

Abstract
Objective/Hypothesis
Recent studies indicate that vestibular schwannomas (VS) rely on PI3-kinase/AKT activation to promote cell proliferation and survival; therefore, targeting AKT may provide new therapeutic options. We have previously shown that AR42, a novel histone deacetylase inhibitor, potently suppresses VS growth in vitro at doses correlating with AKT inactivation. The objectives of the current study were translational: 1) to examine the end biologic effects of AR42 on tumor growth in vivo, 2) to validate AKT as its in vivo molecular target, 3) to determine whether AR42 penetrates the blood brain barrier (BBB), and 4) to study AR42’s pharmacotoxicity profile.
Study Design
In vivo mouse studies
Methods
AR42 was dosed orally in murine schwannoma allografts and human vestibular schwannoma xenografts. MRI was used to quantify changes in tumor volume while intracellular molecular targets were analyzed using immunohistochemistry. Blood-brain barrier (BBB) penetration was assayed, and both blood chemistry measurements and histology studies were used to evaluate toxicity.
Results
Growth of schwannoma implants was dramatically decreased by AR42 at doses correlating with AKT dephosphorylation, cell cycle arrest, and apoptosis. AR42 penetrated the BBB, and wild-type mice fed AR42 for 6-months behaved normally and gained weight appropriately. Blood chemistry studies and organ histology performed after 3- and 6-months of AR42 treatment demonstrated no clinically significant abnormalities.
Conclusions
AR42 suppresses schwannoma growth at doses correlating with AKT pathway inhibition. This orally bioavailable drug penetrates the BBB, is well tolerated, and represents a novel candidate for translation to human VS clinical trials.
Keywords: neurofibromatosis type 2, NF2, merlin, vestibular schwannoma, AKT, HDAC, histone deacetylase inhibitors, HDAC inhibitors (HDACi), AR42, cell cycle arrest, apoptosis
Background
Vestibular schwannomas (VS), intracranial tumors arising from Schwann cells of the 8th cranial nerve, are disease-defining neoplasms in patients with neurofibromatosis type 2 (NF2). This autosomal dominant syndrome occurs due to the bi-allelic loss of the NF2 gene on chromosome 22, which encodes a tumor suppressor protein named merlin (moesin, ezrin, and radixin-like protein).1,2 While patients with germ-line NF2 gene mutations develop bilateral VS, unilateral sporadic tumors are more common (95%). Most vestibular schwannomas are histologically benign; however, they can cause sensorineural hearing loss, balance abnormalities, tinnitus, vertigo, facial paralysis, hydrocephalus, and even death due to their critical intracranial location.3 Current treatment options are excision via craniotomy and/or stereotactic radiation therapy. Surgical risks include spinal fluid leaks, meningitis, intracranial hemorrhage, coma and death, whereas use of stereotactic radiation raises concerns for malignant transformation and delayed-onset, radiation-induced skull base malignancies.4,5 Malignant vestibular schwannomas (triton tumors) are highly aggressive and uniformly lethal.6 No medical therapies are currently FDA-approved for VS; therefore, the clinical impact of developing novel drug treatments is self-evident.
Normal, differentiated cells become quiescent upon sensing cell-cell contact.7-9 At confluence, the NF2 gene product merlin is thought to be a critical regulator of this contact-dependent inhibition to proliferation, but mechanistic details regarding merlin’s tumor suppressor function remain elusive.10,11 Recent studies suggest that merlin links cell adhesion to transmembrane receptor signaling12 by controlling the availability and function of growth-promoting receptors at the cell surface.13 Merlin’s interactions with adaptor proteins such as NHERF1/EBP50 (Na–H+ exchange regulatory factor 1; ERM-binding protein of 50 kDa)14,15 may explain why numerous intracellular growth promoting pathways such as phosphatidylinositol 3-kinase (PI3-kinase)/AKT, mitogen-activated protein kinase (MAPK), p21 activated kinase (PAK)/JNK, and others can be deregulated by merlin loss.16 Deregulated AKT signaling is tumorigenic in many human malignancies.17 A longstanding hypothesis in our laboratory has been that vestibular schwannomas also rely on aberrant PI3-kinase/AKT activation to promote cell proliferation and survival. Prior work has shown that the absence of merlin results in post-translational activation of the PI3-kinase/AKT pathway and that upstream inhibition of AKT phosphorylation using a PDK1 inhibitor suppresses schwannoma growth.18-20
Histone deacetylase inhibitors (HDACi), a novel class of antitumor agents, were initially thought to suppress tumor growth via chromatin remodeling and epigenetic effects. Phylogenetic studies, however, demonstrated that all four classes of HDACs preceded evolution of histones,21 and over 50 transcription factors, DNA repair enzymes, signal transduction mediators, chaperone proteins and structural proteins have now been identified as HDAC substrates.22 AR42 (formerly OSU-HDAC42; Arno Therapeutics, Parsippany, NJ) is a phenylbutyrate-derived HDACi that inhibits AKT downstream from PI3K and PDK1 through protein phosphatase 1-mediated AKT dephosphorylation.23 We recently confirmed that AR42 suppresses schwannoma cell proliferation in vitro at doses correlating with AKT dephosphorylation, cell cycle arrest, and apoptosis.24 The current in vivo work was designed to facilitate AR42’s transition to human VS clinical trials. We sought to 1) examine the end biologic effects of AR42 on tumor growth in vivo, 2) validate AKT as an in vivo molecular target; 3) determine whether AR42 penetrates the blood brain barrier (BBB); and 4) study AR42’s pharmacotoxicity profile.
Materials and Methods
Drug compounds
Pharmaceutical grade AR42 was provided by Arno Therapeutics, Inc. through a materials transfer agreement. The authors have no financial relationships with the company.
Tissue acquisition and establishment of schwannoma cell cultures
Longstanding Institutional Review Board (IRB) approved protocols for the acquisition of surgically removed VS specimens are in place. VS specimens obtained from the operating room are placed in Dulbecco’s modified Eagle’s medium (DMEM) (Invitrogen, Carlsbad, CA) immediately after surgical resection and cut into fragments prior to implantation into immunodeficient mice. Mouse Nf2−/− schwannoma cells are obtained from tumors that develop spontaneously in P0Cre; Nf2flox/flox mice having conditional Nf2 inactivation in Schwann cells.25 They are cultured on dishes coated with poly-D-lysine/laminin (PDLL) (Sigma–Aldrich, St. Louis, MO) and maintained in DMEM/10% FBS containing 10 ng/mL recombinant human NRG1-β1/HRG1-β1 EGF domain (heregulin, R&D Systems, Minneapolis, MN) and 0.2 μM forskolin (Sigma).
Establishment of schwannoma xenografts/allografts
Institutional Animal Care and Use Committee (IACUC) protocols for the establishment of mouse xenografts/allografts are in place. Severe combined immunodeficient (SCID-ICR; Harlan Laboratories, Indianapolis, IN) mice are anesthetized with inhalational isoflurane during implantation of cells or tissues. Nf2−/− schwannoma allografts are then established by aseptically preparing one flank of each SCID mouse, suspending Nf2−/− schwannoma cells in Matrigel (BD Biosciences, San Jose, CA), and injecting approximately 0.2 mL of cell suspension subcutaneously near the sciatic nerve (flank injection; Figure 1A). For implantation of VS specimens, an incision is made along one proximal thigh while the contralateral leg is left un-dissected as an internal control. The biceps femoris muscle and sciatic nerve are identified in order to implant vestibular schwannoma tissues en bloc near the nerve.
Figure 1. Overview of in vivo research plan for AR42 treatment.


(A) Flow diagram showing the protocol for AR42-treated schwannoma allografts/xenografts. Representative photos of SCID mice implanted with Nf2−/− P0Cre mouse schwannoma cells and resultant tumor formation.
(B) Flow diagram showing the protocol for pharmacodynamics and pharmacotoxicity studies.
Animal experiments
AR42-containing rodent chow was generated by Research Diets, Inc. (New Brunswick, NJ) to deliver an approximate target dose of 25 mg/kg/day/mouse. Wild-type female FVB/N mice (The Jackson Laboratory, Bar Harbor, Maine) were fed drug-impregnated rodent chow for pharmacotoxicology and drug penetration studies while female SCID-ICR mice were utilized for xenograft experiments. Control mice in all experiments received normal rodent diets (Harlan Teklad, Indianapolis, IN). Both drug-treated and control mice were checked weekly, and at each time point, the cages were cleaned and water ports were charged. The total amount of food in each cage was weighed, and the amount of food consumed during the previous week was calculated. Each mouse was weighed separately. The total chow consumed per cage was divided by the number of days between checks, and this value was then divided by the number of mice per cage to calculate the amount of mouse chow consumed per mouse per day. Multiplying by the concentration of drug per gram of food, the amount of drug consumed per mouse per day was derived.
1. AR42-treated P0Cre Nf2−/− schwannoma allografts (Figure 1A)
P0Cre Nf2−/− schwannoma cells (300,000 cells in 0.2 mL of a 25% Matrigel solution) were injected subcutaneously into one flank of each SCID mouse using an 18-gauge needle. The contralateral leg was utilized as control. Starting on post-implantation day 3, the mice were divided into two groups: 1) control mice were fed normal rodent chow and 2) treatment mice were fed drug-impregnated rodent chow formulated to deliver approximately 25 mg/kg AR42 daily. The mice were imaged at 14, 23, and 33 days following injection using a 9.4 T/cm MRI scanner. Following the final imaging sequence, all mice were sacrificed; their flank tumors were harvested, fixed in 10% neutral buffered formalin and embedded in paraffin. Five-micron tissue sections were obtained. Standard hematoxylin–eosin staining was performed as well as immunostaining for cleaved caspase-3 (#9661L) and phosphoSer473-AKT (D9E, #4060) (both from Cell Signaling Technology, Danvers, MA). Negative controls were treated with the same immunostaining protocol, except without the primary antibody.
2. AR42-treated human vestibular schwannoma xenografts (Figure 1A)
Vestibular schwannoma xenografts were surgically established in SCID mice utilizing a flank incision. The contralateral non-operated limb was used as an internal control. All animals were fed normal rodent diets until 1-month MRI scans were obtained for each mouse. Tumor volumes calculated from this set of images were used as baseline measurements. Subsequently, experimental animals were fed AR42-impregnated rodent chow formulated to deliver 25 mg/kg of drug per day for a total of 2 months while control animals were fed normal diets. A second MRI was obtained 3-months post-implantation (i.e., after 2-months of drug treatment in the experimental group), and differences in tumor size were quantified volumetrically. Tumor explants were stained for p-AKT and cleaved caspase-3 using the same antibodies noted above. Ki-67 immunostaining was also performed (Ki-67 Clone SP6 rabbit monoclonal antibody #RM-9106 from Thermo Scientific, Rockford, IL). Negative controls were treated with the same immunostaining protocol, except without the primary antibody.
3. MRI analysis
All imaged mice were anesthetized with 2.5% isoflurane mixed with 1 liter per minute carbogen and maintained with 1-1.5% isoflurane during imaging. They were placed prone on a temperature-controlled mouse bed and inserted into a 35 mm diameter quadrature volume coil. The imaging was performed using a Bruker Biospin 94/30 magnet (Bruker Biospin, Karlsruhe, Germany). The respiration and temperature of the animals were monitored during the course of the experiment using a Small Animal Monitoring and Gating system (Model 1025, Small Animals Instruments, Inc., Stony Brook, NY). Axial T1-weighted (TR=1500 ms, TE=7.5 ms, Rare Factor=4, navgs=4) and T2-weighted RARE images (TR=4200 ms, TE=12 ms, Rare Factor=8, navgs=4) were acquired over the entire region of the tumor. The acquisition parameters for both the T1- and T2-weighted multi-slice scans were as follows: FOV = 30 mm × 30 mm, slice thickness = 1 mm, matrix size = 256 × 256. An expert observer delineated a region-of-interest (ROI) that included the tumor. This was manually outlined (software developed in-house) using the T1- and T2-weighted images in order to calculate tumor volumes (i.e., pixel size outlined ROI slice thickness).
AR42 pharmacotoxicity studies (Figure 1B)
A total of 29 female FVB/N wild-type mice (14 control mice and 15 AR42-treated mice) were utilized for pharmacotoxicity studies. Blood chemistry data were obtained from fourteen untreated mice in order to establish strain-specific normal laboratory values. Eight AR42-treated mice were submitted for organ histology and blood chemistries after they had been dosed for 3-months with AR42. An additional seven AR42-treated mice were submitted for analysis after 6-months of oral AR42 dosing. For comparative normal histology, two control animals each (fed normal chow) also underwent survey organ histology at the 3- and 6-month time points.
1. Clinical and anatomic pathology
Complete postmortem evaluations were performed on recently euthanized wild-type female FVB/N mice fed AR42-impregnated chow or control rodent chow for 3- and 6-months. Body (pre-bleed) and selected post-bleed organ weights were recorded. All tissues (including heart; trachea and lungs; thymus; aorta; kidney and urinary bladder; liver and gall bladder; spleen; pancreas; salivary glands; lymph nodes; gastrointestinal tract; ovaries and uterus; thyroid, adrenal and pituitary glands; sciatic nerve and brain; and skin with mammary gland and adipose tissue) were fixed in 10% neutral buffered formalin with the exception of the sternum, vertebral column with spinal cord and rear legs which were fixed in Decalcifier I (Surgipath, Richmond, IL) for 48 hours. These tissues were processed and embedded in paraffin. Sections (4 μm) were stained with hematoxylin and eosin (H&E), and evaluated with an Olympus BX45 light microscope with attached DP25 digital camera (B & B Microscopes Limited, Pittsburgh, PA). Representative images from heart, liver, muscle, kidney and brain sections were collected.
2. Hematology and blood chemistry studies
Blood from recently euthanized wild-type female FVB/N mice fed AR42-impregnated chow or control rodent chow for appropriate durations was collected by percutaneous cardiac puncture. Complete blood counts with 5-part white blood cell differentials were performed on a portion of EDTA anti-coagulated whole blood (FORCYTE auto-sampler 10, Oxford Science, Inc., Oxford, CT). Following coagulation of the remaining whole blood at room temperature for 30 min., the clotted blood was centrifuged at 3000 rpm for 5-10 min. at 4°C. Biochemical profiles (including alkaline phosphatase; alanine aminotransferase; aspartate aminotransferase; gamma glutamyl transferase; total protein; albumin; globulin; total bilirubin; blood urea nitrogen; creatinine; cholesterol; triglycerides; glucose; calcium; sodium; potassium; chloride; phosphorus; amylase; lipase; and creatine kinase) were performed on serum samples (VetACE, Alfa Wasserman, West Caldwell, NJ).
AR42 pharmacokinetics and disposition in brain tissues (Figure 1B)
Three control and six AR42-treated female FVB/N wild-type mice were submitted for drug penetration studies in plasma and normal brain tissue after 3-months of continuous oral dosing via AR42-fortified chow. Plasma AR42 levels were determined using a validated LC-MS/MS method [Cheng, Liu et al 2006 & Cheng, Jones, 2007, both AACR Annual Meeting Abstracts] and AR42 levels in brain tissues were determined by adapting this method to brain tissues homogenates. Briefly, approximately 50 mg of the frozen mouse brain tissue was transferred to a 2 mL micro-centrifuge tube and 10 μL of internal standard solution (IS) (10 μg/mL hesperetin) was added. The solution was homogenized in an ice bath and the homogenate was mixed with 200 μL of lysis buffer followed by centrifugation. The supernatant was extracted with 1 mL ethyl acetate and the organic layer was evaporated to dryness. The residue was reconstituted in 100 μL 50% acetonitrile and a 20 μL aliquot of supernatant was injected for LC-MS/MS analysis. The LC-MS/MS system was a Thermo TSQ Quantum triple quadrupole mass spectrometer equipped with an electrospray ionization source, a Shimadzu LC-20AD HPLC pump and an LC-20AC auto-sampler. The sample was separated on a Thermo Betabasic C8 column (50 mm × 2.1 mm) with isocratic 80% methanol containing 0.2% formic acid at the flow rate of 0.2 mL/min. Finally, AR42 and the IS were detected by multiple reactions monitoring the following paired ion transitions: m/z 313.00>133.10 and m/z 303.10>153.10.
RESULTS
AR42 inhibits P0Cre Nf2−/− schwannoma allografts in vivo at doses correlating with decreased AKT phosphorylation
SCID mice harboring P0Cre Nf2−/− schwannoma allografts were orally treated with AR42-impregnated rodent chow, targeting a dose of 25 mg/kg/day. Both control and treated mice were imaged 14, 23, and 33 days following injection, and tumors were sized volumetrically. Between the first and third imaging time points, the mean increase in tumor volume for the control (untreated) group was 12690% (95% CI 9920% - 15444%) while the increase in tumor volumes for AR42-treated mice was 3296% (95% CI 829% - 5762%) (Figure 2A & B). This striking decrease in allograft growth rates for drug treated tumors was statistically significant (p=0.001). Histopathology and immunohistochemistry were also performed in tumor explants for in vivo molecular target analysis. With fewer darkly staining, hyper-chromatic nuclei, H&E stained sections demonstrated relaxed chromatin structure after AR42 treatment ((Figure 3A). On average, fewer mitotic figures were also observed in AR42-exposed P0Cre Nf2−/− schwannoma allografts (3 per high power field) than in controls (5 per high power field). Immunohistochemistry found significant decreases in p-AKT staining in drug-treated animals (Figure 3B), and increased cleavage of procaspase-3 (Figure 3C) after AR42 exposure suggesting AR42-induced apoptosis. Average cell counts for p-AKT labeling using four high power fields (HPFs) from each P0Cre schwannoma allograft found that 41 cells labeled with p-AKT antibody vs. 9 cells for the AR42 treatment group. The analysis of cleaved caspase-3 found an average of 8 labeled cells in control explants, whereas 19 cells labeled in the AR42 treated animals. Ki-67 staining (images not shown) was robust in both groups due to the rapidly proliferating nature of this cell line. Differences in proliferative index for control versus treated implants were not discernable by the naked eye, even at 600x magnification.
Figure 2. AR42 dramatically reduced the growth of Nf2−/− P0Cre mouse schwannoma allografts.


(A) SCID-ICR mice were implanted with P0Cre mouse schwannoma cells, fed either normal rodent chow or AR42-containing chow, and imaged on days 14, 23, and 33 post-implantation. Tumor volumes calculated using high-field MRI scans and plotted for each individual mouse revealed a dramatic drop in tumor volume in AR42-fed mice versus mice fed normal chow (P=0.001). (B) Representative MR images from a control mouse and an AR42-fed mouse.
Figure 3. P0Cre schwannoma allografts treated with AR42 demonstrated relaxed chromatin, reduced p-AKT staining, and increased apoptosis.



Tumors from the mice described in Figure 2 were excised after the final MRI imaging sequence, fixed in formalin, and paraffin embedded. (A) Sections analyzed by hematoxylin and eosin consistently revealed lighter nuclear staining, suggestive of relaxed euchromatin structure. (B) Immunostaining was then performed for phospho-Ser473-AKT, which revealed a marked drop in staining for AR42-treated tumors compared to control tumors. The arrow in the control tumor section points to an example cell with high p-AKT positivity. (C) Cleaved caspase-3 immunostaining was used to evaluate apoptosis. Control tumors had noticeably less apoptotic labeling compared with AR42-treated tumors.
AR42 suppresses growth of human vestibular schwannoma xenografts
SCID mice harboring human vestibular schwannoma xenografts were orally treated with either control rodent food or AR42-impregnated rodent chow targeting a dose of 25 mg/kg/day. Representative MR images for each group are presented in Figure 4C. Four of 10 control VS xenografts grew during the 2-month imaging interval, while 4 persisted (minimal decrease in size) and one tumor shrank (Figure 4A). Statistical analysis found a 6% mean increase in xenograft volume for the untreated group with 95% confidence interval of -10% to 22%. In the AR42-treated animals, 9 of 10 xenografts shrank dramatically while one implant persisted with minimal change in tumor size between scans (Figure 4B). A 28% mean decrease in tumor volume was observed with 95% confidence interval of -45% to -12% change. This confidence interval did not cross the zero-point, indicating that tumor shrinkage could be expected with >95% confidence in AR42-treated implants. Additionally, the confidence intervals between control and treatment groups did not overlap, suggesting statistically significant differences between their means. Unpaired t-test analysis confirmed that these differences between mean tumor volumes (+6% in controls versus -28% for AR42 treatment) were statistically significant (p=0.006). Immunostaining for p-AKT, cleaved pro-caspase 3, and Ki-67 was performed on several tumor explants (Figure 5), and qualitative examination of several randomly chosen high power fields from treated and control slides was completed. Overall, the immunostaining for all three antibodies was less dramatic than for P0Cre allografts. On average, VS controls had 6 p-AKT labeled cells vs. 3-labeled cells for AR42 treated VS xenografts (per high power field). Cleaved caspase-3 immunostaining was absent in the control VS explant slides and was sporadically found in the treatment group (one cell per 4 high power fields). While there was only isolated cleaved caspase 3 staining for the AR42 treated group, a more dramatic decline in Ki67 staining was seen in the drug treated animals. The control tumor explants had 7 labeled cells versus 2 labeled cells in the AR42 group (per high power field). Taken together, this data suggests that while both cell cycle arrest and apoptosis was observed in VS after AR42 exposure, the former is predominant.
Figure 4. AR42 decreased tumor volumes in human vestibular schwannoma xenografts.



SCID-ICR mice were implanted in the left flank with fresh human VS specimens. A month following tumor implantation, the mice were given a baseline MRI and fed either normal rodent chow or AR42-containing chow. A final MRI sequence was taken 2 months afterward. (A) The change in volume for each mouse in the control group was plotted, showing a 6% mean increase in tumor volume across all mice. (B) Waterfall plot representing the change in volume for each mouse in the AR42-fed group revealed an overall decrease in tumor volume for the group (mean decrease ~ 28%). (C) Representative MR images from a control mouse and an AR42-fed mouse.
Figure 5. AR42 exposure decreased p-AKT staining and Ki-67 labeling in treated human VS xenografts.

VS tumors from the mice described in Figure 4 were excised after the final MRI imaging sequence, fixed in formalin, and paraffin embedded. Immunostaining performed for phospho-Ser473-AKT revealed an ~ 50% decline in p-AKT staining for AR42-treated tumors versus control sections. Likewise, Ki-67 labeling revealed that AR42 treatment caused an ~ 50% decrease in the proliferative index. Staining sections with cleaved caspase-3 antibody was performed analysis to evaluate apoptosis. AR42-treated tumors had modestly more cleaved caspase-3 positivity compared to control tumors, but the labeling was sparse.
Pharmacotoxicity studies in animals receiving AR42 over 3- and 6-month dosing schedules found no clinically overt abnormalities
After the first month, the cohort of AR42-treated FVB/N mice reliably ingested enough chow to achieve the targeted drug dose. All wild-type FVB/N mice gained weight and veterinary staff noted normal social and grooming behavior during this study. Compared with controls (Figure 6A), a small drop in weight was observed for mice in the AR42 treatment group, which recovered quickly after the first 7-10 days (Figure 6B).
Figure 6. Long-term AR42 dosing in mice achieved the targeted daily drug dose and did not affect weight gain.
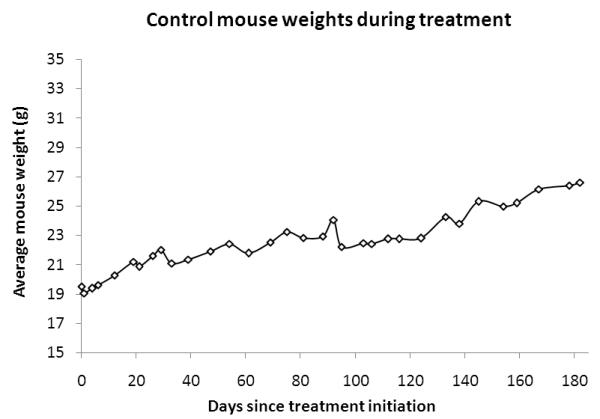
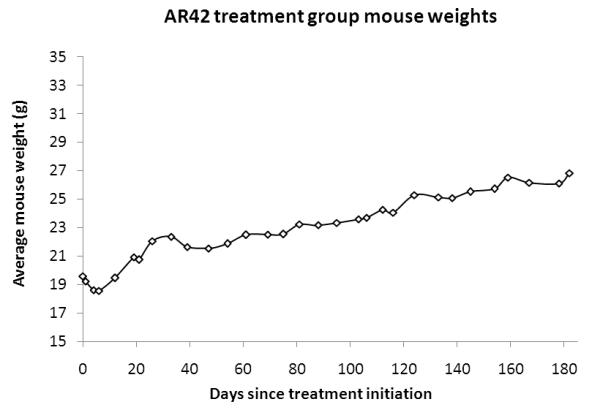
Wild-type female FVB/N mice were fed either normal or AR42 chow for 3 to 6 months. Both (A) control mice and (B) AR42-fed mice gained weight at comparable rates. A small weight drop was initially observed in the AR42 group when chow feeding was initiated, but this weight loss recovered rapidly.
Multi-organ histology in animals that received AR42 over 3- and 6-month dosing schedules found no clinically significant abnormalities
After euthanasia, gross and microscopic analysis of kidney, heart, skeletal muscle, and cerebral cortex specimens found no architectural differences between control and drug-fed mice (Figure 7). Nearly all mice fed AR42 had some centrilobular hepatocellular hypertrophy at 6-months, a finding not present at 3 months. Both control and AR42-treated mice demonstrated macroscopic evidence of cystic ovarian follicles, which are common findings in the reproductive tract of aged female mice and do not represent pre-neoplastic lesions. Two of five AR42-treated mice demonstrated microscopic evidence of lymphoid depletion in the thymus at 6 months, which was not noted in the 3-month treatment group.
Figure 7. AR42 was well tolerated with no clinically significant abnormalities in organ tissue histology identified during 6-month of oral dosing.

The FVB/N mice described in Figure 6 were analyzed for histopathology across multiple organs. Shown are representative sections from the cerebral cortex, cardiac muscle, skeletal muscle, kidney, and liver. Mice fed AR42 chow for 3-months showed no detectable differences relative to control mice. Mice treated with AR42 for 6-months displayed centrilobular hepatocellular hypertrophy, a common adaptive response to xenobiotics.
Hematology and blood chemistry studies in animals that received AR42 over 3- and 6-month dosing schedules found no clinically significant abnormalities
White blood cell counts (WBC; with differential), hemoglobin, hematocrit, platelet counts, kidney function studies, liver enzymes, and bilirubin were determined in control (wild-type) FVB/N mice, 3-month AR42-treated mice, and mice treated with AR42 for 6-months (Figure 8A). No significant differences were observed between controls and 3-month treated mice; however, mice treated with AR42 for 6 months had a lower WBC count than controls (Figure 8B). Analysis of WBC differentials found that this difference was accounted for by an absolute lymphopenia. Neutropenia was not observed, and all mice, including those housed outside the ultra-sterile barrier facility, survived the study period without clinical evidence of infection.
Figure 8. Mice fed AR42 for 6-months showed mild lymphopenia.


FVB/N mice treated with AR42 for 3- and 6-months underwent hematology and blood chemistry analyses. (A) No significant differences were observed between control mice and mice fed AR42 for 3-months. Mice treated with AR42 for 6-months had lower WBC counts than controls. Data are shown as the mean value and 95% confidence intervals. (B) Differential WBC analysis showed that the low WBC counts seen in 6-month AR42-fed mice were due to an absolute lymphopenia. Data are again shown as the mean value and 95% confidence intervals.
Orally administered AR42 crosses the blood-brain barrier
To validate AR42 as a potential treatment for intracranial neoplasms, blood-brain barrier penetration assays were performed using 6 wild-type FVB/N mice treated for 3 months. These mice gained weight normally (Figure 9A) and demonstrated no clinical evidence of toxicity. While drug-impregnated rodent chow was formulated to deliver a daily dose of 25 mg/kg of AR42 per mouse, we found that the daily AR42 intake was variable, ranging from a minimum of 25.3 mg/kg/d to a maximum of 37.5 mg/kg/d. The average dose of AR42 for the entire 3-month experiment was 34.4 mg/kg/d (Figure 9B). Pharmacokinetic analysis found significant variability in the drug concentration for both plasma and brain tissue (Figure 9C); however, AR42 was detectable in all brain tissue specimens analyzed.
Figure 9. Orally administered AR42 crosses the blood-brain barrier (BBB).
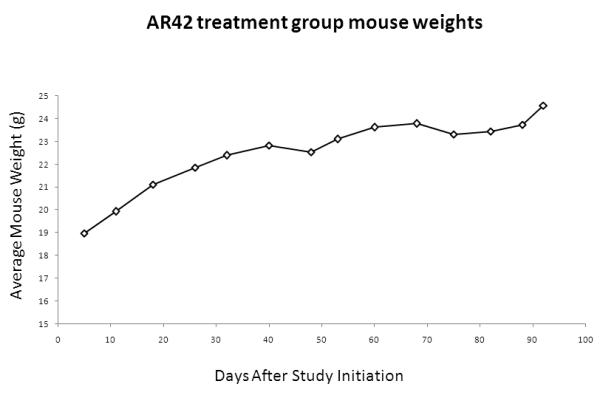
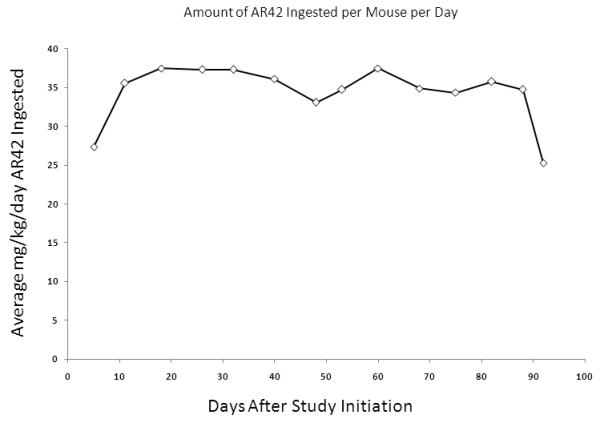

FVB/N mice were fed AR42 for 3 months and blood-brain barrier penetration assays were performed. (A) All mice in this study gained weight and displayed no clinical signs of toxicity. (B) The estimated mean AR42 dose was 34.4 mg/kg/day for this group. (C) AR42 was detected in the brains of all mice analyzed but concentrations were highly variable.
Discussion
Recent evidence linking aberrant deacetylation of histone and non-histone proteins to cell proliferation has provided strong impetus for the development of histone deacetylase inhibitors (HDACi) as cancer therapeutics. Structurally diverse natural and synthetic inhibitors to the zinc-containing catalytic site of these enzymes have been isolated. Most HDACi in clinical development inhibit both Class I and Class II HDACs and fall into several broad groups based on chemical structure: (1) hydroxamic acids, such as trichostatin A, (2) cyclic tetrapeptides and the depsipeptides, (3) benzamides, (4) electrophilic ketones, and (5) aliphatic acid compounds such as phenylbutyrate and valproic acid.26,27 Normal cells seem relatively resistant to HDACi treatment, while tumor cells undergo growth arrest, cell differentiation and/or cell death.28-30 Postulated mechanisms of action for this class of novel drugs include modulation of gene expression through chromatin remodeling,31 induction of p21 and p27 mediated cell cycle arrest,32-40 inhibition of the molecular chaperone protein HSP90,41-44 disruption of aggresome-mediated protein degradation,45 cytoskeletal disruption due to abnormal tubulin assembly,46-50 induction of programmed cell death,51-55 disruption of intracellular signal transduction,23,56-60 induction of oxidative injury,61-63 and inhibition of angiogenesis.64-70
AR42 (OSU-HDAC42; Arno Therapeutics) is a high potency phenylbutyrate-derived histone deacetylase inhibitor that suppresses tumor growth, at least in part, by reshuffling protein phosphatase-1, which then results in downstream AKT dephosphorylation/inactivation.23 We have recently confirmed that AR42 potently inhibits primary VS (inhibitory concentration 50% [IC50] ~ 500 nM) and Nf2−/− murine schwannoma (IC50 ~ 250 nM) cell growth at doses correlating with decreases in p-AKT.24 Total AKT protein levels remained unchanged in that study, suggesting that HDACi therapy did not alter AKT gene expression through epigenetic effects. Also, stable total AKT protein levels in schwannoma cells indicated that HDACi mediated acetylation of lysine residues on molecular chaperones such as HSP90 did not result in targeting AKT for degradation.71-75,24
Several groups have shown that post-translational activation of the PI3-kinase/AKT pathway is common in vestibular schwannomas and that AKT inhibition may be a viable treatment strategy.18-20 Abnormal PI3-kinase/AKT activation has been found in both NF2-associated and sporadic schwannoma cells but not in primary human Schwann cells.18,76 NF2/merlin deficiency in vestibular schwannomas activates AKT signaling by several distinct but potentially complementary mechanisms. First, merlin helps mediate contact dependent-inhibition of growth12 by controlling the availability and function of several cell surface receptors13 such as the epidermal growth factor receptor (EGFR), ErbB2, and the platelet-derived growth factor (PDGF) receptor.14,15 Through its interactions with adaptor proteins such as NHERF1,14,15 merlin may facilitate context-dependent receptor signaling.12,14,16,76,77 Second, Rong and colleagues have demonstrated that the NF2 gene product merlin can inhibit AKT activation by binding PIKE-L (PI3-kinase enhancer long isoform).78 PIKE-L is a GTPase that normally couples PI3-kinase to metabotropic glutamate receptors (mGluRs).79 The binding of normally functioning merlin to PIKE-L prevents this protein’s association with and activation of PI3K. Third, direct AKT-merlin interactions may contribute to abnormal AKT activation in schwannomas.80-82
Once activated, AKT can target downstream modulators of apoptosis such as GSK3β, IRS-1 (insulin receptor substrate-1), PDE-3B (phosphodiesterase-3B), BAD, caspase-9, Forkhead proteins, NF-κB, mTOR, nitric oxide synthase (NOS), Raf protein kinase, and BRCA1. AKT phosphorylation of BAD and the Forkhead transcription factors FKHR/FKHRL1 promotes their binding to 14-3-3 proteins and sequestration within the cytoplasm, rendering them inactive.83-85 AKT can directly phosphorylate and inhibit caspase-9, a mediator of intrinsic apoptosis. 86 AKT also activates IκB kinases, which phosphorylate IκB and target this protein for degradation; this action frees NF-κB from IκB-mediated cytoplasmic sequestration and permits NF-κB nuclear translocation, where it promotes transcription of pro-survival genes.87 AKT prevents nuclear accumulation of β-catenin by phosphorylating and inhibiting its upstream regulator GSK3β.88 β-catenin, part of the Wnt signaling pathway, is normally phosphorylated by GSK3β and targeted for degradation; however, GSK3β inhibition allows β-catenin to build up in the cytoplasm, translocate into the nucleus, and interact with the TCF/LEF family of transcription factors to initiate expression of anti-apoptosis genes. AKT also phosphorylates and inactivates tuberin (TSC2), which results in the downstream activation of mTOR (normally inhibited by the TSC1/TSC2 complex).89 mTOR activation increases protein synthesis by simultaneously activating p70 ribosomal protein S6 kinase (p70S6K) and inhibiting the translational repressor 4E-binding protein 1 (4E-BP1).90,91
AKT activation also can foster tumor growth by promoting cell cycle progression. First, GSK3β phosphorylates cyclin D1 and targets it for proteasomal degradation; thus, AKT-mediated inhibition of GSK3β promotes the stability of cyclin D1.92 Second, AKT inhibits the transcription of and/or promotes degradation of p27Kip1, a CDK inhibitor that blocks the activity of the G1 and S-phase complexes cyclin E-CDK2, cyclin D-CDK4 and cyclin A-CDK2. AKT phosphorylates the FOXO Forkhead transcription factors, sequestering them in the cytoplasm and blocking FOXO-mediated transcription of p27Kip1.93 AKT also directly phosphorylates p27Kip1, causing p27Kip1 to be retained in the cytosol,94 where can undergo E3 ubiquitin ligase-mediated degradation.95 Third, AKT activation may inhibit p53-mediated cell cycle checkpoints through phosphorylation and sequestration of p21cip1/waf1 and through MDM2-mediated inhibition of p53.96,97 AKT phosphorylation of MDM2 (murine double minute 2) on serine 166 and serine 186 is necessary for MDM2 translocation into the nucleus, where promotes p53 degradation.97,98 AKT also phosphorylates p21cip1/waf1 at threonine 145, resulting in cytoplasmic localization.98,99 Finally, PI3-kinase/AKT inhibition has been shown to promote G2/M arrest in several models.100 This effect may be mediated by AKT phosphorylation of BRCA1 (breast cancer type 1 susceptibility protein)101 or through Chk1, a kinase that inhibits the CDK-activating protein cdc25.102
The anti-proliferative effects of HDACi therapy are known to be drug specific, cell-type dependent and context specific.103 Our previous in vitro results found that IC50 doses of AR42 preferentially caused cell cycle arrest in cultured human schwannomas, whereas increased doses and prolonged drug treatment resulted in apoptosis.24 Based on this data, we hypothesized that prolonged AR42 exposure in vivo would result predominantly in apoptosis. The current study found this to be the case only for the more rapidly growing murine schwannoma implants, but human vestibular schwannoma xenografts primarily underwent cell cycle arrest. AR42 suppressed tumor growth at doses correlating with decreased AKT phosphorylation in both sets of schwannoma grafts but more dramatically so in the Nf2−/− murine schwannoma allografts. Also, analysis of cleaved caspase-3 immunostaining found clear evidence of apoptosis in AR42-treated Nf2−/− schwannoma allografts. In contrast, the anti-proliferative effects of AR42 in VS xenografts appeared to involve primarily cell cycle arrest and only a minimal increase in apoptosis. Decreased Ki-67 and increased cleaved caspase-3 immunostaining were both observed, but the former was more apparent.
To experimentally determine whether AR42 is truly cytostatic or cytotoxic to vestibular schwannomas, a dedicated in vivo time course study looking at intracellular cell death mechanisms after varying periods of drug exposure needs be performed using an animal model. Another, perhaps more informative approach would be to perform a Phase 0 human clinical trial looking at intra-tumoral drug concentrations and molecular endpoints in patients asked to ingest AR42 for several weeks prior to planned surgical resection of their tumors. The latter would be meaningful as a prelude to Phase 2 efficacy studies in humans.
Results from our murine pharmacotoxicity studies indicate that AR42 was well tolerated during both 3-month and 6-month dosing regimens. Histologic evaluation of heart; trachea and lungs; thymus; aorta; kidney and urinary bladder; liver and gall bladder; spleen; pancreas; salivary glands; lymph nodes; gastrointestinal tract; ovaries and uterus; thyroid, adrenal and pituitary glands; sciatic nerve and brain; and skin with mammary gland and adipose tissues found no structural abnormalities. The only consistent morphologic lesion was mild hepatocellular hypertrophy, characterized histologically by cytoplasmic changes in the centrilobular to midzonal regions of the liver; this was only seen in mice fed AR42 for 6-months. Centrilobular hypertrophy is a common adaptive response to a range of stimuli, including pregnancy and lactation, hormonal fluctuations, dietary constituents, infections, and exposures to xenobiotics. It is typically caused by changes in mitochondria, smooth endoplasmic reticulum or peroxisomes, and a common cause is induction of the P450 microsomal system. While most constitutively expressed P450s and other phase I metabolism enzymes have a predominant centrilobular localization, this hypertrophic response is not considered a toxic effect.
Hematologic and biochemical laboratory studies were performed in mice administered AR42 during 3- and 6-month regimens. Mice variably displayed mild biochemical abnormalities in ALT, AST, and GGT; increased glucose; increased total bilirubin; increased phosphorus; increased triglycerides; decreased amylase; decreased calcium; decreased cholesterol; and increased or decreased creatine kinase; however, differences between treated and control mice were not statistically significant. Variability in laboratory values between mice is common; for example, GGT levels vary widely with values upwards of 14 U/L being found in seemingly “normal” mice. Moderately decreased WBC counts, primarily due to lymphopenia, was the only statistically significant difference between control mice and those fed AR42 for 6 months. This finding was not present at the 3-month treatment window, and importantly, no mice had overt signs of infection during either 3- or 6-months of AR42 treatment. In 2008, Sargeant and colleagues81 dosed mice with AR42 for up to 18 weeks and reported similar findings: completely reversible leukopenia and anemia; increased serum AST, total bilirubin, albumin and total protein; and decreased serum cholesterol, alkaline phosphatase and potassium. Also consistent with our study, Sargeant and coworkers also found no significant changes in body weight or other indicators of poor general health.
According to the NIH Clinical Trials website (http://www.clinicaltrials.gov), there are currently only four active clinical trials targeting NF2-associated tumors. Three involve already FDA approved agents such as Lapatinib (GlaxoSmithKline; Research Triangle Park, North Carolina) for vestibular schwannomas, Bevacizumab/Avastin™ (Roche, Basel, Switzerland) for vestibular schwannomas/meningiomas and Sunitinib (Pfizer, New York, NY) for recurrent/progressive meningiomas while the fourth clinical trial looks at PTC299 (PTC Therapeutics; South Plainfield, NJ), a “novel, orally administered, small molecule designed to inhibit the production of VEGF by targeting the post-transcriptional processes that regulate VEGF synthesis.” Based on our current study results demonstrating schwannoma-specific preclinical efficacy, detailed molecular target analysis, and in vivo safety profiling, we propose that AR42 is an excellent candidate for translation to VS clinical trials. Discussions are currently being had with the pharmaceutical company licensing AR42 as well as the FDA in order to move in that direction. As AR42 was reliably detected in brain tissues of all orally dosed mice, its use as therapy for intracranial tumors is appropriate. We believe that with sustained oral dosing in human clinical trials, the in vitro IC50 for vestibular schwannomas (approximately 500 nM) will likely be achieved.
Conclusions
AR42 suppresses schwannoma growth at doses correlating with AKT pathway inhibition. This orally bioavailable drug penetrates the BBB, is well tolerated, and represents a novel candidate for translation to human VS clinical trials.
Acknowledgments
We sincerely thank: Drs. D. Bradley Welling and Long-Sheng Chang for their mentorship and use of lab space; Dr. Marco Giovannini and INSERM for the P0Cre;Nf2flox/flox mice; Drs. D. Bradley Welling and Edward Dodson for VS specimens; the staff of the OSU Comparative Pathology and Mouse Phenotyping Shared Resource; the staff of the OSU Pathology Core Facility; Dr. Kimerly Powell for small animal MRI imaging and volumetric tumor analysis; Dr. Matthew Ringel for the use of laboratory space to perform many of these experiments; and the Cooperative Human Tissue Network of The Ohio State University Comprehensive Cancer Center—funded by the National Cancer Institute—processed tissue samples. We would especially like to thank Beth Miles-Markley, the OSU Neurotology Research Coordinator, for assisting with the complex logistics of this in vivo study.
Funding/Financial Disclosures: National Institute of Deafness and Other Communication Disorders [ABJ – K08 DC009644-01A1; DBW – R01 DC005985 and 3R01DC005985-05S11], American Hearing Research Foundation [MLB – GRT00014844], Children’s Tumor Foundation [ABJ – DDI 2007-05--2], and the Triological Society [ABJ – GRT00011359]
Footnotes
Conflict of interest – None
References
- 1.Rouleau GA, Merel P, Lutchman M, et al. Alteration in a new gene encoding a putative membrane-organizing protein causes neuro-fibromatosis type 2. Nature. 1993;363:515–521. doi: 10.1038/363515a0. [DOI] [PubMed] [Google Scholar]
- 2.Trofatter JA, MacCollin MM, Rutter JL, et al. A novel moesin-, ezrin-, radixin-like gene is a candidate for the neurofibromatosis 2 tumor suppressor. Cell. 1993;72:791–800. doi: 10.1016/0092-8674(93)90406-g. [DOI] [PubMed] [Google Scholar]
- 3.Welling DB, Packer MD, Chang LS. Molecular studies of vestibular schwannomas: a review. Current opinion in otolaryngology & head and neck surgery. 2007;15:341–346. doi: 10.1097/MOO.0b013e3282b97310. [DOI] [PubMed] [Google Scholar]
- 4.Evans DG, Birch JM, Ramsden RT, Sharif S, Baser ME. Malignant transformation and new primary tumours after therapeutic radiation for benign disease: substantial risks in certain tumour prone syndromes. Journal of medical genetics. 2006;43:289–294. doi: 10.1136/jmg.2005.036319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Balasubramaniam A, Shannon P, Hodaie M, Laperriere N, Michaels H, Guha A. Glioblastoma multiforme after stereotactic radiotherapy for acoustic neuroma: case report and review of the literature. Neuro-oncology. 2007;9:447–453. doi: 10.1215/15228517-2007-027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Son EI, Kim IM, Kim SP. Vestibular schwannoma with malignant transformation: a case report. Journal of Korean medical science. 2001;16:817–821. doi: 10.3346/jkms.2001.16.6.817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Levine EM, Becker Y, Boone CW, Eagle H. Contact Inhibition, Macromolecular Synthesis, and Polyribosomes in Cultured Human Diploid Fibroblasts. Proc Natl Acad Sci U S A. 1965;53:350–356. doi: 10.1073/pnas.53.2.350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Levine BB. Studies on Delayed Hypersensitivity. I. Inferences on the Comparative Binding Affinities of Antibodies Mediating Delayed and Immediate Hypersensitivity Reactions in the Guinea Pig. J Exp Med. 1965;121:873–888. doi: 10.1084/jem.121.6.873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Eagle H, Levine EM, Boone CW. Cellular Growth, Contact Inhibition, and Macromolecular Synthesis. Science. 1965;148:665. doi: 10.1126/science.148.3670.665. [DOI] [PubMed] [Google Scholar]
- 10.Evans DG. Neurofibromatosis type 2 (NF2): a clinical and molecular review. Orphanet J Rare Dis. 2009;4:16. doi: 10.1186/1750-1172-4-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Eldridge R. Central neurofibromatosis with bilateral acoustic neuroma. Adv Neurol. 1981;29:57–65. [PubMed] [Google Scholar]
- 12.Curto M, McClatchey AI. Nf2/Merlin: a coordinator of receptor signalling and intercellular contact. Br J Cancer. 2008;98:256–262. doi: 10.1038/sj.bjc.6604002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Maitra S, Kulikauskas RM, Gavilan H, Fehon RG. The tumor suppressors Merlin and Expanded function cooperatively to modulate receptor endocytosis and signaling. Curr Biol. 2006;16:702–709. doi: 10.1016/j.cub.2006.02.063. [DOI] [PubMed] [Google Scholar]
- 14.Curto M, Cole BK, Lallemand D, Liu CH, McClatchey AI. Contact-dependent inhibition of EGFR signaling by Nf2/Merlin. J Cell Biol. 2007;177:893–903. doi: 10.1083/jcb.200703010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.James MF, Beauchamp RL, Manchanda N, Kazlauskas A, Ramesh V. A NHERF binding site links the betaPDGFR to the cytoskeleton and regulates cell spreading and migration. J Cell Sci. 2004;117:2951–2961. doi: 10.1242/jcs.01156. [DOI] [PubMed] [Google Scholar]
- 16.McClatchey AI, Fehon RG. Merlin and the ERM proteins--regulators of receptor distribution and signaling at the cell cortex. Trends Cell Biol. 2009;19:198–206. doi: 10.1016/j.tcb.2009.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Altomare DA, Testa JR. Perturbations of the AKT signaling pathway in human cancer. Oncogene. 2005;24:7455–7464. doi: 10.1038/sj.onc.1209085. [DOI] [PubMed] [Google Scholar]
- 18.Jacob A, Lee TX, Neff BA, Miller S, Welling B, Chang LS. Phosphatidylinositol 3-kinase/AKT pathway activation in human vestibular schwannoma. Otology & Neurotology. 2008;29:58–68. doi: 10.1097/mao.0b013e31816021f7. [DOI] [PubMed] [Google Scholar]
- 19.Lee TX, Packer MD, Huang J, et al. Growth inhibitory and anti-tumour activities of OSU-03012, a novel PDK-1 inhibitor, on vestibular schwannoma and malignant schwannoma cells. Eur J Cancer. 2009;45:1709–1720. doi: 10.1016/j.ejca.2009.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hilton DA, Ristic N, Hanemann CO. Activation of ERK, AKT and JNK signalling pathways in human schwannomas in situ. Histopathology. 2009;55:744–749. doi: 10.1111/j.1365-2559.2009.03440.x. [DOI] [PubMed] [Google Scholar]
- 21.Gregoretti IV, Lee YM, Goodson HV. Molecular evolution of the histone deacetylase family: functional implications of phylogenetic analysis. J Mol Biol. 2004;338:17–31. doi: 10.1016/j.jmb.2004.02.006. [DOI] [PubMed] [Google Scholar]
- 22.Minucci S, Pelicci PG. Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. Nat Rev Cancer. 2006;6:38–51. doi: 10.1038/nrc1779. [DOI] [PubMed] [Google Scholar]
- 23.Chen CS. Histone Acetylation-independent Effect of Histone Deacetylase Inhibitors on Akt through the Reshuffling of Protein Phosphatase 1 Complexes. Journal of Biological Chemistry. 2005;280:38879–38887. doi: 10.1074/jbc.M505733200. [DOI] [PubMed] [Google Scholar]
- 24.Bush ML, Oblinger J, Brendel V, et al. AR42, a novel histone deacetylase inhibitor, as a potential therapy for vestibular schwannomas and meningiomas. Neuro-Oncology. 2011 doi: 10.1093/neuonc/nor072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Giovannini M, Robanus-Maandag E, van der Valk M, et al. Conditional biallelic Nf2 mutation in the mouse promotes manifestations of human neurofibromatosis type 2. Genes Dev. 2000;14:1617–1630. [PMC free article] [PubMed] [Google Scholar]
- 26.Drummond DC, Marx C, Guo Z, et al. Enhanced pharmacodynamic and antitumor properties of a histone deacetylase inhibitor encapsulated in liposomes or ErbB2-targeted immunoliposomes. Clin Cancer Res. 2005;11:3392–3401. doi: 10.1158/1078-0432.CCR-04-2445. [DOI] [PubMed] [Google Scholar]
- 27.Drummond DC, Noble CO, Kirpotin DB, Guo Z, Scott GK, Benz CC. Clinical development of histone deacetylase inhibitors as anticancer agents. Annu Rev Pharmacol Toxicol. 2005;45:495–528. doi: 10.1146/annurev.pharmtox.45.120403.095825. [DOI] [PubMed] [Google Scholar]
- 28.Dokmanovic M, Clarke C, Marks PA. Histone deacetylase inhibitors: overview and perspectives. Mol Cancer Res. 2007;5:981–989. doi: 10.1158/1541-7786.MCR-07-0324. [DOI] [PubMed] [Google Scholar]
- 29.Dokmanovic M, Marks PA. Prospects: histone deacetylase inhibitors. J Cell Biochem. 2005;96:293–304. doi: 10.1002/jcb.20532. [DOI] [PubMed] [Google Scholar]
- 30.Kelly WK, Marks PA. Drug insight: Histone deacetylase inhibitors--development of the new targeted anticancer agent suberoylanilide hydroxamic acid. Nat Clin Pract Oncol. 2005;2:150–157. doi: 10.1038/ncponc0106. [DOI] [PubMed] [Google Scholar]
- 31.Xu WS, Parmigiani RB, Marks PA. Histone deacetylase inhibitors: molecular mechanisms of action. Oncogene. 2007;26:5541–5552. doi: 10.1038/sj.onc.1210620. [DOI] [PubMed] [Google Scholar]
- 32.Sasakawa Y, Naoe Y, Inoue T, et al. Effects of FK228, a novel histone deacetylase inhibitor, on tumor growth and expression of p21 and c-myc genes in vivo. Cancer Lett. 2003;195:161–168. doi: 10.1016/s0304-3835(03)00184-8. [DOI] [PubMed] [Google Scholar]
- 33.Sasakawa Y, Naoe Y, Inoue T, et al. Effects of FK228, a novel histone deacetylase inhibitor, on human lymphoma U-937 cells in vitro and in vivo. Biochem Pharmacol. 2002;64:1079–1090. doi: 10.1016/s0006-2952(02)01261-3. [DOI] [PubMed] [Google Scholar]
- 34.Nimmanapalli R, Fuino L, Stobaugh C, Richon V, Bhalla K. Cotreatment with the histone deacetylase inhibitor suberoylanilide hydroxamic acid (SAHA) enhances imatinib-induced apoptosis of Bcr-Abl-positive human acute leukemia cells. Blood. 2003;101:3236–3239. doi: 10.1182/blood-2002-08-2675. [DOI] [PubMed] [Google Scholar]
- 35.Abramova MV, Pospelova TV, Nikulenkov FP, Hollander CM, Fornace AJ, Jr., Pospelov VA. G1/S arrest induced by histone deacetylase inhibitor sodium butyrate in E1A + Ras-transformed cells is mediated through down-regulation of E2F activity and stabilization of beta-catenin. J Biol Chem. 2006;281:21040–21051. doi: 10.1074/jbc.M511059200. [DOI] [PubMed] [Google Scholar]
- 36.Archer SY, Johnson J, Kim HJ, et al. The histone deacetylase inhibitor butyrate downregulates cyclin B1 gene expression via a p21/WAF-1-dependent mechanism in human colon cancer cells. Am J Physiol Gastrointest Liver Physiol. 2005;289:G696–703. doi: 10.1152/ajpgi.00575.2004. [DOI] [PubMed] [Google Scholar]
- 37.Gui CY, Ngo L, Xu WS, Richon VM, Marks PA. Histone deacetylase (HDAC) inhibitor activation of p21WAF1 involves changes in promoter-associated proteins, including HDAC1. Proc Natl Acad Sci U S A. 2004;101:1241–1246. doi: 10.1073/pnas.0307708100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Han JW, Ahn SH, Kim YK, et al. Activation of p21(WAF1/Cip1) transcription through Sp1 sites by histone deacetylase inhibitor apicidin: involvement of protein kinase C. J Biol Chem. 2001;276:42084–42090. doi: 10.1074/jbc.M106688200. [DOI] [PubMed] [Google Scholar]
- 39.Han JW, Ahn SH, Park SH, et al. Apicidin, a histone deacetylase inhibitor, inhibits proliferation of tumor cells via induction of p21WAF1/Cip1 and gelsolin. Cancer Res. 2000;60:6068–6074. [PubMed] [Google Scholar]
- 40.Hecker RM, Amstutz RA, Wachtel M, Walter D, Niggli FK, Schafer BW. p21 Downregulation is an important component of PAX3/FKHR oncogenicity and its reactivation by HDAC inhibitors enhances combination treatment. Oncogene. 2010 doi: 10.1038/onc.2010.145. [DOI] [PubMed] [Google Scholar]
- 41.Bali P, Pranpat M, Bradner J, et al. Inhibition of histone deacetylase 6 acetylates and disrupts the chaperone function of heat shock protein 90: a novel basis for antileukemia activity of histone deacetylase inhibitors. J Biol Chem. 2005;280:26729–26734. doi: 10.1074/jbc.C500186200. [DOI] [PubMed] [Google Scholar]
- 42.Banerji U. Heat shock protein 90 as a drug target: some like it hot. Clin Cancer Res. 2009;15:9–14. doi: 10.1158/1078-0432.CCR-08-0132. [DOI] [PubMed] [Google Scholar]
- 43.Glaser KB. HDAC inhibitors: clinical update and mechanism-based potential. Biochem Pharmacol. 2007;74:659–671. doi: 10.1016/j.bcp.2007.04.007. [DOI] [PubMed] [Google Scholar]
- 44.Solit DB, Rosen N. Hsp90: a novel target for cancer therapy. Curr Top Med Chem. 2006;6:1205–1214. doi: 10.2174/156802606777812068. [DOI] [PubMed] [Google Scholar]
- 45.Kawaguchi Y, Kovacs JJ, McLaurin A, Vance JM, Ito A, Yao TP. The deacetylase HDAC6 regulates aggresome formation and cell viability in response to misfolded protein stress. Cell. 2003;115:727–738. doi: 10.1016/s0092-8674(03)00939-5. [DOI] [PubMed] [Google Scholar]
- 46.Balasubramanian S, Ramos J, Luo W, Sirisawad M, Verner E, Buggy JJ. A novel histone deacetylase 8 (HDAC8)-specific inhibitor PCI-34051 induces apoptosis in T-cell lymphomas. Leukemia. 2008;22:1026–1034. doi: 10.1038/leu.2008.9. [DOI] [PubMed] [Google Scholar]
- 47.Haggarty SJ, Koeller KM, Wong JC, Grozinger CM, Schreiber SL. Domain-selective small-molecule inhibitor of histone deacetylase 6 (HDAC6)-mediated tubulin deacetylation. Proc Natl Acad Sci U S A. 2003;100:4389–4394. doi: 10.1073/pnas.0430973100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hubbert C, Guardiola A, Shao R, et al. HDAC6 is a microtubule-associated deacetylase. Nature. 2002;417:455–458. doi: 10.1038/417455a. [DOI] [PubMed] [Google Scholar]
- 49.Matsuyama A, Shimazu T, Sumida Y, et al. In vivo destabilization of dynamic microtubules by HDAC6-mediated deacetylation. EMBO J. 2002;21:6820–6831. doi: 10.1093/emboj/cdf682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang Y, Li N, Caron C, et al. HDAC-6 interacts with and deacetylates tubulin and microtubules in vivo. EMBO J. 2003;22:1168–1179. doi: 10.1093/emboj/cdg115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ahn MY, Jung JH, Na YJ, Kim HS. A natural histone deacetylase inhibitor, Psammaplin A, induces cell cycle arrest and apoptosis in human endometrial cancer cells. Gynecol Oncol. 2008;108:27–33. doi: 10.1016/j.ygyno.2007.08.098. [DOI] [PubMed] [Google Scholar]
- 52.D’Acunto CW, Carratu A, Rodriquez M, Taddei M, Parente L, Petrella A. LGP1, A histone deacetylase inhibitor analogue of FR235222, sensitizes promyelocytic leukaemia U937 cells to TRAIL-mediated apoptosis. Anticancer Res. 2010;30:887–894. [PubMed] [Google Scholar]
- 53.Insinga A, Monestiroli S, Ronzoni S, et al. Inhibitors of histone deacetylases induce tumor-selective apoptosis through activation of the death receptor pathway. Nat Med. 2005;11:71–76. doi: 10.1038/nm1160. [DOI] [PubMed] [Google Scholar]
- 54.Kulp SK, Chen CS, Wang DS, Chen CY. Antitumor effects of a novel phenylbutyrate-based histone deacetylase inhibitor, (S)-HDAC-42, in prostate cancer. Clin Cancer Res. 2006;12:5199–5206. doi: 10.1158/1078-0432.CCR-06-0429. [DOI] [PubMed] [Google Scholar]
- 55.Shankar S, Srivastava RK. Histone deacetylase inhibitors: mechanisms and clinical significance in cancer: HDAC inhibitor-induced apoptosis. Adv Exp Med Biol. 2008;615:261–298. doi: 10.1007/978-1-4020-6554-5_13. [DOI] [PubMed] [Google Scholar]
- 56.Barter MJ, Pybus L, Litherland GJ, et al. HDAC-mediated control of ERK- and PI3K-dependent TGF-beta-induced extracellular matrix-regulating genes. Matrix Biol. 2010 doi: 10.1016/j.matbio.2010.05.002. [DOI] [PubMed] [Google Scholar]
- 57.Kim YK, Seo DW, Kang DW, Lee HY, Han JW, Kim SN. Involvement of HDAC1 and the PI3K/PKC signaling pathways in NF-kappaB activation by the HDAC inhibitor apicidin. Biochem Biophys Res Commun. 2006;347:1088–1093. doi: 10.1016/j.bbrc.2006.06.196. [DOI] [PubMed] [Google Scholar]
- 58.Kobayashi Y, Ohtsuki M, Murakami T, et al. Histone deacetylase inhibitor FK228 suppresses the Ras-MAP kinase signaling pathway by upregulating Rap1 and induces apoptosis in malignant melanoma. Oncogene. 2006;25:512–524. doi: 10.1038/sj.onc.1209072. [DOI] [PubMed] [Google Scholar]
- 59.Ozaki K, Kosugi M, Baba N, et al. Blockade of the ERK or PI3K-Akt signaling pathway enhances the cytotoxicity of histone deacetylase inhibitors in tumor cells resistant to gefitinib or imatinib. Biochem Biophys Res Commun. 2010;391:1610–1615. doi: 10.1016/j.bbrc.2009.12.086. [DOI] [PubMed] [Google Scholar]
- 60.Yu X, Guo ZS, Marcu MG, et al. Modulation of p53, ErbB1, ErbB2, and Raf-1 expression in lung cancer cells by depsipeptide FR901228. J Natl Cancer Inst. 2002;94:504–513. doi: 10.1093/jnci/94.7.504. [DOI] [PubMed] [Google Scholar]
- 61.Butler LM, Zhou X, Xu WS, et al. The histone deacetylase inhibitor SAHA arrests cancer cell growth, up-regulates thioredoxin-binding protein-2, and down-regulates thioredoxin. Proc Natl Acad Sci U S A. 2002;99:11700–11705. doi: 10.1073/pnas.182372299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ungerstedt JS, Sowa Y, Xu WS, et al. Role of thioredoxin in the response of normal and transformed cells to histone deacetylase inhibitors. Proc Natl Acad Sci U S A. 2005;102:673–678. doi: 10.1073/pnas.0408732102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Xu W, Ngo L, Perez G, Dokmanovic M, Marks PA. Intrinsic apoptotic and thioredoxin pathways in human prostate cancer cell response to histone deacetylase inhibitor. Proc Natl Acad Sci U S A. 2006;103:15540–15545. doi: 10.1073/pnas.0607518103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cao ZA, Bass KE, Balasubramanian S, et al. CRA-026440: a potent, broad-spectrum, hydroxamic histone deacetylase inhibitor with antiproliferative and antiangiogenic activity in vitro and in vivo. Mol Cancer Ther. 2006;5:1693–1701. doi: 10.1158/1535-7163.MCT-06-0042. [DOI] [PubMed] [Google Scholar]
- 65.Chou CW, Chen CC. HDAC inhibition upregulates the expression of angiostatic ADAMTS1. FEBS Lett. 2008;582:4059–4065. doi: 10.1016/j.febslet.2008.10.048. [DOI] [PubMed] [Google Scholar]
- 66.Kim SH, Ahn S, Han JW, et al. Apicidin is a histone deacetylase inhibitor with anti-invasive and anti-angiogenic potentials. Biochem Biophys Res Commun. 2004;315:964–970. doi: 10.1016/j.bbrc.2004.01.149. [DOI] [PubMed] [Google Scholar]
- 67.Kwon HJ, Kim MS, Kim MJ, Nakajima H, Kim KW. Histone deacetylase inhibitor FK228 inhibits tumor angiogenesis. Int J Cancer. 2002;97:290–296. doi: 10.1002/ijc.1602. [DOI] [PubMed] [Google Scholar]
- 68.Mie Lee Y, Kim SH, Kim HS, et al. Inhibition of hypoxia-induced angiogenesis by FK228, a specific histone deacetylase inhibitor, via suppression of HIF-1alpha activity. Biochem Biophys Res Commun. 2003;300:241–246. doi: 10.1016/s0006-291x(02)02787-0. [DOI] [PubMed] [Google Scholar]
- 69.Qian DZ, Kato Y, Shabbeer S, et al. Targeting tumor angiogenesis with histone deacetylase inhibitors: the hydroxamic acid derivative LBH589. Clin Cancer Res. 2006;12:634–642. doi: 10.1158/1078-0432.CCR-05-1132. [DOI] [PubMed] [Google Scholar]
- 70.Williams RJ. Trichostatin A, an inhibitor of histone deacetylase, inhibits hypoxia-induced angiogenesis. Expert Opin Investig Drugs. 2001;10:1571–1573. doi: 10.1517/13543784.10.8.1571. [DOI] [PubMed] [Google Scholar]
- 71.Basso AD, Solit DB, Chiosis G, Giri B, Tsichlis P, Rosen N. Akt forms an intracellular complex with heat shock protein 90 (Hsp90) and Cdc37 and is destabilized by inhibitors of Hsp90 function. J Biol Chem. 2002;277:39858–39866. doi: 10.1074/jbc.M206322200. [DOI] [PubMed] [Google Scholar]
- 72.Fujita N, Sato S, Ishida A, Tsuruo T. Involvement of Hsp90 in signaling and stability of 3-phosphoinositide-dependent kinase-1. J Biol Chem. 2002;277:10346–10353. doi: 10.1074/jbc.M106736200. [DOI] [PubMed] [Google Scholar]
- 73.Kovacs JJ, Murphy PJ, Gaillard S, et al. HDAC6 regulates Hsp90 acetylation and chaperone-dependent activation of glucocorticoid receptor. Mol Cell. 2005;18:601–607. doi: 10.1016/j.molcel.2005.04.021. [DOI] [PubMed] [Google Scholar]
- 74.Rao R, Fiskus W, Yang Y, et al. HDAC6 inhibition enhances 17-AAG--mediated abrogation of hsp90 chaperone function in human leukemia cells. Blood. 2008;112:1886–1893. doi: 10.1182/blood-2008-03-143644. [DOI] [PubMed] [Google Scholar]
- 75.Sato S, Fujita N, Tsuruo T. Modulation of Akt kinase activity by binding to Hsp90. Proc Natl Acad Sci U S A. 2000;97:10832–10837. doi: 10.1073/pnas.170276797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ammoun S, Flaiz C, Ristic N, Schuldt J, Hanemann CO. Dissecting and targeting the growth factor-dependent and growth factor-independent extracellular signal-regulated kinase pathway in human schwannoma. Cancer Res. 2008;68:5236–5245. doi: 10.1158/0008-5472.CAN-07-5849. [DOI] [PubMed] [Google Scholar]
- 77.Ammoun S, Cunliffe CH, Allen JC, et al. ErbB/HER receptor activation and preclinical efficacy of lapatinib in vestibular schwannoma. Neuro Oncol. 2010 doi: 10.1093/neuonc/noq012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Rong R, Tang X, Gutmann DH, Ye K. Neurofibromatosis 2 (NF2) tumor suppressor merlin inhibits phosphatidylinositol 3-kinase through binding to PIKE-L. Proc Natl Acad Sci U S A. 2004;101:18200–18205. doi: 10.1073/pnas.0405971102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Rong R, Ahn JY, Huang H, et al. PI3 kinase enhancer-Homer complex couples mGluRI to PI3 kinase, preventing neuronal apoptosis. Nat Neurosci. 2003;6:1153–1161. doi: 10.1038/nn1134. [DOI] [PubMed] [Google Scholar]
- 80.Liao Y, Hung MC. Physiological regulation of Akt activity and stability. Am J Transl Res. 2010;2:19–42. [PMC free article] [PubMed] [Google Scholar]
- 81.Okada M, Wang Y, Jang SW, Tang X, Neri LM, Ye K. Akt phosphorylation of merlin enhances its binding to phosphatidylinositols and inhibits the tumor-suppressive activities of merlin. Cancer Res. 2009;69:4043–4051. doi: 10.1158/0008-5472.CAN-08-3931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ye K. Phosphorylation of merlin regulates its stability and tumor suppressive activity. Cell Adh Migr. 2007;1:196–198. doi: 10.4161/cam.1.4.5192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Yaffe MB, Rittinger K, Volinia S, et al. The structural basis for 14-3-3:phosphopeptide binding specificity. Cell. 1997;91:961–971. doi: 10.1016/s0092-8674(00)80487-0. [DOI] [PubMed] [Google Scholar]
- 84.Franke TF, Cantley LC. Apoptosis. A Bad kinase makes good. Nature. 1997;390:116–117. doi: 10.1038/36442. [DOI] [PubMed] [Google Scholar]
- 85.Nakamura N, Ramaswamy S, Vazquez F, Signoretti S, Loda M, Sellers WR. Forkhead transcription factors are critical effectors of cell death and cell cycle arrest downstream of PTEN. Mol Cell Biol. 2000;20:8969–8982. doi: 10.1128/mcb.20.23.8969-8982.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Cardone MH, Roy N, Stennicke HR, et al. Regulation of cell death protease caspase-9 by phosphorylation. Science. 1998;282:1318–1321. doi: 10.1126/science.282.5392.1318. [DOI] [PubMed] [Google Scholar]
- 87.Kane LP, Shapiro VS, Stokoe D, Weiss A. Induction of NF-kappaB by the Akt/PKB kinase. Curr Biol. 1999;9:601–604. doi: 10.1016/s0960-9822(99)80265-6. [DOI] [PubMed] [Google Scholar]
- 88.Cross DA, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature. 1995;378:785–789. doi: 10.1038/378785a0. [DOI] [PubMed] [Google Scholar]
- 89.Potter CJ, Pedraza LG, Xu T. Akt regulates growth by directly phosphorylating Tsc2. Nat Cell Biol. 2002;4:658–665. doi: 10.1038/ncb840. [DOI] [PubMed] [Google Scholar]
- 90.Tee AR, Fingar DC, Manning BD, Kwiatkowski DJ, Cantley LC, Blenis J. Tuberous sclerosis complex-1 and -2 gene products function together to inhibit mammalian target of rapamycin (mTOR)-mediated downstream signaling. Proc Natl Acad Sci U S A. 2002;99:13571–13576. doi: 10.1073/pnas.202476899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Manning BD, Tee AR, Logsdon MN, Blenis J, Cantley LC. Identification of the tuberous sclerosis complex-2 tumor suppressor gene product tuberin as a target of the phosphoinositide 3-kinase/akt pathway. Mol Cell. 2002;10:151–162. doi: 10.1016/s1097-2765(02)00568-3. [DOI] [PubMed] [Google Scholar]
- 92.Diehl JA, Cheng M, Roussel MF, Sherr CJ. Glycogen synthase kinase-3beta regulates cyclin D1 proteolysis and subcellular localization. Genes Dev. 1998;12:3499–3511. doi: 10.1101/gad.12.22.3499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Burgering BM, Medema RH. Decisions on life and death: FOXO Forkhead transcription factors are in command when PKB/Akt is off duty. J Leukoc Biol. 2003;73:689–701. doi: 10.1189/jlb.1202629. [DOI] [PubMed] [Google Scholar]
- 94.Shin I, Yakes FM, Rojo F, et al. PKB/Akt mediates cell-cycle progression by phosphorylation of p27(Kip1) at threonine 157 and modulation of its cellular localization. Nat Med. 2002;8:1145–1152. doi: 10.1038/nm759. [DOI] [PubMed] [Google Scholar]
- 95.Mamillapalli R, Gavrilova N, Mihaylova VT, et al. PTEN regulates the ubiquitin-dependent degradation of the CDK inhibitor p27(KIP1) through the ubiquitin E3 ligase SCF(SKP2) Curr Biol. 2001;11:263–267. doi: 10.1016/s0960-9822(01)00065-3. [DOI] [PubMed] [Google Scholar]
- 96.Mayo LD, Dixon JE, Durden DL, Tonks NK, Donner DB. PTEN protects p53 from Mdm2 and sensitizes cancer cells to chemotherapy. J Biol Chem. 2002;277:5484–5489. doi: 10.1074/jbc.M108302200. [DOI] [PubMed] [Google Scholar]
- 97.Mayo LD, Donner DB. A phosphatidylinositol 3-kinase/Akt pathway promotes translocation of Mdm2 from the cytoplasm to the nucleus. Proc Natl Acad Sci U S A. 2001;98:11598–11603. doi: 10.1073/pnas.181181198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Zhou BP, Liao Y, Xia W, Zou Y, Spohn B, Hung MC. HER-2/neu induces p53 ubiquitination via Akt-mediated MDM2 phosphorylation. Nat Cell Biol. 2001;3:973–982. doi: 10.1038/ncb1101-973. [DOI] [PubMed] [Google Scholar]
- 99.Zhou BP, Liao Y, Xia W, Spohn B, Lee MH, Hung MC. Cytoplasmic localization of p21Cip1/WAF1 by Akt-induced phosphorylation in HER-2/neu-overexpressing cells. Nat Cell Biol. 2001;3:245–252. doi: 10.1038/35060032. [DOI] [PubMed] [Google Scholar]
- 100.Kandel ES, Skeen J, Majewski N, et al. Activation of Akt/protein kinase B overcomes a G(2)/m cell cycle checkpoint induced by DNA damage. Mol Cell Biol. 2002;22:7831–7841. doi: 10.1128/MCB.22.22.7831-7841.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Altiok S, Batt D, Altiok N, et al. Heregulin induces phosphorylation of BRCA1 through phosphatidylinositol 3-Kinase/AKT in breast cancer cells. J Biol Chem. 1999;274:32274–32278. doi: 10.1074/jbc.274.45.32274. [DOI] [PubMed] [Google Scholar]
- 102.Shtivelman E, Sussman J, Stokoe D. A role for PI 3-kinase and PKB activity in the G2/M phase of the cell cycle. Curr Biol. 2002;12:919–924. doi: 10.1016/s0960-9822(02)00843-6. [DOI] [PubMed] [Google Scholar]
- 103.Rosato RR, Grant S. Histone deacetylase inhibitors: insights into mechanisms of lethality. Expert Opin Ther Targets. 2005;9:809–824. doi: 10.1517/14728222.9.4.809. [DOI] [PubMed] [Google Scholar]