

Abstract
Background
At present five evolutionary hypotheses have been proposed to explain the great variability of the genomic GC content among and within genomes: the mutational bias, the biased gene conversion, the DNA breakpoints distribution, the thermal stability and the metabolic rate. Several studies carried out on bacteria and teleostean fish pointed towards the critical role played by the environment on the metabolic rate in shaping the base composition of genomes. In mammals the debate is still open, and evidences have been produced in favor of each evolutionary hypothesis. Human genes were assigned to three large functional categories (as well as to the corresponding functional classes) according to the KOG database: (i) information storage and processing, (ii) cellular processes and signaling, and (iii) metabolism. The classification was extended to the organisms so far analyzed performing a reciprocal Blastp and selecting the best reciprocal hit. The base composition was calculated for each sequence of the whole CDS dataset.
Results
The GC3 level of the above functional categories was increasing from (i) to (iii). This specific compositional pattern was found, as footprint, in all mammalian genomes, but not in frog and lizard ones. Comparative analysis of human versus both frog and lizard functional categories showed that genes involved in the metabolic processes underwent the highest GC3 increment. Analyzing the KOG functional classes of genes, again a well defined intra-genomic pattern was found in all mammals. Not only genes of metabolic pathways, but also genes involved in chromatin structure and dynamics, transcription, signal transduction mechanisms and cytoskeleton, showed an average GC3 level higher than that of the whole genome. In the case of the human genome, the genes of the aforementioned functional categories showed a high probability to be associated with the chromosomal bands.
Conclusions
In the light of different evolutionary hypotheses proposed so far, and contributing with different potential to the genome compositional heterogeneity of mammalian genomes, the one based on the metabolic rate seems to play not a minor role. Keeping in mind similar results reported in bacteria and in teleosts, the specific compositional patterns observed in mammals highlight metabolic rate as unifying factor that fits over a wide range of living organisms.
Background
As recently stated by Meyer and collaborators "structure and organization of genomes belongs among the key questions of genome biology" [1]. One of the most crucial and largely debated questions is centered on the nature of the forces driving the base compositional variation among genomes. At present as much as five evolutionary hypotheses have been proposed to explain the great variability of the genomic GC content, which can be split in two groups, on the bases of the nature of the forces driving the genome evolution, i.e. intra- or extra-cellular [2]. The former, including the mutational bias, the biased gene conversion (BGC) and the DNA breakpoints distribution (BPR) hypotheses, is mainly founded on stochastic events arising during intracellular processes, such as DNA replication, repair and recombination. The latter, including the thermal stability and the metabolic rate hypotheses, take into account the role of adaptive processes resulting from the interaction of the organism with the surrounding environment.
In the frame of the neutral theory, the mutational bias hypothesis [3-5] was first proposed to explain the great variation of the genomic GC content among bacteria, and later on extended to higher vertebrates [6]. In the same frame, the BGC is based essentially on a synergy between recombination events and biased DNA repair system [7-9]. The BPR hypothesis considers that evolutionary rearrangement of breakages happen with a uniform propensity along the genome. Growing body of evidence shows a heterogeneous distribution of breakpoints in mammalian genomes, occurring more frequently in the GC-rich regions, harboring replication origin sites and characterized by high transcriptional activity [10].
In the frame of the adaptive point of view, several environmental factors significantly correlated with the DNA base composition, have been reported in bacteria: competition for metabolic resources [11], anaerobiosis [12], endosymbiosis [13], environments/habitats [2], growth temperature [14], and "aerobic respiration" [15]. In particular, the last two papers stressed the effect on the genomic GC content of the main factors affecting the environmental dynamics: temperature and metabolism. According to the thermodynamic hypothesis, an increment of environmental or body temperature triggers a GC increment, stabilizing DNA, RNA and proteins [16]. The hypothesis based on the metabolic rate was grounded on two DNA features, bendability [17] and nucleosome formation potential [18], both significantly correlated with the GC content. More precisely, a higher DNA bendability and a decrement of the nucleosome formation potential have been reported to be both favored by an increment of the GC content [17,18]. Accordingly, GC-richest genomic regions showed a high transcriptional activity [19,20].
Preliminary analysis of human genes grouped in functional classes according to the KOG database [21,22] showed a biased distribution of the GC3 level that was significantly higher in the functional classes of genes involved in metabolic processes [23]. In the present paper, the analysis of the KOG categories and functional classes of genes was extended to thirteen completely sequenced mammalian genomes, as well as to amphibian (X. tropicalis) and reptile (A. carolinensis) genomes.
Current results confirmed previous conclusions [23], further stressing the role of metabolic rate in shaping the mammalian genome organization. Indeed, a compositional hierarchy among functional categories was found, and the GC3 content of the genes involved in metabolic processes was the highest in all mammalian genomes so far analyzed. Interestingly, the mammalian compositional pattern was absent in the amphibian and the reptile genomes. The finding opened critical evolutionary questions on the compositional transition from "cold- to warm-blooded vertebrates" [24-26], more precisely from amphibian/reptile to mammals, and will be discussed in the light of the current hypotheses about genome compositional variability.
Interestingly, in all mammals the functional classes that recurrently showed a GC3 level higher than that of the whole genome were, apart from those involved in metabolic processes, those involved in: Chromatin structure and dynamics, Transcription, Signal transduction mechanisms and Cytoskeleton. In the human genome, the aforesaid functional classes showed a high probability to cluster in the GC-richest chromosomal bands. In the light of current literature this organization could reflect the needs of a coordinated response to stressing stimuli altering the normal metabolic rate.
Methods
In the KOG database http://www.ncbi.nlm.nih.gov/COG/[21,22] human proteins were grouped in functional classes (denoted by capital letters in square brackets), in turn grouped in three large categories, namely: (i) information storage and processing; (ii) cellular processes and signaling; (iii) metabolism. The corresponding protein and coding sequences (CDS) were retrieved from NCBI http://www.ncbi.nlm.nih.gov using a batch entrez function. Proteins classified in more than one class were removed from further analysis. Genes whose function was predicted only [R] or unknown [S], representing about 19% of the all dataset were removed from further analyses. In order to avoid statistical bias, the functional classes represented by less than a hundred sequences, namely [M], [N] and [Y], accounting overall for less than one percent of the all dataset, were also removed. For sake of simplicity square brackets denoting the functional classes were not used in the other sections of the present paper.
The whole set of human CDS, as well as that of the following species (in alphabetical order): Anolis carolinensis, Bos taurus, Dasypus novemcinctus, Equus caballus, Gorilla gorilla, Loxodonta africana, Monodelphis domestica, Mus musculus, Ornithorhynchus anatinus, Oryctolagus cuniculus, Pteropus vampyrus, Pongo pygmaeus, Spermophilus tridecemlineatus, Tursiops truncatus and Xenopus tropicalis were retrieved from the Ensemble database http://www.ensembl.org. CDS were classified in the KOG functional classes through the orthology with the human proteins. In other words, for each mammalian genome each gene acquired the same KOG classification of the corresponding human gene after the identification of the orthologous pair. In order to identify orthologs, a Perl script, essentially performing reciprocal Blastp [27] (e-value < 1e-05) and selecting the best reciprocal hit (BRH), was compiled.
Flanking regions (2000 bp flanking at 5'and 3' the transcript) and intronic sequences of KOG human genes were retrieved respectively form Ensemble http://www.ensembl.org using Biomart tools, and from UCSC http://genome.ucsc.edu/.
CodonW (1.4.4) was used to calculate the GC content (i.e. the molar ratio of guanine plus cytosine) of coding and non-coding sequences, as well as the GC3 content (i.e. the molar ratio of guanine plus cytosine the third codon positions) of CDS. The average of GC3 level was calculated for each sequence of each genome so far analyzed. In order to determine the statistical significance of the differences in GC3 content between the three main categories of genes, a two-tale Mann-Whitney test was performed.
The de Finetti's diagram was used to assess the compositional/spatial distribution of the three categories in different genomes (see Additional file 1 for a detailed description of the analysis). Shortly, for each organism the whole GC3 range was split in three intervals of equal size, denoted as Low, Medium and High, respectively. The number of functional classes belonging to the three categories were counted in each interval and normalized to 1 for plotting.
For each species the average GC3 level of each functional class was compared with that of the genome (i.e. the average of the GC3 level calculated using all the available coding sequences of the species), and statistical significance was assessed by the t-Student's test, with Bonferroni's correction (α = 0.05) for multiple-comparisons. The data were showed as Butterfly plot.
Human CDS from each KOG class were assigned to the different compositional band types (L1+, L1-, H3-, H3+ bands) previously identified in the human chromosomes [28,29].
Results
In the KOG database [21,22] human genes were classified in 25 functional classes, denoted by capital letters in square brackets, and grouped in three main categories: i) information storage and processing, represented by five functional classes; ii) cellular processes and signaling, represented by ten functional classes; and iii) metabolism, represented by eight functional classes. The three categories, from now on denoted as Blue, Black, and Red for sake of simplicity, accounted, respectively, for 22%, 42% and 16% of the all KOG dataset. The complete list of the functional classes, the corresponding number of genes and the average GC3 level of each functional class were reported in Table 1. The average GC3 content of the human genome (58.5%) was very close to that of the KOG dataset (58.4%). Within the human genome, the average GC3 of the three categories was significantly different (Figure 1). Indeed, the GC3 of Red category was significantly higher than that of both Black and Blue ones (p < 2.2 × 10-16 and p < 4.2 × 10-8, respectively). In turn, the GC3 of the Black category was significantly higher than that of Blue one (p < 1.6 × 10-6). In short, in the human genome the GC3 content of the three functional categories showed the following trend: Blue < Black < Red (Figure 1).
Table 1.
Functional classification of human genes
| KOG classes | Categories | # | GC3 | s.d. |
|---|---|---|---|---|
| INFORMATION STORAGE AND PROCESSING | ||||
| [A] | RNA processing and modification | 600 | 0.517 | 0.151 |
| [B] | Chromatin structure and dynamics | 224 | 0.610 | 0.172 |
| [J] | Translation, ribosomal structure and biogenesis | 1273 | 0.545 | 0.117 |
| [K] | Transcription | 1137 | 0.619 | 0.170 |
| [L] | Replication, recombination and repair | 300 | 0.546 | 0.154 |
| CELLULAR PROCESSES AND SIGNALING | ||||
| [D] | Cell cycle control, cell division, chromosome partitioning | 267 | 0.552 | 0.160 |
| [M] | Cell wall/membrane/envelope biogenesis | 63 | 0.576 | 0.164 |
| [N] | Cell motility | 26 | 0.586 | 0.163 |
| [O] | Posttranslational modification, protein turnover, chaperones | 1471 | 0.557 | 0.151 |
| [T] | Signal transduction mechanisms | 2214 | 0.616 | 0.158 |
| [U] | Intracellular trafficking, secretion, and vesicular transport | 685 | 0.571 | 0.161 |
| [V] | Defense mechanisms | 1023 | 0.527 | 0.172 |
| [W] | Extracellular structures | 284 | 0.588 | 0.150 |
| [Y] | Nuclear structure | 17 | 0.534 | 0.150 |
| [Z] | Cytoskeleton | 801 | 0.638 | 0.158 |
| METABOLISM | ||||
| [C] | Energy production and conversion | 403 | 0.576 | 0.153 |
| [E] | Amino acid transport and metabolism | 499 | 0.618 | 0.154 |
| [F] | Nucleotide transport and metabolism | 187 | 0.588 | 0.151 |
| [G] | Carbohydrate transport and metabolism | 469 | 0.606 | 0.150 |
| [H] | Coenzyme transport and metabolism | 102 | 0.563 | 0.135 |
| [I] | Lipid transport and metabolism | 410 | 0.595 | 0.154 |
| [P] | Inorganic ion transport and metabolism | 402 | 0.646 | 0.161 |
| [Q] | Secondary metabolites biosynthesis, transport and catabolism | 191 | 0.591 | 0.155 |
| POORLY CHARACTERIZED | ||||
| [R] | General function prediction only | 1889 | 0.593 | 0.162 |
| [S] | Function unknown | 1171 | 0.568 | 0.162 |
| Total number of genes | 16118 | 0.584 | 0.160 | |
(#) Number of genes
Figure 1.

The histogram shows the average GC3 content in the three functional categories of the human genome: (i) information storage and processing (Blue bar); (ii) cellular processes and signaling (Black bar); (iii) metabolism (Red bar). For each histogram bar the Standard error is reported.
Using the best reciprocal hit (BRH) approach the genes of mammalian genomes representing the following orders: primates (G. gorilla and P. pygmaeus), rodentia (M. musculus, and S. tridecemlineatus), lagomorpha (O. cuniculus) artiodactyla (B. taurus) perissodactyla (E. caballus), chiroptera (P. vampyrus), cetacea (T. truncatus), proboscidea (L. africana), cingulata (D. novemcinctus), didelphimorphia (M. domestica) and monotremata (O. anatinus) were classified in the KOG functional classes through the orthology with the human ones. The same approach was used to classify the genes of the species representing the order of anura (X. tropicalis) and squamata (A. carolinensis) genes. For each genome the whole number of available genomic CDS, the subset of genes classified according to the KOG database, the amount of genes belonging to the Blue, Black and Red categories, as well as the corresponding GC3 levels and the standard deviation, were reported in Table 2. Interestingly, the same trend of the GC3 content observed in the human genome, i.e. Blue < Black < Red, was found in almost all mammalian genomes. In the case of both platypus (M. domestica) and opossum (O. anatinus) genomes (Figure 2) the GC3 content of the Red category was significantly the highest, but no significant differences were observed comparing the GC3 content of the Black and the Blue categories (see Additional file 2).
Table 2.
Average GC3 levels, standard deviation and gene number of KOG's functional categories
| KOG** | BLUE* | BLACK* | RED* | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Organism | GC3 | s.d. | # | GC3 | s.d. | # | GC3 | s.d. | # | GC3 | s.d. | # | |
| Mammals | H. sapiens | 0.584 | 0.159 | 12942 | 0.568 | 0.154 | 3564 | 0.584 | 0.163 | 6745 | 0.604 | 0.155 | 2663 |
| G. Gorilla | 0.609 | 0.162 | 6268 | 0.593 | 0.166 | 1491 | 0.609 | 0.166 | 3357 | 0.626 | 0.148 | 1420 | |
| P. pygmaeus | 0.594 | 0.164 | 7455 | 0.583 | 0.167 | 1766 | 0.593 | 0.166 | 4012 | 0.611 | 0.154 | 1677 | |
| M. musculus | 0.606 | 0.114 | 7505 | 0.596 | 0.127 | 1780 | 0.605 | 0.122 | 4032 | 0.617 | 0.101 | 1693 | |
| O. cuniculus | 0.630 | 0.175 | 5413 | 0.609 | 0.179 | 1296 | 0.629 | 0.177 | 2867 | 0.653 | 0.164 | 1250 | |
| S. tridecemilineatus | 0.565 | 0.154 | 5455 | 0.542 | 0.157 | 1325 | 0.567 | 0.155 | 2900 | 0.584 | 0.144 | 1230 | |
| B. taurus | 0.630 | 0.167 | 7139 | 0.613 | 0.171 | 1706 | 0.632 | 0.169 | 3794 | 0.642 | 0.155 | 1639 | |
| E. caballus | 0.609 | 0.164 | 7102 | 0.594 | 0.170 | 1646 | 0.608 | 0.165 | 3840 | 0.626 | 0.153 | 1613 | |
| P. vampyrus | 0.605 | 0.164 | 6780 | 0.590 | 0.170 | 1638 | 0.607 | 0.165 | 3607 | 0.619 | 0.155 | 1535 | |
| T. truncatus | 0.618 | 0.167 | 6812 | 0.602 | 0.172 | 1635 | 0.617 | 0.169 | 3634 | 0.635 | 0.155 | 1543 | |
| L. africana | 0.583 | 0.152 | 5704 | 0.575 | 0.157 | 1413 | 0.582 | 0.153 | 3007 | 0.595 | 0.141 | 1284 | |
| D. novemcinctus | 0.585 | 0.180 | 5358 | 0.563 | 0.181 | 1310 | 0.585 | 0.182 | 2832 | 0.607 | 0.173 | 1216 | |
| O. anatinus | 0.648 | 0.166 | 5287 | 0.646 | 0.169 | 1319 | 0.646 | 0.166 | 2734 | 0.657 | 0.161 | 1234 | |
| M. domestica | 0.533 | 0.145 | 3598 | 0.529 | 0.153 | 1641 | 0.531 | 0.144 | 3598 | 0.542 | 0.137 | 1578 | |
| Reptiles | A. cardiensis | 0.539 | 0.159 | 5959 | 0.535 | 0.158 | 3063 | 0.546 | 0.1655 | 1498 | 0.539 | 0.153 | 1398 |
| Amphibians | X. tropicalis | 0.500 | 0.112 | 3584 | 0.499 | 0.112 | 1753 | 0.499 | 0.1174 | 961 | 0.501 | 0.108 | 870 |
(*) Blue, Black and Red refers to the gene classification of KOG database (see Methods)
(**) Genes orthologous to KOG human genes
(#) Number of genes
Figure 2.

The histogram shows the average GC3 content in the three functional categories in all analyzed genomes. Color code of histogram bars is as in Figure 1. For each histogram bar standard error is reported. Genome legend: M. domestica (1), S. tridecemlineatus (2), L. africana (3), D. novemcinctus (4), H. sapiens (5), P. pygmaeus (6), P. vampyrus (7), E. caballus (8), M. musculus (9), G. gorilla (10), T. truncatus (11), B. taurus (12), O. cuniculus (13), O. anatinus (14), A. carolinensis (15) and X. tropicalis (16).
In the amphibian and reptile genomes the GC3 content of the Red category was never significantly different from that of the Black and Blue categories, thus the mammalian pattern (Blue < Black < Red) was not observed (Figure 2 and Additional file 2).
In order to compare across the different genomes the impact of the Blue < Black < Red relation, a descriptive analysis of the distribution of the functional categories over a GC3 range was performed. The results were reported in the de Finetti's diagram (Figure 3), showing the distance of a given point from a given side accounting for the frequency of a given category in one of the three GC3 ranges, namely Low, Medium and High. Obviously, short distances accounted for low frequencies. In the diagram: i) the Red category was rarely present in the Low GC3 range, being mainly confined to a restricted part of the diagram space, i.e. the upper right side; ii) no overlap was observed between the spatial distribution of the Red with that of the Blue category; and iii) partial overlap was observed between the Blue and Black categories which did not show specific distribution, although rarely present in the High GC3 range. The significance of assumption was tested performing 1000 class permutations and observing the diagram distribution of the red classes (see Additional file 1). The probability to reproduce by chance the confinement of the Red category in the upper right side of the diagram was estimate to be p < 1.76 × 10-2. In contrast to mammals, the spatial distribution of the three categories was not significantly different in frog and lizard genomes (data not shown).
Figure 3.

de Finetti's diagram shows the spatial distribution of the three functional categories: (i) information storage and processing (Blue dots); (ii) cellular processes and signaling (Black dots); (iii) metabolism (Red dots). Numbers close to dots refer to the occurrence of overlapping genomes.
A comparative compositional analysis of the functional categories between human and frog (H/F), and between human and lizard (H/L) was performed. More precisely the GC3 increment (ΔGC3) was investigated. In both comparisons, positive values of ΔGC3 were observed in the human categories (Figure 4). Interestingly, in both H/F and H/L comparisons the highest ΔGC3 increment was observed in the Red category. Infact, ΔGC3 of the Red category was significantly higher than that of the Black (p < 4.72 × 10-4 and p < 1.14 × 10-2, respectively for the H/F and H/L), and significantly higher than that of the Blue category (p < 4.56 × 10-5 and p < 2.27 × 10-16, respectively for the H/F and H/L).
Figure 4.

The histogram shows the average GC3 increment in the three functional categories comparing human vs frog (H/F) and human vs lizard (H/L). Color code of histogram bars is as in Figure 1. In the H/F comparison the standard error for the Blue, Black and Red bars were, respectively, 5.1 × 10-3, 3.7 × 10-3 and 5.5 × 10-3. In the H/L comparison the standard error for the Blue, Black and Red bars were, respectively, 3.8 × 10-3, 2.8 × 10-3 and 3.9 × 10-3.
In order to shed light on the genome organization of the species so far analyzed, within each genome the average GC3 of each KOG functional class (see Table 1 for detailed definition) was investigated. More precisely, the difference between the GC3 levels of each functional class against that of the corresponding genome was calculated. Clustering negative and positive values a so-called butterfly plot for each genome was obtained. An overview of the all butterfly plots and detailed representations of each genome were reported in (Additional files 3, 4, 5, 6, 7). At first glance, the butterfly plots of the mammalian genomes showed an unbalance distribution of the bars (Additional files 3, 4, 5, 6, 7). Indeed, the bars of the Blue and Black categories where mainly in the negative side of the histogram, whereas those of the Red category where mainly in the positive side. In human, for instance, only two over five blue classes (namely B and K) and three the over seven black classes (namely T and Z) were in the positive side of the butterfly plot (Figure 5, panel C). On the contrary, six over eight Red classes (namely F, C, I, G, Q, E and P) were in the positive side of the butterfly plot (Figure 5, panel C). This picture was recurrently found among mammalian genomes (Additional files 3, 4, 5, 6, 7). The B functional class, for instance, was in the positive side in 88% of the mammalian genomes, whereas the classes G, K, E, Z and P were in the positive side of the butterfly plot in 100% of the cases (Figure 5, panel C). A t-Student's test with Bonferroni's correction (α = 0.05) was performed. The following functional classes: K (Blue), T and Z (Black) and G, E and P (Red), turned out to have an average GC3 level significantly higher than that of the whole genome (labeled by an asterisk in Figure 5, panel C). The butterfly plots of X. tropicalis and A. carolinensis were also reported (Figure 5; panel A and B, respectively). Regarding the reptile genome only two functional classes, namely the K and the Z classes showed an average GC3 level significantly higher than the genomic one (Figure 5, panel B), while none of the frog functional classes turned out to be significantly different (Figure 5, panel A).
Figure 5.

The butterfly plot of frog (panel A), lizard (panel B) and human functional classes (panel C). Color code of histogram bars is as in Figure 1. Asterisks show bars with an average GC3 content significantly higher than the genomic one (Bonferroni's test, α = 0.05).
The distribution of the human genes of each KOG class in the four chromosomal band types, namely L1+, L1-, H3- and H3+, characterized by increasing GC content [29], was analyzed (Figure 6). As general rule, the gene frequency was increasing as the GC content of the chromosomal band type was increasing. However, regarding the genes belonging to the Blue category, the maximum of the gene frequency was observed in the H3- bands, whereas that of Black and Red categories was in the H3+ (Figure 6). The χ2 - test showed an association between the three functional categories (Blue, Black and Red) and the two chromosomal bands types (H3- and H3+) far from the randomness, p < < 10-10. Moreover, using the one side Z-test as test for proportion it was found that: i) regarding the Blue category, the gene frequency of the functional classes B and J was significantly higher in H3- (p < 2.8 × 10-2 and p < 2.4 × 10-2, respectively), that of class A showed the same trend (but at the limit of significance, p < 7.0 × 10-2), whereas the functional classes K and L, although having the same trend, did not show significant p-values; ii) regarding the Black category, three out of seven functional classes, namely T, W and O, were significantly higher in the H3+ bands (p < 2.8 × 10-10, p < 1.0 × 10-6 and p < 5.6 × 10-2, respectively), whereas the remaining classes showed no trend; finally iii) regarding the Red category, five out of eight functional classes showed a significantly different distribution between the chromosomal bands H3- and H3+, but a non uniform trend was observed. More precisely, the functional classes E, F and P were significantly higher in H3+ (p < 1.8 × 10-3, 1.9 × 10-2 and 5.5 × 10-3, respectively), while G and Q showed opposite trend (p < 1.7 × 10-2 and 3.5 × 10-2, respectively). Dividing the functional classes in two groups, i.e. positive and negative, according to the position of the corresponding functional class in the butterfly plot of Figure 5 (panel C), small differences were found between the two groups in the L1+ and L1- bands (about 2%), whereas in the H3- and H3+ bands the differences increased up to 5% and 7%, respectively. In short, the probability to find genes of the positive group in the H3+ bands was significantly higher than in the L1+ bands (Z-test one tail, p < 10-2).
Figure 6.

The histogram shows the gene frequency in the four types of human chromosomal bands for each KOG functional class of the Blue, Black and Red KOG categories.
Discussion
Since the pioneering Ikemura's papers [30,31], the GC3 content, accounting for the base composition at the third (wobble) position in a codon, has been generally associated mainly with the codon usage and with the tRNA content. Furthermore, studies primarily performed on the human genome, showed that GC3 should be considered a keystone parameter to understand genome evolution. Indeed, GC3 turned out to be significantly correlated with the amino acid frequencies, i.e. GC1 + 2, as well as with the GC content of non-coding regions, i.e. introns and flanking regions [32-36]. Recent attempts to disregard the pivotal role of the GC3 parameter in understanding the genome organization [37], failed to take into consideration that "the use of indirect methods can lead to apparently conflicting conclusions" [38]. Recently, the role of the GC3 parameter as genome marker was further confirmed by the unexpected finding of correlations with genome size and body mass of mammals [39]. The subset of KOG human genes analyzed in the present paper followed, as expected, the well assessed rules first described in the 90's [32-36]. These rules held not only for the whole set of genes, but also when the genes were grouped in the KOG functional classes (see Additional file 8 for statistical reports).
In order to shed light on the debate around the evolutionary forces shaping the base composition among and within genomes [4,6-10,16-18], genes were classified in the three functional categories [namely: (i) information storage and processing (Blue); (ii) cellular processes and signaling (Black); (iii) metabolism (Red)] and their base compositional properties were analyzed. The results showed that within mammalian genomes the three functional categories were characterized by a different GC3 content, following the pattern Blue < Black < Red (Figure 2 and 3). It is worth to stress that, regarding platypus and opossum, no significant differences were observed comparing the Black vs. the Blue category (see Additional file 1). No pattern was found in the reptile and amphibian genomes (Figure 2).
Do current hypotheses explain the above finding?
It is worth to bring to mind that the keystone of the biased gene conversion hypothesis (BGC) was the strong correlation between hot spot recombination sites and GC content, establishing a cause/effect link of the first over the second parameter [7]. Consequently, the genomic impact of the BGC would be an increment of the GC content detectable at non-synonymous sites, synonymous sites, flanking and intronic sequences [9]. Since the BGC was reported to mimic perfectly natural selection [9], the compositional correlations holding in the human genome [32-36], including those reported in Additional file 8 could not be considered evidences for natural selection hypothesis.
In the light of the BGC hypothesis, the Blue < Black < Red pattern (observed in the majority of mammalian genomes) could have been explained as the result of the star-like phylogeny of mammals [40]. However, comparative genome analyses showed that hot spot recombination sites are "highly mobile" and therefore not phylogenetically related [41]. A result further supported by the studies conducted on the fast-evolving DNA-binding domain of PRDM9, identified as a major hotspot determinant of recombination. Indeed, the sequences and the number of PRDM9 domains were reported to vary a lot among species (reviewed in [42]), The lack of the Blue < Black < Red pattern in both frog and lizard and its appearance in mammalian genomes at present stands unclear. Although BGC received support from the analysis of the short sequences HARs and HACNSs in the human genome [43], considering that BGC was reported to be a widespread process affecting all genomes [9], the hypothesis was unable to explain the base compositional variability among bacterial genomes [44].
An interesting alternative hypothesis to the BGC was that proposed by Lemaitre and colleagues based on the analysis of the DNA breakpoint regions (BPR) [10]. Very recently, indeed, a 3D analysis of BPR showed that "two loci distant in the human genome but adjacent in the mouse genome are significantly more often observed in close proximity in the human nucleus than expected" [45]. The conservation of the Blue < Black < Red pattern among mammals, that started to diverge about 100 Mya [40], could probably be explained by the fact that 3D chromatin structure could be conserved over long evolutionary distances [45]. The time of divergence between amniotes and amphibian and between mammals and lizard was estimated to be several orders of magnitude greater than that of mammalian radiation (340-370Mya [46] and ~310Mya [40], respectively). Therefore explaining why the pattern was not conserved in reptiles and amphibians. However, according to the BPR hypothesis, evolutionary rearrangement breakages happen with a uniform propensity along the genome [10], leaving unexplained how the Blue < Black < Red pattern, absent in frog and lizard, could have been evolved in mammals. Moreover, as far as we know, no evidence has been produced to explain the base compositional variability among bacterial genomes in the light of the BPR hypothesis.
The critical query (Blue < Black < Red pattern) could be explained, on the contrary, by both thermal stability and metabolic rate hypotheses [16-18]. Indeed, in situ hybridization experiments performed on both human and amphibian nuclei (i.e. Rana esculenta), showed a comparable chromatin organization [47,48]. In both genomes, GC-poorest regions were found in closed chromatin structures localized at the nuclear periphery, while GC-richest ones were found in open chromatin structures localized more internally the nuclei [47,48]. According to the above reports, the different living temperature experienced by amphibians and mammals, could induce an increment of the GC content in mammals, in order to stabilize the open chromatin structures [16]. On the other hand, an increment of the metabolic rate, well known to be higher in mammals, should induce an increment of the GC content to increase DNA bendability, on one hand, and decrease nucleosome formation potential, on the other, to face an increment of transcriptional activity [17,18]. To this regard it should be recalled that along human chromosome the GC content and the gene expression profiles showed a positive correlation [19].
Temperature and metabolic rate are well known to be strongly correlated [49]. Therefore, disentangle the two variables would be not an easy task in the light of present data, also considering that terrestrial animals are living in an environment where oxygen is not a limiting factor. The problem was recently addressed analyzing the genomes of organisms living in aquatic habitats were the available oxygen in the environment is limited by the Henry's law. The analyses of teleostean fish genomes showed that: i) the genomic GC content of polar fish was higher than that of tropical fish; ii) that a positive and significant correlation holds between GC content and metabolic rate; and iii) a negative correlation was found between environmental temperature and GC content [50,51]. The problem was tackled in the present paper analyzing the orthologous pairs of human/frog (H/F) and human/lizard (H/L) genes. In both cases, the highest ΔGC3 turned out to take place in the Red category, that is the functional category grouping genes involved in metabolic processes (Figure 4). Although not resolving the dichotomy between temperature and metabolic rate (both increasing, indeed, from frog to human [52]) the result was congruent with the conclusion drawn out from the comparison of teleostean fish genomes [50,51].
The detailed investigation on the distribution of the KOG functional classes revealed that the Blue < Black < Red pattern was even more multifaceted. Indeed, in the positive side of the human butterfly plot, apart the majority of Red bars, the B and K blue bars, as well as the T and Z black bars were also observed (Figure 5, panel C). The above picture was not confined to the human genome, but commonly found in all mammals. Indeed, the B and T classes were in the positive side of the butterfly plot in the 93% of the cases, whereas the K and Z classes reached the 100% of the cases (Figure 5, panel C). The occurrence of the bars belonging to the Red category ranged from 86% of the Q class to 100% of the G, E and P classes. Needless to say, the pattern was not found in the frog and lizard genomes. All the considerations formerly drawn out in the light of the different evolutionary hypotheses regarding the Blue < Black < Red pattern, applied even more radically to the pattern of functional classes clustering in the positive side of all mammalian butterfly plots (Additional files 3, 4, 5, 6, 7 and Figure 5, panel C), showing a different chromosomal distribution.
The above result deserves a more detailed argumentation. As reported in Table 1 the genes belonging to the four classes were involved in the following task: Chromatin structure and dynamics (B), Transcription (K), Signal transduction mechanisms (T) and Cytoskeleton (Z). The fact that the GC3 content of genes belonging to the B and K classes was not surprising, since an increment of the metabolic rate affects transcription process and chromatin structure, as discussed above. More inscrutable was the result regarding the T and Z classes. Recently, an interesting paper was published on the effect of estrogen exposure in mice brain, inducing an increment of the expression level of a discrete number of genes [53]. From their results it is possible to derive that beside a 39% of genes involved in metabolic processes, 18% belonged to the Z class and 25% to the T one, whereas only 6% of the genes belonged to the category grouping genes involved in information storage and processing. Szego's and colleagues report [53] was an interesting preliminary approach, pointing towards further investigations on the link between genome organization and the physiological reaction to stressing stimuli increasing the metabolic rate. Interestingly, gene clusters for metabolic pathways have been reported also in plants (reviewed in [54]).
Conclusions
All the different evolutionary hypotheses proposed till now surely contribute, with different weight, to the compositional variability observed among and within organisms [55]. Few, however, seem to fit with the very wide range from prokaryotes to eukaryotes. Indeed, recent analysis showed that mutational bias cannot explain genome composition in bacteria, reviewed in [56]. The BGC hypothesis, supported by the data produced on sequences HARs and HACNSs in mammals [43], also failed to explain the base compositional variability among bacterial genomes [44], and hardly explains the present results. The BPR hypothesis was very promising, especially in the light of the studies carried out on conservation of the 3D chromatin structure over long evolutionary distances [45]. However, still remain to clarify the mechanism leading to the observed patterns in mammals.
Regarding the thermodynamic hypothesis, the extensive studies carried out on bacterial genomes has been matter of debate [57-60]. Within bacterial families a significant positive correlation between growth temperature and GC content was observed in 9 out of 20 families [58]. However, the positive correlation failed to be observed in teleostean fish genomes, where a negative one was found indeed [50,51]. Unfortunately, present data neither shed light in favor nor against the effect of temperature on the compositional transition from amphibian/reptile to mammals [24-26]. On the contrary, the metabolic rate hypothesis [17,18] not only explained both the transition [61] and the shifting mode of evolution of vertebrate genomes [50,51], but also the within genome patterns showed in the present paper. Moreover, a correlation between metabolic rate and GC % has been found also in bacteria [12,15], as well as among teleostean fish [50,51]. It is worth to bring to mind that, although the metabolic rate hypothesis is in the frame of the adaptive hypotheses, most probably there is no need to invoke the effect of the positive selection. Indeed, the shift of the of threshold for the "best-fit GC content" could account for the genome compositional shift observed comparing teleostean fish living in different habitats [50]. Natural selection has been also proposed to explain the great compositional heterogeneity of the human genome [62].
Abbreviations
BGC: biased gene conversion; bp: base pair; BPR: DNA breakpoint distribution; BRH: best reciprocal hit; GC: molar ratio of guanine plus cytosine; GC1 + 2: molar ratio of guanine plus cytosine at first and second positions; GC3: molar ratio of guanine plus cytosine at third codon positions; ΔGC3: GC3 increment; GCi: molar ratio of guanine plus cytosine in intronic sequences; HACNS: human-accelerated conserved non-coding sequences; HAR: human-accelerated region; KOG: clusters of orthologous groups for eukaryotic complete genomes.
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
LB carried out KOG analysis and drafted the manuscript. AC retrieved and analyzed non-coding sequences. CA performed and described statistical analyses. CF assigned CDS to the chromosomal band types. SS contributed to the design of the work and to the general discussion. GD envisages the design and coordination of the study and wrote the manuscript. All authors read and approved the final manuscript.
Supplementary Material
The de Finetti's diagram.
p-values of the Mann-Whitney test among categories.
Butterfly plot of mammalian order: primates.
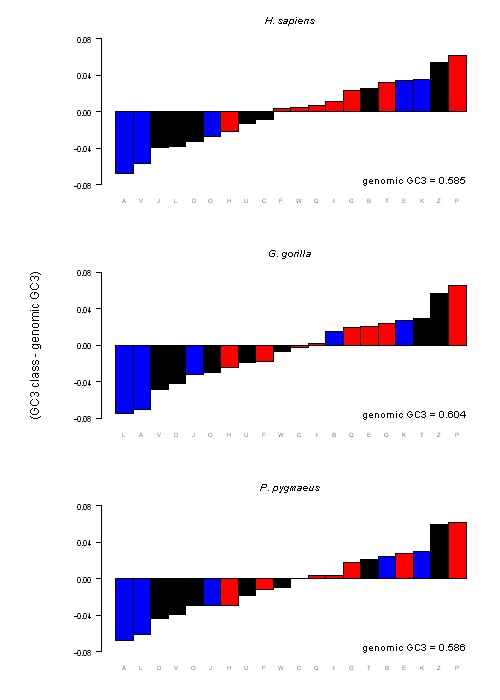{kind=link}
Butterfly plot of mammalian order: rodentia and lagomorpha.
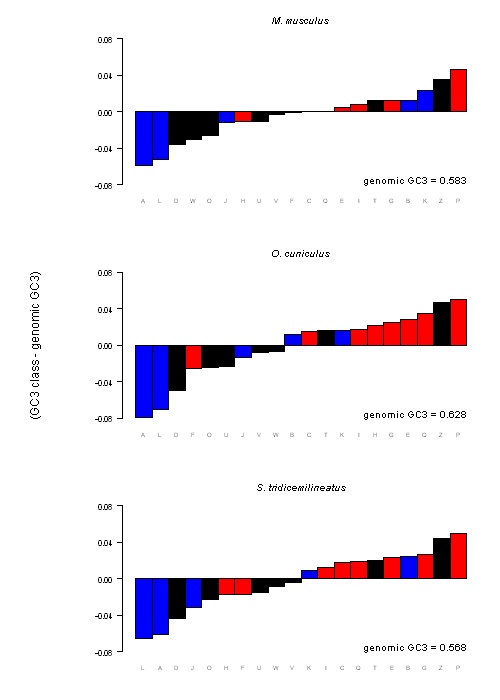{kind=link}
Butterfly plot of mammalian order: artiodactyla, perissodactyla, chiroptera, cetacea.
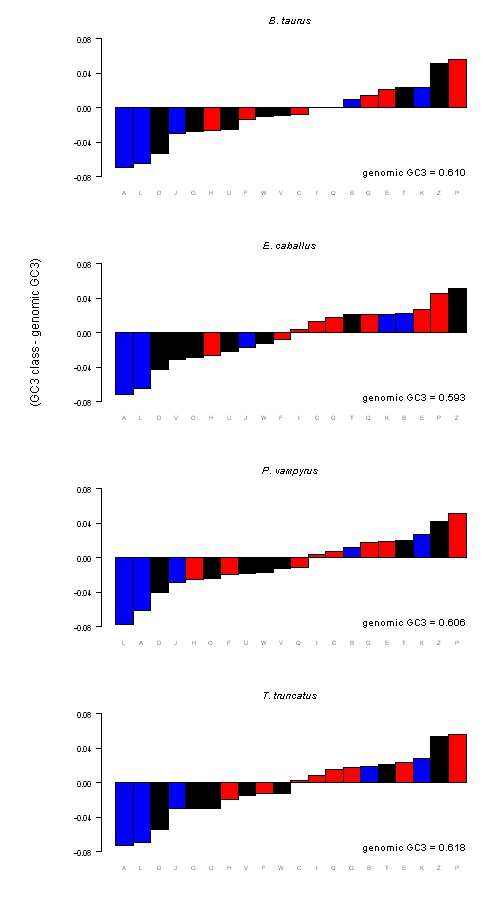{kind=link}
Butterfly plot of mammalian order: didelphimorpha and monotremata.
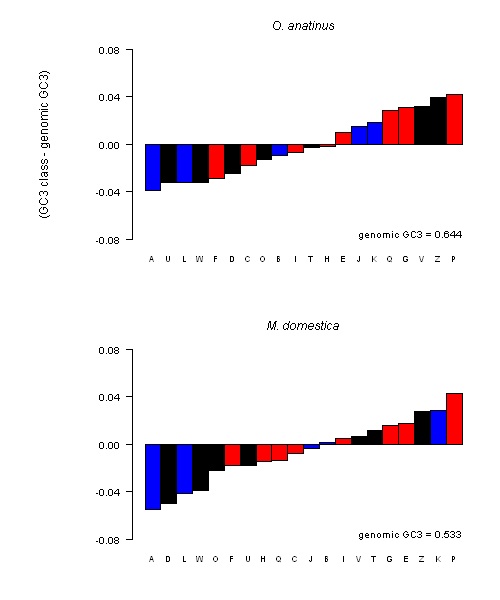{kind=link}
Butterfly plot of mammalian order: proboscidea and cingulata.
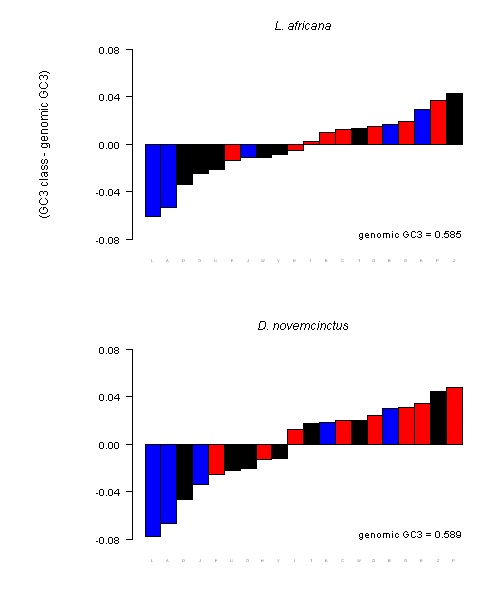{kind=link}
Statistical Summary of KOG genes.
Contributor Information
Luisa Berná, Email: luisa.berna@szn.it.
Ankita Chaurasia, Email: ankita.chaurasia@szn.it.
Claudia Angelini, Email: c.angelini@iac.cnr.it.
Concetta Federico, Email: federico@unict.it.
Salvatore Saccone, Email: saccosal@unict.it.
Giuseppe D'Onofrio, Email: donofrio@szn.it.
Acknowledgements
Thanks are due to Claudio Agnisola, for critically reading the manuscript, Guillermo Lamolle, for the butterfly plots, and Fernando Alvarez-Valin, for the bioinformatics facilities of the Facultad de Ciencias, Universidad de la República (Uruguay).
References
- Salzburger W, Steinke D, Braasch I, Meyer A. Genome desertification in eutherians: can gene deserts explain the uneven distribution of genes in placental mammalian genomes? J Mol Evol. 2009;69(3):207–216. doi: 10.1007/s00239-009-9251-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foerstner KU, von Mering C, Hooper SD, Bork P. Environments shape the nucleotide composition of genomes. EMBO Rep. 2005;6(12):1208–1213. doi: 10.1038/sj.embor.7400538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freese E. On the evolution of base composition of DNA. J Theor Biol. 1962;3:82–101. doi: 10.1016/S0022-5193(62)80005-8. [DOI] [Google Scholar]
- Sueoka N. On the genetic basis of variation and heterogeneity of DNA base composition. Proc Natl Acad Sci USA. 1962;48:582–592. doi: 10.1073/pnas.48.4.582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobry JR, Sueoka N. Asymmetric directional mutation pressures in bacteria. Genome Biol. 2002;3(10) doi: 10.1186/gb-2002-3-10-research0058. research0058-research0058.0014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sueoka N. Directional mutation pressure and neutral molecular evolution. Proc Natl Acad Sci USA. 1988;85(8):2653–2657. doi: 10.1073/pnas.85.8.2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eyre-Walker A. Recombination and mammalian genome evolution. Proc R Soc Lond B. 1993;252:237–243. doi: 10.1098/rspb.1993.0071. [DOI] [PubMed] [Google Scholar]
- Galtier N, Duret L. Adaptation or biased gene conversion? Extending the null hypothesis of molecular evolution. Trends Genet. 2007;23(6):273–277. doi: 10.1016/j.tig.2007.03.011. [DOI] [PubMed] [Google Scholar]
- Duret L, Galtier N. Biased gene conversion and the evolution of mammalian genomic landscapes. Annu Rev Genomics Hum Genet. 2009;10:285–311. doi: 10.1146/annurev-genom-082908-150001. [DOI] [PubMed] [Google Scholar]
- Lemaitre C, Zaghloul L, Sagot MF, Gautier C, Arneodo A, Tannier E, Audit B. Analysis of fine-scale mammalian evolutionary breakpoints provides new insight into their relation to genome organization. BMC Genomics. 2009;10:335. doi: 10.1186/1471-2164-10-335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocha EP, Danchin A. Base composition bias might result from competition for metabolic resources. Trends Genet. 2002;18(6):291–294. doi: 10.1016/S0168-9525(02)02690-2. [DOI] [PubMed] [Google Scholar]
- Naya H, Romero H, Zavala A, Alvarez B, Musto H. Aerobiosis increases the genomic guanine plus cytosine content (GC%) in prokaryotes. J Mol Evol. 2002;55(3):260–264. doi: 10.1007/s00239-002-2323-3. [DOI] [PubMed] [Google Scholar]
- Woolfit M, Bromham L. Increased rates of sequence evolution in endosymbiotic bacteria and fungi with small effective population sizes. Mol Biol Evol. 2003;20(9):1545–1555. doi: 10.1093/molbev/msg167. [DOI] [PubMed] [Google Scholar]
- Musto H, Naya H, Zavala A, Romero H, Alvarez-Valin F, Bernardi G. Genomic GC level, optimal growth temperature, and genome size in prokaryotes. Biochem Biophys Res Commun. 2006;347(1):1–3. doi: 10.1016/j.bbrc.2006.06.054. [DOI] [PubMed] [Google Scholar]
- Romero H, Pereira E, Naya H, Musto H. Oxygen and guanine-cytosine profiles in marine environments. J Mol Evol. 2009;69(2):203–206. doi: 10.1007/s00239-009-9230-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernardi G. Sructural and Evolutionary Genomics. Natural Selection in Genome Evolution. Amsterdam: Elsevier; 2004. [Google Scholar]
- Vinogradov AE. Bendable genes of warm-blooded vertebrates. Mol Biol Evol. 2001;18(12):2195–2200. doi: 10.1093/oxfordjournals.molbev.a003766. [DOI] [PubMed] [Google Scholar]
- Vinogradov AE. Noncoding DNA, isochores and gene expression: nucleosome formation potential. Nucleic Acids Res. 2005;33(2):559–563. doi: 10.1093/nar/gki184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Versteeg R, van Schaik BD, van Batenburg MF, Roos M, Monajemi R, Caron H, Bussemaker HJ, van Kampen AH. The human transcriptome map reveals extremes in gene density, intron length, GC content, and repeat pattern for domains of highly and weakly expressed genes. Genome Res. 2003;13(9):1998–2004. doi: 10.1101/gr.1649303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arhondakis S, Auletta F, Torelli G, D'Onofrio G. Base composition and expression level of human genes. Gene. 2004;325:165–169. doi: 10.1016/j.gene.2003.10.009. [DOI] [PubMed] [Google Scholar]
- Tatusov RL, Natale DA, Garkavtsev IV, Tatusova TA, Shankavaram UT, Rao BS, Kiryutin B, Galperin MY, Fedorova ND, Koonin EV. The COG database: new developments in phylogenetic classification of proteins from complete genomes. Nucleic Acids Res. 2001;29(1):22–28. doi: 10.1093/nar/29.1.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatusov RL, Fedorova ND, Jackson JD, Jacobs AR, Kiryutin B, Koonin EV, Krylov DM, Mazumder R, Mekhedov SL, Nikolskaya AN. et al. The COG database: an updated version includes eukaryotes. BMC Bioinformatics. 2003;4:41. doi: 10.1186/1471-2105-4-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Onofrio G, Ghosh TC, Saccone S. Different functional classes of genes are characterized by different compositional properties. FEBS Lett. 2007;581(30):5819–5824. doi: 10.1016/j.febslet.2007.11.052. [DOI] [PubMed] [Google Scholar]
- Bernardi G, Bernardi G. Compositional transitions in the nuclear genomes of cold-blooded vertebrates. J Mol Evol. 1990;31:282–293. doi: 10.1007/BF02101123. [DOI] [PubMed] [Google Scholar]
- D'Onofrio G, Jabbari K, Musto H, Alvarez-Valin F, Cruveiller S, Bernardi G. Evolutionary genomics of vertebrates and its implications. Ann N Y Acad Sci. 1999;870:81–94. doi: 10.1111/j.1749-6632.1999.tb08867.x. [DOI] [PubMed] [Google Scholar]
- D'Onofrio G, Ghosh TC. The compositional transition of vertebrate genomes: an analysis of the secondary structure of the proteins encoded by human genes. Gene. 2005;345(1):27–33. doi: 10.1016/j.gene.2004.11.037. [DOI] [PubMed] [Google Scholar]
- Delaney S, Butler G, Lam C, Thiel L. Three Improvements to the BLASTP Search of Genome Databases. SSDBM Proceedings of the 12th International Conference on Scientific and Statistical Database Management. 2000. pp. 14–24.
- Federico C, Andreozzi L, Saccone S, Bernardi G. Gene density in the Giemsa bands of human chromosomes. Chromosome Res. 2000;8(8):737–746. doi: 10.1023/A:1026797522102. [DOI] [PubMed] [Google Scholar]
- Costantini M, Clay O, Federico C, Saccone S, Auletta F, Bernardi G. Human chromosomal bands: nested structure, high-definition map and molecular basis. Chromosoma. 2007;116(1):29–40. doi: 10.1007/s00412-006-0078-0. [DOI] [PubMed] [Google Scholar]
- Ikemura T. Correlation between the abundance of Escherichia coli transfer RNAs and the occurrence of the respective codons in its protein genes. J Mol Biol. 1981;146:1–21. doi: 10.1016/0022-2836(81)90363-6. [DOI] [PubMed] [Google Scholar]
- Ikemura T. Codon usage and tRNA content in unicellular and multicellular organisms. Mol Biol Evol. 1985;2(1):13–34. doi: 10.1093/oxfordjournals.molbev.a040335. [DOI] [PubMed] [Google Scholar]
- D'Onofrio G, Mouchiroud D, Aïssani B, Gautier C, Bernardi G. Correlations between the compositional properties of human genes, codon usage, and amino acid composition of proteins. J Mol Evol. 1991;32(6):504–510. doi: 10.1007/BF02102652. [DOI] [PubMed] [Google Scholar]
- Aïssani B, D'Onofrio G, Mouchiroud D, Gardiner K, Gautier C, Bernardi G. The compositional properties of human genes. J Mol Evol. 1991;32(6):493–503. doi: 10.1007/BF02102651. [DOI] [PubMed] [Google Scholar]
- Mouchiroud D, D'Onofrio G, Aissani B, Macaya G, Gautier C, Bernardi G. The distribution of genes in the human genome. Gene. 1991;100:181–187. doi: 10.1016/0378-1119(91)90364-h. [DOI] [PubMed] [Google Scholar]
- Ikemura T, Aota S. Global variation in G+C content along vertebrate genome DNA. Possible correlation with chromosome band structures. J Mol Biol. 1988;203:1–13. doi: 10.1016/0022-2836(88)90086-1. [DOI] [PubMed] [Google Scholar]
- D'Onofrio G, Bernardi G. A universal compositional correlation among codon position. Gene. 1992;110:81–88. doi: 10.1016/0378-1119(92)90447-W. [DOI] [PubMed] [Google Scholar]
- Elhaik E, Landan G, Graur D. Can GC content at third-codon positions be used as a proxy for isochore composition? Mol Biol Evol. 2009;26(8):1829–1833. doi: 10.1093/molbev/msp100. [DOI] [PubMed] [Google Scholar]
- Clay OK, Bernardi G. GC3 of genes can be used as a proxy for isochore base composition: A reply to Elhaik et al. Mol Biol Evol. 2010;28(1):21–23. doi: 10.1093/molbev/msq222. [DOI] [PubMed] [Google Scholar]
- Romiguier J, Ranwez V, Douzery EJ, Galtier N. Contrasting GC-content dynamics across 33 mammalian genomes: relationship with life-history traits and chromosome sizes. Genome Res. 2010;20(8):1001–1009. doi: 10.1101/gr.104372.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S, Hedges SB. A molecular time scale for vertebrate evolution. Nature. 1998;392:917–920. doi: 10.1038/31927. [DOI] [PubMed] [Google Scholar]
- Huang SW, Friedman R, Yu N, Yu A, Li WH. How strong is the mutagenicity of recombination in mammals? Mol Biol Evol. 2005;22(3):426–431. doi: 10.1093/molbev/msi025. [DOI] [PubMed] [Google Scholar]
- Hochwagen A, Marais GA. Meiosis: a PRDM9 guide to the hotspots of recombination. Curr Biol. 2010;20(6):R271–274. doi: 10.1016/j.cub.2010.01.048. [DOI] [PubMed] [Google Scholar]
- Duret L, Galtier N. Comment on "Human-specific gain of function in a developmental enhancer". Science. 2009;323(5915):714. doi: 10.1126/science.1165848. author reply 714. [DOI] [PubMed] [Google Scholar]
- Hildebrand F, Meyer A, Eyre-Walker A. Evidence of selection upon genomic GC-content in bacteria. PLoS Genet. 2010;6(9):e1001107. doi: 10.1371/journal.pgen.1001107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veron AS, Lemaitre C, Gautier C, Lacroix V, Sagot MF. Close 3D proximity of evolutionary breakpoints argues for the notion of spatial synteny. BMC Genomics. 2011;12(1):303. doi: 10.1186/1471-2164-12-303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paton RL, Smithson TR, Clack JA. An amniote-like skeleton from the Early Carboniferous of Scotland. Nature. 1999;398:508–513. doi: 10.1038/19071. [DOI] [Google Scholar]
- Saccone S, Federico C, Solovei I, Croquette MF, Della Valle G, Bernardi G. Identification of the gene-richest bands in human prometaphase chromosomes. Chromosome Res. 1999;7(5):379–386. doi: 10.1023/A:1009220131225. [DOI] [PubMed] [Google Scholar]
- Federico C, Scavo C, Cantarella CD, Motta S, Saccone S, Bernardi G. Gene-rich and gene-poor chromosomal regions have different locations in the interphase nuclei of cold-blooded vertebrates. Chromosoma. 2006;115(2):123–128. doi: 10.1007/s00412-005-0039-z. [DOI] [PubMed] [Google Scholar]
- Kleiber M. Body size and metabolism. Hilgardia. 1932;6:315–353. [Google Scholar]
- Uliano E, Chaurasia A, Bernà L, Agnisola C, D'Onofrio G. Metabolic rate and genomic GC. What we can learn from teleost fish. Marine Genomics. 2010;3:29–34. doi: 10.1016/j.margen.2010.02.001. doi:10.1016/j.margen.2010.1002.1001. [DOI] [PubMed] [Google Scholar]
- Chaurasia A, Uliano E, Bernà L, Agnisola C, D'Onofrio G. In: Does Habitat Affect the Genomic GC Content? A Lesson from Teleostean Fish: A Mini Review. Fish Ecology, Sean P. Dempsey, editor. Huppage, NY (USA) Nova Science Publishers; 2011. pp. 61–80. [Google Scholar]
- Altman PL, Dittmer DS. In: Fed Am Soc Exp Biol. Altman PL, Dittmer DS, editor. III. Bethesda, Mariland (USA); 1974. Biology Data Book; pp. 1613–1645. [Google Scholar]
- Szego EM, Kekesi KA, Szabo Z, Janaky T, Juhasz GD. Estrogen regulates cytoskeletal flexibility, cellular metabolism and synaptic proteins: A proteomic study. Psychoneuroendocrinology. 2009;35(6):807–819. doi: 10.1016/j.psyneuen.2009.11.006. [DOI] [PubMed] [Google Scholar]
- Osbourn A. Gene clusters for secondary metabolic pathways: an emerging theme in plant biology. Plant Physiol. 2010;154(2):531–535. doi: 10.1104/pp.110.161315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pozzoli U, Menozzi G, Fumagalli M, Cereda M, Comi GP, Cagliani R, Bresolin N, Sironi M. Both selective and neutral processes drive GC content evolution in the human genome. BMC Evol Biol. 2008;8:99. doi: 10.1186/1471-2148-8-99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocha EP, Feil EJ. Mutational patterns cannot explain genome composition: are there any neutral sites in the genomes of bacteria? PLoS Genet. 2010;6(9):e1001104. doi: 10.1371/journal.pgen.1001104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galtier N, Lobry JR. Relationships between genomic G+C content, RNA secondary structures, and optimal growth temperature in prokaryotes. J Mol Evol. 1997;44(6):632–636. doi: 10.1007/PL00006186. [DOI] [PubMed] [Google Scholar]
- Musto H, Naya H, Zavala A, Romero H, Alvarez-Valin F, Bernardi G. Correlations between genomic GC levels and optimal growth temperatures in prokaryotes. FEBS Lett. 2004;573(1-3):73–77. doi: 10.1016/j.febslet.2004.07.056. [DOI] [PubMed] [Google Scholar]
- Wang HC, Susko E, Roger AJ. On the correlation between genomic G+C content and optimal growth temperature in prokaryotes: Data quality and confounding factors. Biochem Biophys Res Commun. 2006;342(3):681–684. doi: 10.1016/j.bbrc.2006.02.037. [DOI] [PubMed] [Google Scholar]
- Musto H, Naya H, Zavala A, Romero H, Alvarez-Valin F, Bernardi G. The correlation between genomic G+C and optimal growth temperature of prokaryotes is robust: a reply to Marashi and Ghalanbor. Biochem Biophys Res Commun. 2005;330(2):357–360. doi: 10.1016/j.bbrc.2005.02.133. [DOI] [PubMed] [Google Scholar]
- Vinogradov AE, Anatskaya OV. Genome size and metabolic intensity in tetrapods: a tale of two lines. Proc Biol Sci. 2006;273(1582):27–32. doi: 10.1098/rspb.2005.3266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernardi G. The neoselectionist theory of genome evolution. Proc Natl Acad Sci USA. 2007;104(20):8385–8390. doi: 10.1073/pnas.0701652104. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The de Finetti's diagram.
p-values of the Mann-Whitney test among categories.
Butterfly plot of mammalian order: primates.
Butterfly plot of mammalian order: rodentia and lagomorpha.
Butterfly plot of mammalian order: artiodactyla, perissodactyla, chiroptera, cetacea.
Butterfly plot of mammalian order: didelphimorpha and monotremata.
Butterfly plot of mammalian order: proboscidea and cingulata.
Statistical Summary of KOG genes.