terminal binding site binds UmuD in an ‘arms up’ or ‘arms down’ conformation (Figure 6C), UmuD was cross-linked using the homobifunctional thiol-specific reagent BMH in the presence or absence of α (Figure 6B). The UmuD dimer has two cysteine residues, one on each arm, which are represented by a star in Figure 6C. In order to cross-link with BMH, the two cysteine residues need to be in close proximity (within ∼13 Å) to each other. If the arms are bound to the body in the ‘arms down’ conformation, the two Cys residues are too far apart to be cross-linked; the arms will only be efficiently cross-linked if they are unbound from the body. As a result, this cross-linking assay is ideal for looking at the conformational dynamics of the arms. In order to prevent contamination with the UmuD′ cleavage product, the UmuD-S60A non-cleavable variant was used. It was observed that in the presence of the α truncation α917–1160, the amount of cross-linked UmuD-S60A dimer compared to the total UmuD-S60A present in the reaction was significantly reduced compared to cross-linking of UmuD-S60A alone (Figure 6B). This suggests that the C-terminus of α binds UmuD in an ‘arms down’ conformation. The presence of the α truncation α1–280 showed a similar but less dramatic reduction of cross-linking efficiency (Figure 6B).
The C-terminal binding site favors full-length UmuD
To determine whether the interaction between α and UmuD is selective for UmuD or UmuD′, binding constants were determined for the interaction of UmuD variants with full-length α, and the truncations α1–280 and α917–1160 (Table 1). When wild-type UmuD is purified, there is typically some amount of contaminating UmuD′. Moreover, cleavage can occur upon incubation or interaction with other proteins, so it is nearly always a complicating factor with wild-type UmuD. Furthermore, mixtures of UmuD and UmuD′ form UmuDD′ heterodimers (67); therefore, UmuD with some UmuD′ present will be a mixture of several dimeric species. Consequently, to determine the binding constant between full-length α and UmuD uncontaminated with UmuD′, the non-cleavable UmuD variant UmuD-S60A was used. The binding constant between UmuD-S60A and full-length α was determined to be 10.6 ± 2.9 µM. With UmuD′, the binding constant was 10.9 ± 1.6 µM, suggesting that full-length α does not favor UmuD′ or UmuD.
Table 1.
Equilibrium dissociation constants Kd (µM) for binding of α truncations to UmuD variants
| UmuD variants | Full-length α | α1–280 | α917–1160 |
|---|---|---|---|
| UmuD | 1.1 ± 0.6 | 7.3 ± 1.4 | 13.9 ± 5.1 |
| UmuD-S60A | 10.6 ± 2.9 | 10.3 ± 4.3 | 0.7 ± 0.3 |
| UmuD′ | 10.9 ± 1.6 | 8.6 ± 1.0 | 3.8 ± 0.9 |
| UmuD3A | 8.7 ± 1.5 | 9.3 ± 3.2 | 3.4 ± 1.0 |
| UmuD′-S60A/UmuD-S60A | 4.6 ± 0.9 |
Binding affinity was also analyzed between the α truncation α917–1160, containing the proposed C-terminal binding site (residues 956–975), and the UmuD variants UmuD-S60A and UmuD′. As shown in Table 1, α917–1160 binds significantly more strongly to UmuD-S60A (0.7 ± 0.3 µM) than to UmuD′ (3.8 ± 0.9 µM), suggesting that the C-terminal binding site selectively favors full-length UmuD over the cleavage product, UmuD′. On the other hand, the α truncation α1–280, containing the proposed N-terminal binding site, does not distinguish between full-length UmuD and UmuD′, exhibiting similar binding constants with UmuD-S60A (10.3 ± 4.3 µM) and UmuD′ (8.6 ± 1.0 µM) (Table 1).
UmuD3A is a full-length UmuD variant that exhibits properties of UmuD′ (27). UmuD3A contains three mutations (T14A, L17A, F18A) located in the N-terminal arms of the protein, which disrupt packing of the arms against the C-terminal globular domain and cause UmuD3A to adopt a more UmuD′-like conformation (27,56,64). As expected, UmuD3A has similar affinity for the α variants as UmuD′ (Table 1). Because both UmuD′ and UmuD3A have a more exposed globular domain than wild-type UmuD, it seems that the α C-terminus preferentially binds UmuD when the arms are bound to the C-terminal globular domain of UmuD, which is consistent with our findings in the cross-linking experiments with BMH (Figure 6B).
The distinguishably different binding constants for wild-type UmuD compared to those of UmuD-S60A and UmuD′ are consistent with our observation that preparations of wild-type UmuD may contain some UmuD′. To probe this further, we assembled a mixture of 1:1 UmuD-S60A:UmuD′-S60A (Table 1) to somewhat resemble the conditions of wild-type UmuD. Because it is known that a monomer of UmuD can cleave the arm of its partner (68), we used the active site mutation S60A in both UmuD and UmuD′ to prevent any further cleavage. Under these conditions, the Kd was determined to be 4.6 ± 0.9 µM, which is intermediate between the Kds determined with UmuD compared to those with UmuD′ or UmuD-S60A.
UmuD disrupts the DNA Pol III α–β complex
Our observation that UmuD binds α at a site (residues 956–975) that is near the β-binding site on α (residues 920–924) prompted us to investigate the effect of the umuD gene products on the DNA pol III α–β complex. The β clamp and the α subunit were labeled with donor and acceptor fluorophores, respectively, as shown in Figure 7C, and FRET was monitored in the presence or absence of UmuD, UmuD′ or UmuD-S60A (Figure 7A and B). As expected, FRET was observed when the donor on the β clamp was excited in the presence of acceptor-labeled α subunit (Figure 7A), indicating that the two proteins interact with each other. Addition of UmuD to the reaction resulted in higher emission intensity from the donor, indicating a decrease in FRET efficiency and consistent with disruption of the α–β complex. The effect was more apparent with the addition of UmuD-S60A, the full-length non-cleavable variant of UmuD. This suggests that UmuD is able to disrupt the interaction between the α subunit and the β clamp. In comparison, no difference in FRET efficiency was observed in the presence of UmuD′ (Figure 7A and B), suggesting that UmuD′ is not able to disrupt the α–β complex. These findings are consistent with our observation that the binding site for UmuD on the C-terminal domain of α, near where β also binds to α, binds full-length UmuD (UmuD-S60A) with higher affinity than it binds UmuD′. Taken together, these observations indicate that specifically full-length UmuD is capable of disrupting the α–β complex (Figure 7C).
Figure 7.

Fluorescence resonance energy transfer analysis shows that UmuD disrupts the interaction between the α polymerase, labeled with Alexa Fluor 647 C2-maleimide, and the β processivity clamp, labeled with Alexa Fluor 488 C5-maleimide. (A) FRET was reduced in the presence of UmuD and UmuD-S60A but not in the presence of UmuD′. FRET between β and α was reduced in the presence of UmuD-S60A in a concentration-dependent manner, from 1 to 40 μM UmuD-S60A (not shown). (B) Bar graph showing FRET efficiency at each condition; representative of five trials. The error bars represent the standard deviation of at least three trials. (C) Schematic of FRET experiment showing that the FRET observed upon interaction of β and α is reduced with the addition of full-length UmuD, but not UmuD'. As UmuD interacts with both α and β at competing sites, the α–β interaction could be disrupted by interaction of UmuD with either protein. Other modes of interaction are possible, in which UmuD induces a conformational change in one or both proteins that inhibits binding, for example, but since UmuD interacts with α at the same site where β interacts with α and UmuD interacts with β at a site overlapping where α interacts with β (69), direct competition is the most straightforward model.
DISCUSSION
The disruption of replication fork progression caused by DNA damage necessitates TLS polymerases having access to the primer terminus at the site of the damage in order to carry out replication of damaged DNA. Two models have been proposed to explain how replicative and TLS polymerases exchange: the ‘toolbelt’ model and the dynamic processivity model. It has been shown that the α subunit and pol IV can interact with the β clamp simultaneously (70), allowing replication to alternate between the two DNA polymerases without the need for them to dissociate from the β clamp (the ‘toolbelt’ model). In such a model, when a DNA lesion is encountered, α stalls and a Y family polymerase takes over replication (30). Once the DNA lesion has been bypassed, α once again resumes replication. On the other hand, the dynamic processivity model (71) suggests that the replicating polymerase is constantly being exchanged with other polymerases without affecting overall processivity. DNA pol IV (DinB), a TLS polymerase, has been shown to inhibit DNA pol III replication, which may facilitate dissociation of the pol III α subunit and allow TLS to occur (59).
It has been shown that UmuDC inhibits DNA replication (49,50), allowing time for accurate DNA repair processes to occur (50). There is also evidence that the umuD gene products play a direct role in regulating access to the replication fork by interacting with the α, β and ε subunits of DNA pol III (58). The interaction between UmuD and the β clamp (27) is believed to contribute to a primitive DNA damage checkpoint (58). In this report, we investigate the interaction between the α subunit and the umuD gene products, UmuD and UmuD′. Our observations suggest that another contribution to the DNA damage checkpoint is likely to be the disruption of the α–β complex by UmuD.
In this work, we identified two UmuD binding sites on α (Figure 3A): one in the N-terminal domain (residues 1–280) and one in the C-terminal domain (residues 956–975). It has been shown that UmuD and UmuD′ differentially bind the α and β subunits, where UmuD′ interacts more strongly with α than with β and UmuD interacts more strongly with β than with α (58). A comparison of the binding constant between UmuD and β (5.5 ± 0.8 µM) (27) with the binding constant for UmuD-S60A and wild-type α (10.6 ± 2.9 µM) (Table 1), supports the previous suggestion, based on affinity chromatography with cell extracts, that UmuD binds more tightly to the β clamp. On the other hand, the interaction between α and the umuD gene products is complicated by our observation of two UmuD binding sites on α and because the C-terminal binding site favors binding to full-length UmuD. Notably, the Kds we determined for a variety of UmuD and α constructs (ranging from 0.7–14 μM, Table 1) are similar to those obtained between UmuD and β and between UmuD and DinB (27,57). The cellular concentration of UmuD and UmuD′ ranges from 0.25 to 1 μM depending on whether cells are SOS induced (72), which suggests that UmuD and α can interact in vivo.
The C-terminal domain of α appears to be a hub for protein and DNA interactions. This apparent regulatory domain contains the binding site for the β clamp (residues 920–924) (22), and a binding site for τ (residues 1120–1160) (7,19), a subunit of the clamp loader complex (Figure 1A). This domain is also believed to contain an OB fold, which has been shown to bind preferentially to ssDNA and act as a sensor detecting ssDNA upstream from the primer terminus (17,73). We observed that the C-terminal domain of α (residues 917–1160) favors binding to full-length UmuD (Table 1), and that full-length UmuD is able to disrupt the binding of α to β. On the other hand, less is known about the N-terminal domain of α. Apart from binding UmuD, as we have demonstrated here, this domain has also been shown to bind the ε subunit (15). Unfortunately, it is difficult to probe possible direct effects of UmuD on the α–ε interaction because of the difficulty of acquiring purified, soluble ε.
The umuD gene products have ‘distinct and temporally separated’ roles in the DNA damage response, first by taking part in a primitive DNA damage checkpoint by inhibiting DNA replication and then by activating TLS (50). We observed that FRET between α and β was reduced with the addition of full-length UmuD, suggesting that the interaction between α and β is disrupted specifically by UmuD (Figure 7C). FRET efficiency was not affected by the addition of UmuD′. This selective disruption of the α–β complex by full-length UmuD together with the preferential binding of full-length UmuD by the C-terminal domain of α suggests a specific role for full-length UmuD in polymerase management. Because UmuD is the predominant species at the beginning of the SOS response, the displacement of α from the β clamp most likely occurs before the appearance of DNA pol V (UmuD′C).
How can α differentiate between UmuD and UmuD′ at the molecular level? The primary difference between UmuD and UmuD′ is the absence of the N-terminal 24 amino acid residues in UmuD′ suggesting that the N-terminal arms are involved in the interaction between α and UmuD. As shown here, several lines of evidence, including thermal melting and cross-linking analysis, support a model in which α binds UmuD in an ‘arms down’ conformation. Similarly, the interaction between UmuD and the β clamp has been shown to involve both the N-terminal arms and the C-terminal globular domain of UmuD (51); furthermore, the β clamp can bind to a UmuD variant that contains an artificial disulfide bridge (C24-F94C) locking the arms to the globular domain (51). Taken together, previous and current work suggests that both α and the β clamp bind UmuD in a similar fashion in which the N-terminal arms are bound to the C-terminal globular domain. Collectively, our findings suggest that the already extensive role of UmuD in responses to DNA damage includes releasing α from the β clamp.
FUNDING
New Faculty Award from the Camille & Henry Dreyfus Foundation to (P.J.B.); the National Science Foundation (CAREER Award, grant number MCB-0845033 to P.J.B.); the Northeastern University Office of the Provost. PJB is a Cottrell Scholar of the Research Corporation for Science Advancement. Funding for open access charge: National Science Foundation CAREER Award MCB-0845033.
Conflict of interest statement. None declared.
ACKNOWLEDGEMENTS
The authors thank Prof. April Gu (Northeastern University) and laboratory members for providing time and training to use their RT-PCR instrument for the thermal-shift assays. They also thank Prof. Zhaohui Zhou (Northeastern University) for use of the fluorimeter, Dr. Ronaldo Mohana-Borges (Instituto de Biofísica Carlos Chagas Filho, UFRJ, Brazil) for the LexA expression plasmid, and Dr. Meindert Lamers and Prof. Jon Kuriyan (UC Berkeley) for the plasmids expressing truncations of α. The authors thank members of the Beuning Laboratory, especially Jaylene N. Ollivierre and Jana Sefcikova, for assistance.
REFERENCES
- 1.Maki H, Maki S, Kornberg A. DNA polymerase III holoenzyme of Escherichia coli. IV. The holoenzyme is an asymmetric dimer with twin active sites. J. Biol. Chem. 1988;263:6570–6578. [PubMed] [Google Scholar]
- 2.Kelman Z, O’Donnell M. DNA polymerase III holoenzyme: structure and function of a chromosomal replicating machine. Annu. Rev. Biochem. 1995;64:171–200. doi: 10.1146/annurev.bi.64.070195.001131. [DOI] [PubMed] [Google Scholar]
- 3.Johnson A, O’Donnell M. Cellular DNA replicases: components and dynamics at the replication fork. Annu. Rev. Biochem. 2005;74:283–315. doi: 10.1146/annurev.biochem.73.011303.073859. [DOI] [PubMed] [Google Scholar]
- 4.McHenry CS. DNA replicases from a bacterial perspective. Annu. Rev. Biochem. 2011;80:403–436. doi: 10.1146/annurev-biochem-061208-091655. [DOI] [PubMed] [Google Scholar]
- 5.Onrust R, Finkelstein J, Turner J, Naktinis V, O’Donnell M. Assembly of a chromosomal replication machine: two DNA polymerases, a clamp loader, and sliding clamps in one holoenzyme particle. III. Interface between two polymerases and the clamp loader. J. Biol. Chem. 1995;270:13366–13377. doi: 10.1074/jbc.270.22.13366. [DOI] [PubMed] [Google Scholar]
- 6.Ellison V, Stillman B. Opening of the clamp: an intimate view of an ATP-driven biological machine. Cell. 2001;106:655–660. doi: 10.1016/s0092-8674(01)00498-6. [DOI] [PubMed] [Google Scholar]
- 7.Gao D, McHenry CS. Tau binds and organizes Escherichia coli replication through distinct domains. Partial proteolysis of terminally tagged tau to determine candidate domains and to assign domain V as the alpha binding domain. J. Biol. Chem. 2001;276:4433–4440. doi: 10.1074/jbc.M009828200. [DOI] [PubMed] [Google Scholar]
- 8.Olson MW, Dallmann HG, McHenry CS. DnaX complex of Escherichia coli DNA polymerase III holoenzyme. The chi psi complex functions by increasing the affinity of tau and gamma for delta-delta' to a physiologically relevant range. J. Biol. Chem. 1995;270:29570–29577. [PubMed] [Google Scholar]
- 9.Dallmann HG, Kim S, Pritchard AE, Marians KJ, McHenry CS. Characterization of the unique C terminus of the Escherichia coli tau DnaX protein. Monomeric C-tau binds alpha AND DnaB and can partially replace tau in reconstituted replication forks. J. Biol. Chem. 2000;275:15512–15519. doi: 10.1074/jbc.M909257199. [DOI] [PubMed] [Google Scholar]
- 10.O’Donnell ME. Accessory proteins bind a primed template and mediate rapid cycling of DNA polymerase III holoenzyme from Escherichia coli. J. Biol. Chem. 1987;262:16558–16565. [PubMed] [Google Scholar]
- 11.Lamers MH, Georgescu RE, Lee SG, O’Donnell M, Kuriyan J. Crystal structure of the catalytic alpha subunit of E. coli replicative DNA polymerase III. Cell. 2006;126:881–892. doi: 10.1016/j.cell.2006.07.028. [DOI] [PubMed] [Google Scholar]
- 12.Brautigam CA, Steitz TA. Structural and functional insights provided by crystal structures of DNA polymerases and their substrate complexes. Curr. Opin. Struct. Biol. 1998;8:54–63. doi: 10.1016/s0959-440x(98)80010-9. [DOI] [PubMed] [Google Scholar]
- 13.Rothwell PJ, Waksman G. Structure and mechanism of DNA polymerases. Adv. Protein Chem. 2005;71:401–440. doi: 10.1016/S0065-3233(04)71011-6. [DOI] [PubMed] [Google Scholar]
- 14.Aravind L, Koonin EV. Phosphoesterase domains associated with DNA polymerases of diverse origins. Nucleic Acids Res. 1998;26:3746–3752. doi: 10.1093/nar/26.16.3746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wieczorek A, McHenry CS. The NH2-terminal PHP domain of the alpha subunit of the Escherichia coli replicase binds the epsilon proofreading subunit. J. Biol. Chem. 2006;281:12561–12567. doi: 10.1074/jbc.M513844200. [DOI] [PubMed] [Google Scholar]
- 16.Bailey S, Wing RA, Steitz TA. The structure of T. aquaticus DNA polymerase III is distinct from eukaryotic replicative DNA polymerases. Cell. 2006;126:893–904. doi: 10.1016/j.cell.2006.07.027. [DOI] [PubMed] [Google Scholar]
- 17.McCauley MJ, Shokri L, Sefcikova J, Venclovas C, Beuning PJ, Williams MC. Distinct double- and single-stranded DNA binding of E. coli replicative DNA polymerase III alpha subunit. ACS Chem. Biol. 2008;3:577–587. doi: 10.1021/cb8001107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.López de Saro FJ, Georgescu RE, O’Donnell M. A peptide switch regulates DNA polymerase processivity. Proc. Natl Acad. Sci. USA. 2003;100:14689–14694. doi: 10.1073/pnas.2435454100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jergic S, Ozawa K, Williams NK, Su XC, Scott DD, Hamdan SM, Crowther JA, Otting G, Dixon NE. The unstructured C-terminus of the tau subunit of Escherichia coli DNA polymerase III holoenzyme is the site of interaction with the alpha subunit. Nucleic Acids Res. 2007;35:2813–2824. doi: 10.1093/nar/gkm079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Su XC, Jergic S, Keniry MA, Dixon NE, Otting G. Solution structure of domains IVa and V of the tau subunit of Escherichia coli DNA polymerase III and interaction with the alpha subunit. Nucleic Acids Res. 2007;35:2825–2832. doi: 10.1093/nar/gkm080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.López de Saro FJ, Georgescu RE, Goodman MF, O’Donnell M. Competitive processivity-clamp usage by DNA polymerases during DNA replication and repair. EMBO J. 2003;22:6408–6418. doi: 10.1093/emboj/cdg603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dohrmann PR, McHenry CS. A bipartite polymerase-processivity factor interaction: only the internal beta binding site of the alpha subunit is required for processive replication by the DNA polymerase III holoenzyme. J. Mol. Biol. 2005;350:228–239. doi: 10.1016/j.jmb.2005.04.065. [DOI] [PubMed] [Google Scholar]
- 23.Wieczorek A, McHenry CS. The NH2-terminal php domain of the alpha subunit of the Escherichia coli replicase binds the epsilon proofreading subunit. J. Biol. Chem. 2006;281:12561–12567. doi: 10.1074/jbc.M513844200. [DOI] [PubMed] [Google Scholar]
- 24.Sharma R. Master of Science Thesis. Boston, MA: Northeastern University; 2010. Engineering Escherichia coli DNA Polymerase III α for Translesion Synthesis. [Google Scholar]
- 25.Dohrmann PR, McHenry CS. A bipartite polymerase-processivity factor interaction: only the internal beta binding site of the alpha subunit is required for processive replication by the DNA polymerase III holoenzyme. J. Mol. Biol. 2005;350:228–239. doi: 10.1016/j.jmb.2005.04.065. [DOI] [PubMed] [Google Scholar]
- 26.Gao D, McHenry CS. Tau binds and organizes Escherichia coli replication through distinct domains. Partial proteolysis of terminally tagged tau to determine candidate domains and to assign domain V as the alpha binding domain. J. Biol. Chem. 2001;276:4433–4440. doi: 10.1074/jbc.M009828200. [DOI] [PubMed] [Google Scholar]
- 27.Beuning PJ, Simon SM, Zemla A, Barsky D, Walker GC. A non-cleavable UmuD variant that acts as a UmuD' mimic. J. Biol. Chem. 2006;281:9633–9640. doi: 10.1074/jbc.M511101200. [DOI] [PubMed] [Google Scholar]
- 28.Peat TS, Frank EG, McDonald JP, Levine AS, Woodgate R, Hendrickson WA. Structure of the UmuD' protein and its regulation in response to DNA damage. Nature. 1996;380:727–730. doi: 10.1038/380727a0. [DOI] [PubMed] [Google Scholar]
- 29.Fujii S, Fuchs RP. Interplay among replicative and specialized DNA polymerases determines failure or success of translesion synthesis pathways. J. Mol. Biol. 2007;372:883–893. doi: 10.1016/j.jmb.2007.07.036. [DOI] [PubMed] [Google Scholar]
- 30.Fujii S, Fuchs RP. Defining the position of the switches between replicative and bypass DNA polymerases. EMBO J. 2004;23:4342–4352. doi: 10.1038/sj.emboj.7600438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Higuchi K, Katayama T, Iwai S, Hidaka M, Horiuchi T, Maki H. Fate of DNA replication fork encountering a single DNA lesion during oriC plasmid DNA replication in vitro. Genes Cells. 2003;8:437–449. doi: 10.1046/j.1365-2443.2003.00646.x. [DOI] [PubMed] [Google Scholar]
- 32.Pagés V, Fuchs RP. Uncoupling of leading- and lagging-strand DNA replication during lesion bypass in vivo. Science. 2003;300:1300–1303. doi: 10.1126/science.1083964. [DOI] [PubMed] [Google Scholar]
- 33.Nohmi T. Environmental stress and lesion-bypass DNA polymerases. Annu. Rev. Microbiol. 2006;60:231–253. doi: 10.1146/annurev.micro.60.080805.142238. [DOI] [PubMed] [Google Scholar]
- 34.Sassanfar M, Roberts JW. Nature of the SOS-inducing signal in Escherichia coli. The involvement of DNA replication. J. Mol. Biol. 1990;212:79–96. doi: 10.1016/0022-2836(90)90306-7. [DOI] [PubMed] [Google Scholar]
- 35.Simmons LA, Foti JJ, Cohen SE, Walker GC. Chapter 5.4.3 The SOS Regulatory Network. In: Böck A, Curtiss III R, Kaper JB, Karp PD, Neidhardt FC, Nyström T, Slauch JM, Squires CL, Ussery D, editors. EcoSal-Escherichia coli and Salmonella: Cellular and Molecular Biology. Washington, D.C: ASM Press; 2008. [Google Scholar]
- 36.Jarosz DF, Beuning PJ, Cohen SE, Walker GC. Y-family DNA polymerases in Escherichia coli. Trends Microbiol. 2007;15:70–77. doi: 10.1016/j.tim.2006.12.004. [DOI] [PubMed] [Google Scholar]
- 37.Pata JD. Structural diversity of the Y-family DNA polymerases. Biochim. Biophys. Acta. 2010;1804:1124–1135. doi: 10.1016/j.bbapap.2010.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yang W, Woodgate R. What a difference a decade makes: insights into translesion DNA synthesis. Proc. Natl Acad. Sci. USA. 2007;104:15591–15598. doi: 10.1073/pnas.0704219104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Washington MT, Carlson KD, Freudenthal BD, Pryor JM. Variations on a theme: eukaryotic Y-family DNA polymerases. Biochim. Biophys. Acta. 2010;1804:1113–1123. doi: 10.1016/j.bbapap.2009.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Waters LS, Minesinger BK, Wiltrout ME, D’Souza S, Woodruff RV, Walker GC. Eukaryotic translesion polymerases and their roles and regulation in DNA damage tolerance. Microbiol. Mol. Biol. Rev. 2009;73:134–154. doi: 10.1128/MMBR.00034-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sutton MD, Walker GC. Managing DNA polymerases: coordinating DNA replication, DNA repair, and DNA recombination. Proc. Natl Acad. Sci. USA. 2001;98:8342–8349. doi: 10.1073/pnas.111036998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Goodman MF. Error-prone repair DNA polymerases in prokaryotes and eukaryotes. Annu. Rev. Biochem. 2002;71:17–50. doi: 10.1146/annurev.biochem.71.083101.124707. [DOI] [PubMed] [Google Scholar]
- 43.Pagés V, Fuchs RP. How DNA lesions are turned into mutations within cells? Oncogene. 2002;21:8957–8966. doi: 10.1038/sj.onc.1206006. [DOI] [PubMed] [Google Scholar]
- 44.Reuven NB, Arad G, Maor-Shoshani A, Livneh Z. The mutagenesis protein UmuC is a DNA polymerase activated by UmuD', RecA, and SSB and is specialized for translesion replication. J. Biol. Chem. 1999;274:31763–31766. doi: 10.1074/jbc.274.45.31763. [DOI] [PubMed] [Google Scholar]
- 45.Tang M, Shen X, Frank EG, O’Donnell M, Woodgate R, Goodman MF. UmuD'2C is an error-prone DNA polymerase, Escherichia coli pol V. Proc. Natl Acad. Sci. USA. 1999;96:8919–8924. doi: 10.1073/pnas.96.16.8919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wagner J, Gruz P, Kim SR, Yamada M, Matsui K, Fuchs RP, Nohmi T. The dinB gene encodes a novel E. coli DNA polymerase, DNA pol IV, involved in mutagenesis. Mol. Cell. 1999;4:281–286. doi: 10.1016/s1097-2765(00)80376-7. [DOI] [PubMed] [Google Scholar]
- 47.Ollivierre JN, Fang J, Beuning PJ. The roles of UmuD in regulating mutagenesis. J. Nucleic Acids. 2010 doi: 10.4061/2010/947680. Article ID 947680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sutton MD, Walker GC. umuDC-mediated cold sensitivity is a manifestation of functions of the UmuD2C complex involved in a DNA damage checkpoint control. J. Bacteriol. 2001;183:1215–1224. doi: 10.1128/JB.183.4.1215-1224.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Marsh L, Walker GC. Cold sensitivity induced by overproduction of UmuDC in Escherichia coli. J. Bacteriol. 1985;162:155–161. doi: 10.1128/jb.162.1.155-161.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Opperman T, Murli S, Smith BT, Walker GC. A model for a umuDC-dependent prokaryotic DNA damage checkpoint. Proc Natl Acad. Sci. USA. 1999;96:9218–9223. doi: 10.1073/pnas.96.16.9218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sutton MD, Narumi I, Walker GC. Posttranslational modification of the umuD-encoded subunit of Escherichia coli DNA polymerase V regulates its interactions with the beta processivity clamp. Proc. Natl Acad. Sci. USA. 2002;99:5307–5312. doi: 10.1073/pnas.082322099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sutton MD. Damage signals triggering the Escherichia coli SOS response. In: Seide W, Kow YW, Doetsch PW, editors. DNA Damage and Recognition. New York, NY: Taylor and Francis; 2006. pp. 781–802. [Google Scholar]
- 53.Tang M, Pham P, Shen X, Taylor JS, O’Donnell M, Woodgate R, Goodman MF. Roles of E. coli DNA polymerases IV and V in lesion-targeted and untargeted SOS mutagenesis. Nature. 2000;404:1014–1018. doi: 10.1038/35010020. [DOI] [PubMed] [Google Scholar]
- 54.Simon SM, Sousa FJ, Mohana-Borges R, Walker GC. Regulation of Escherichia coli SOS mutagenesis by dimeric intrinsically disordered umuD gene products. Proc. Natl Acad. Sci. USA. 2008;105:1152–1157. doi: 10.1073/pnas.0706067105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sutton MD, Guzzo A, Narumi I, Costanzo M, Altenbach C, Ferentz AE, Hubbell WL, Walker GC. A model for the structure of the Escherichia coli SOS-regulated UmuD2 protein. DNA Repair (Amst) 2002;1:77–93. doi: 10.1016/s1568-7864(01)00006-4. [DOI] [PubMed] [Google Scholar]
- 56.Ollivierre JN, Budil DE, Beuning PJ. Electron spin labeling reveals the highly dynamic N-terminal arms of the SOS mutagenesis protein UmuD. Mol. BioSyst. 2011;7:3183–3186. doi: 10.1039/c1mb05334e. [DOI] [PubMed] [Google Scholar]
- 57.Godoy VG, Jarosz DF, Simon SM, Abyzov A, Ilyin V, Walker GC. UmuD and RecA directly modulate the mutagenic potential of the Y family DNA polymerase DinB. Mol. Cell. 2007;28:1058–1070. doi: 10.1016/j.molcel.2007.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sutton MD, Opperman T, Walker GC. The Escherichia coli SOS mutagenesis proteins UmuD and UmuD' interact physically with the replicative DNA polymerase. Proc. Natl Acad. Sci. USA. 1999;96:12373–12378. doi: 10.1073/pnas.96.22.12373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Furukohri A, Goodman MF, Maki H. A dynamic polymerase exchange with Escherichia coli DNA polymerase IV replacing DNA polymerase III on the sliding clamp. J. Biol. Chem. 2008;283:11260–11269. doi: 10.1074/jbc.M709689200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Beuning PJ, Simon SM, Godoy VG, Jarosz DF, Walker GC. Characterization of Escherichia coli translesion synthesis polymerases and their accessory factors. Methods Enzymol. 2006;408:318–340. doi: 10.1016/S0076-6879(06)08020-7. [DOI] [PubMed] [Google Scholar]
- 61.Fang J, Engen JR, Beuning PJ. Escherichia coli processivity clamp beta from DNA polymerase III is dynamic in solution. Biochemistry. 2011;50:5958–5968. doi: 10.1021/bi200580b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Little JW, Kim B, Roland KL, Smith MH, Lin LL, Slilaty SN. Cleavage of LexA repressor. Methods Enzymol. 1994;244:266–284. doi: 10.1016/0076-6879(94)44022-0. [DOI] [PubMed] [Google Scholar]
- 63.Ericsson UB, Hallberg BM, Detitta GT, Dekker N, Nordlund P. Thermofluor-based high-throughput stability optimization of proteins for structural studies. Anal. Biochem. 2006;357:289–298. doi: 10.1016/j.ab.2006.07.027. [DOI] [PubMed] [Google Scholar]
- 64.Fang J, Rand KD, Silva MC, Wales TE, Engen JR, Beuning PJ. Conformational dynamics of the Escherichia coli DNA polymerase manager proteins UmuD and UmuD'. J. Mol. Biol. 2010;398:40–53. doi: 10.1016/j.jmb.2010.02.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lee MH, Ohta T, Walker GC. A monocysteine approach for probing the structure and interactions of the UmuD protein. J. Bacteriol. 1994;176:4825–4837. doi: 10.1128/jb.176.16.4825-4837.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kulaeva OI, Wootton JC, Levine AS, Woodgate R. Characterization of the umu-complementing operon from R391. J. Bacteriol. 1995;177:2737–2743. doi: 10.1128/jb.177.10.2737-2743.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Battista JR, Ohta T, Nohmi T, Sun W, Walker GC. Dominant negative umuD mutations decreasing RecA-mediated cleavage suggest roles for intact UmuD in modulation of SOS mutagenesis. Proc. Natl Acad. Sci. USA. 1990;87:7190–7194. doi: 10.1073/pnas.87.18.7190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.McDonald JP, Frank EG, Levine AS, Woodgate R. Intermolecular cleavage by UmuD-like mutagenesis proteins. Proc. Natl Acad. Sci. USA. 1998;95:1478–1483. doi: 10.1073/pnas.95.4.1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Duzen JM, Walker GC, Sutton MD. Identification of specific amino acid residues in the E. coli beta processivity clamp involved in interactions with DNA polymerase III, UmuD and UmuD'. DNA Repair. 2004;3:301–312. doi: 10.1016/j.dnarep.2003.11.008. [DOI] [PubMed] [Google Scholar]
- 70.Indiani C, McInerney P, Georgescu R, Goodman MF, O’Donnell M. A sliding-clamp toolbelt binds high- and low-fidelity DNA polymerases simultaneously. Mol. Cell. 2005;19:805–815. doi: 10.1016/j.molcel.2005.08.011. [DOI] [PubMed] [Google Scholar]
- 71.Yang J, Zhuang Z, Roccasecca RM, Trakselis MA, Benkovic SJ. The dynamic processivity of the T4 DNA polymerase during replication. Proc. Natl Acad. Sci. USA. 2004;101:8289–8294. doi: 10.1073/pnas.0402625101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Woodgate R, Ennis DG. Levels of chromosomally encoded Umu proteins and requirements for in vivo UmuD cleavage. Mol. Gen. Genet. 1991;229:10–16. doi: 10.1007/BF00264207. [DOI] [PubMed] [Google Scholar]
- 73.Georgescu RE, Kurth I, Yao NY, Stewart J, Yurieva O, O’Donnell M. Mechanism of polymerase collision release from sliding clamps on the lagging strand. EMBO J. 2009;28:2981–2991. doi: 10.1038/emboj.2009.233. [DOI] [PMC free article] [PubMed] [Google Scholar]