

1. Introduction
Nature utilizes a common “building block approach” to make nearly all of the molecules found in living systems. In the case of polypeptides, oligonucleotides, and oligosaccharides, this strategy has been successfully replicated in the laboratory to enable efficient, flexible, and fully-automated access to these molecules from readily available bifunctional building blocks.i Because of these advances in synthesis, research in these areas is now primarily focused on discovering, understanding, and optimizing new molecular functions. In stark contrast, the synthesis of “small molecules” remains a relatively inefficient, inflexible, and unsystematized process practiced almost exclusively by highly trained specialists. As a result, synthesis still represents the rate-limiting step in small molecule science.
Importantly, Nature uses the same building block approach to make the vast majority of small molecules, including polyketides, hybrid peptide/polyketides, polyterpenes, fatty acids, and phenylpropanoids.ii Thus, despite their tremendous structural diversity, small molecule natural products share in common substantial inherent modularity. With the goal of more effectively harnessing this modularity and thereby enabling more efficient and flexible access to complex small molecules in the laboratory, we are pursuing the development of a general building block-based approach for small molecule synthesis.iii In the idealized form of this strategy, a collection of off-the-shelf building blocks having all of the required functional groups pre-installed in the correct oxidation states and with the desired stereochemical relationships are sequentially linked using only one reaction iteratively.
Toward this goal, we are developing a platform of N-methyliminodiacetic acid (MIDA) boronate building blocks.3,iv,v,vi,vii,viii,ix,x,xi,xii,xiii MIDA boronates have many highly desirable properties that render them exceptionally useful as synthetic intermediates. They are uniformly air-stable, non-toxic, highly crystalline, and monomeric free-flowing solids that are also fully compatible with silica gel chromatography. The MIDA ligand can be prepared on very large scale for very low cost,xiii is commercially-available, and is fully biodegradable.xiv Many methods now exist for preparing MIDA boronates from a wide range of different starting materials, including boronic acids,3,4,5,6,12,xv haloboranes,5,7 boronic esters,10 trialkoxyborate salts,8-11,13 organohalides,13 organolithium reagents,13 and Grignard reagents.10,11 The MIDA boronate functional group is inert to anhydrous cross-coupling conditions yet can be readily transformed into a fully reactive boronic acid or ester using exceptionally mild conditions.4,10 This pair of features enables the simple, efficient, and highly flexible synthesis of a wide range of complex small molecules via iterative cross-coupling of MIDA-protected haloboronic acids.4,5,6,10,11 MIDA boronates are also inert to a wide range of other reaction conditions, including oxidants, reductants, electrophiles, soft nucleophiles, strong acids, and a wide range of anhydrous bases.6 Combined with their compatibility with silica gel chromatography, these features enable the multi-step preparation of a wide range of otherwise challenging to access boronate building blocks from simple boron-containing starting materials.6,10,11 Rate-controlled hydrolysis of MIDA boronates in situ also represents a general solution for the very important problem of storing and cross-coupling unstable boronic acids.8,13 Finally, a large and growing collection of MIDA boronates are now commercially-available.xvi
With the goal of maximizing the generality of this platform for small molecule synthesis, we aim to transform substructures that commonly appear in a broad range of complex natural products into readily accessible and air-stable bifunctional MIDA boronate building blocks. In this vein, cis-olefins are prevalent in small molecules derived from a wide range of biosynthetic pathways including polyketides, hybrid peptide/polyketides, polyterpenes, fatty acids, and phenylpropanoids (Figure 1).xvii Controlling the stereochemistry during the formation of such double bonds often represents a major challenge in the synthesis of these types of molecules.xviii Pre-installation of this stereochemistry into a readily-accessible bifunctional olefin that can be iteratively functionalized via sequential stereospecific C-C bond forming reactions thus represents a very attractive goal.
Figure 1.
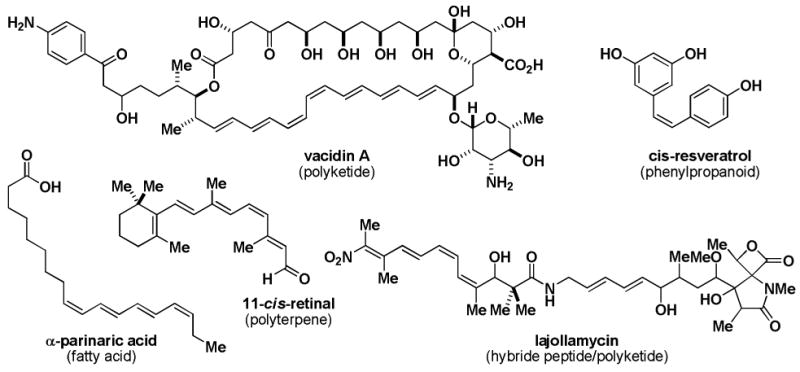
cis-Olefins are found in natural products derived from all major biosynthetic classes.
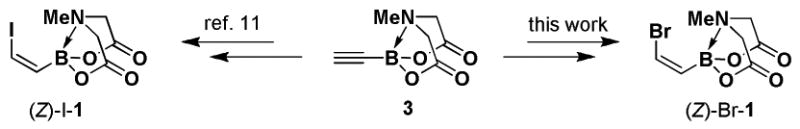
We recently reported a collection of bifunctional iodoalkenyl MIDA boronates in all possible stereoisomeric forms that can provide access to a wide range of stereochemically complex polyene frameworks via selective cross-coupling of the halide terminus.11 A very important component of this platform was (Z)-(2-iodovinyl)-MIDA boronate, (Z)-I-1 (Scheme 1). This bifunctional building block contains the olefin geometry pre-installed in the cis-configuration to allow for the construction of a wide range of polyene motifs containing cis-olefins in a stereocontrolled fashion via stereospecific cross-coupling reactions. While the synthetic utility of (Z)-I-1 proved to be outstanding, its synthesis was cumbersome (requiring multiple rounds of partial reduction and chromatography), low yielding, and not scalable. To enable ready access to such a building block for many diverse applications, we herein describe a convenient, practical, and highly scalable synthesis of an analogous reagent, (Z)-(2-bromovinyl)-MIDA boronate, (Z)-Br-1. We further demonstrate that (Z)-Br-1 can serve as a highly versatile building block for the preparation of a wide range of cis-alkene-containing synthetic targets.
Scheme 1.
2. Results
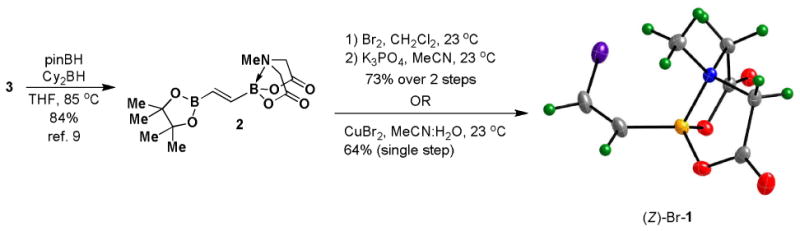
Bromination/elimination reactions provide a convenient method for the synthesis of vinyl halides. In particular, (E)-alkenylboronic esters can be converted to (Z)-alkenyl halides via this type of transformation.xix We have recently reported a convenient synthesis of bisborylated olefin 2 via the hydroboration of ethynyl MIDA boronate 3 (Scheme 2).9 Given the compatibility of MIDA boronates to many common synthetic reagents, we hypothesized that 2 would be a competent starting material for a bromination/elimination sequence, leading to an efficient synthesis of (Z)-Br-1. In the event, bromination of 2 followed by treatment of the resulting vicinal dibromide intermediate with anhydrous K3PO4 in MeCN provides a very convenient route to (Z)-Br-1 in 73% overall yield. This same building block can alternatively be prepared in a one pot procedure by reacting 2 with CuBr2 in aqueous MeCN. Both of these pathways can be performed on the gram scale and (Z)-Br-1 can be isolated in pure form without the use of chromatography.
Scheme 2.
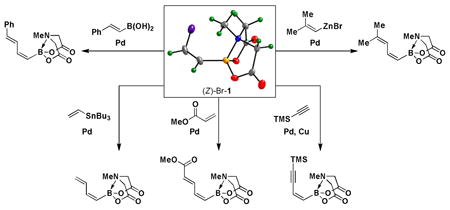
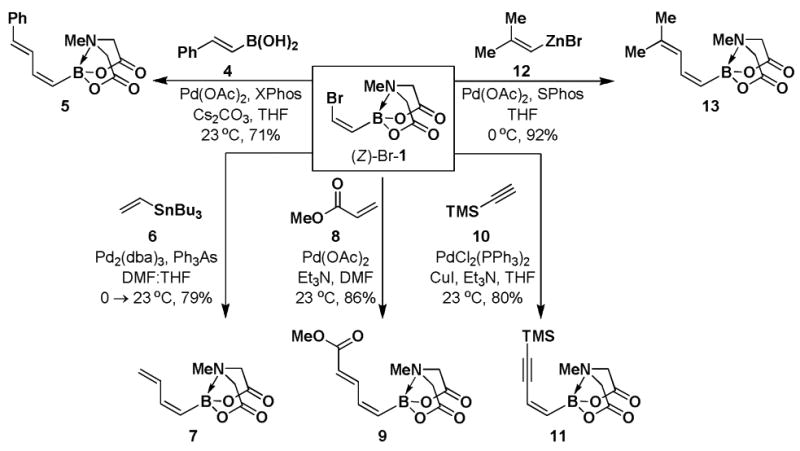
With a pair of simple and readily scalable syntheses of (Z)-Br-1 in hand, we have preliminarily explored its utility in the preparation of a range of new (Z)-alkenyl MIDA boronate building blocks. Like its stereoisomeric counterpart (E)-Br-15, (Z)-Br-1 is a very versatile cross-coupling partner, as shown in Scheme 3. Specifically, Suzuki-Miyaura (SM) cross-coupling with (E)-styrenylboronic acid 4 provided (E,Z)-diene 5. A Stille coupling between (Z)-Br-1 and vinyl stannane 6 provided diene 7. A Heck coupling with methyl acrylate 8 under phosphine-free conditions yielded the unsaturated methyl ester 9 as a single regio- and stereoisomer. Moreover, Sonogashira coupling between (Z)-Br-1 and TMS-acetylene 10 generated enyne 11. Finally, Negishi cross-coupling with 12 yielded MIDA boronate 13. Collectively, the diversity of coupling reactions compatible with (Z)-Br-1 demonstrates that this MIDA boronate possesses substantial utility in the synthesis of (Z)-alkenyl boronate building blocks for small molecule synthesis.
Scheme 3.
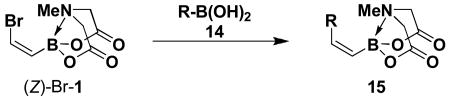
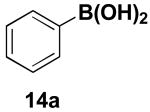
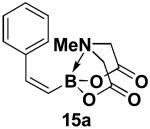
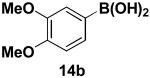
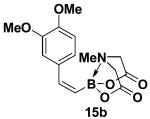
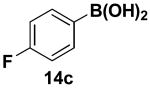
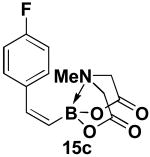
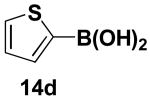
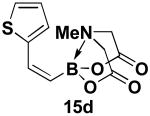
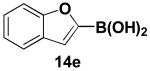
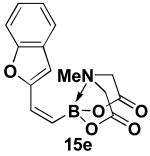
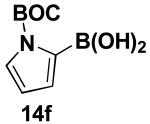
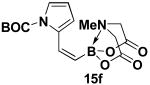
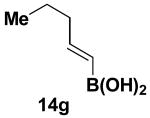
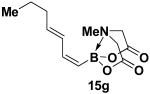
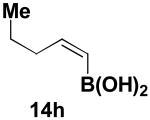
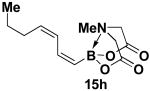
We have further determined that the scope for Suzuki-Miyaura coupling of boronic acids with (Z)-Br-1 is very good, enabling the preparation of a wide range of (Z)-vinyl MIDA boronates (Table 1). Specifically, the cross-coupling of electron neutral, rich, and deficient aryl boronic acids 14a-c (entries 1-3) provide the desired products 15a-c. Additionally, (Z)-Br-1 can be coupled with a series of heteroaryl boronic acids, including 2-thiophene 14d, 2-benzofuran 14e, and 2-pyrrole 14f (entries 4-6). Finally, trans- and cis-pentenyl boronic acid 14g-h can be coupled with (Z)-Br-1 for the synthesis of (E,Z) and (Z,Z) dienes 15g-h in a stereocontrolled fashion (entries 7-8).
Table 1. Suzuki-Miyaura cross-coupling of (Z)-Br-1.
| ||||
|---|---|---|---|---|
|
| ||||
| Entry | 14 | 15 | Conditions | % Isolated yield |
| 1 |
|
|
Pd2(dba)3, XPhos K3PO4, THF, 35 °C | 55% |
| 2 |
|
|
Pd2(dba)3, XPhos K3PO4, THF, 35 °C | 85% |
| 3 |
|
|
Pd2(dba)3, XPhos K3PO4, THF, 35 °C | 64% |
| 4 |
|
|
Pd2(dba)3, P(o-tol)3 Ag2O, THF, 23 °C | 72% |
| 5 |
|
|
Pd2(dba)3, P(o-tol)3 Ag2O, THF, 23 °C | 92% |
| 6 |
|
|
Pd(OAc)2, SPhos K3PO4, THF, 23 °C | 70% |
| 7 |
|
|
Pd(OAc)2, XPhos Cs2CO3, THF, 23 °C | 64% |
| 8 |
|
|
Pd(OAc)2, XPhos Cs2CO3, THF, 23 °C | 60% |
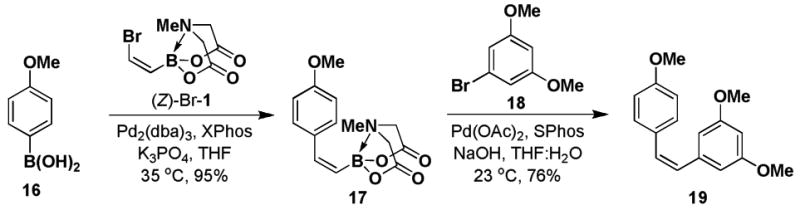
We further explored the utility of (Z)-Br-1 as a substrate for iterative SM coupling reactions. Stilbenoids are small molecule natural products that have been implicated in providing several beneficial effects on human health, including protecting against cardiovascular disease and cancer.xx (Z)-stilbenoids have demonstrated increased anticancer activity in comparison to their corresponding (E)-isomers.xxi In particular, (Z)-3,5,4′-trimethoxystilbene 19 has been shown to be an exceptionally potent analog of the stilbenoid resveratrol.xxii We have completed an iterative cross-coupling-based synthesis of 19 as shown in Scheme 4. The in situ hydrolysis of 17 followed by cross-coupling of the resulting boronic acid with aryl bromide 18 yielded 19 with complete retention of olefin stereochemistry.
Scheme 4.
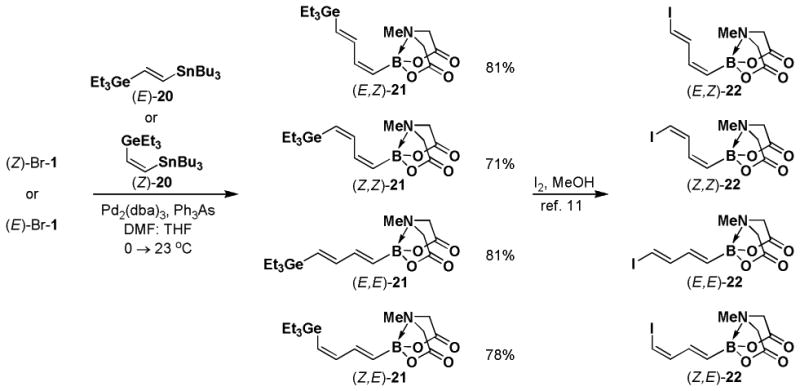
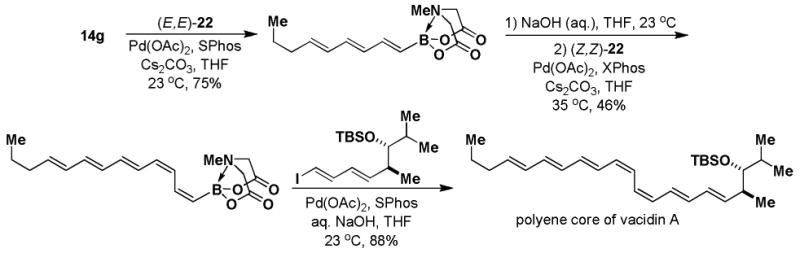
Halopolyenyl MIDA boronates have also proven to be valuable building blocks for the iterative cross-coupling-based synthesis of polyene natural products, including β-parinaric acid, peridinin, and the polyene motifs of amphotericin B and vacidin A.5,10,11 In this vein, we recently reported the synthesis of all possible stereoisomeric forms of iododienyl MIDA boronates via the metal-selective cross-coupling of Sn/Ge bis-metalated olefins followed by the stereospecific iododegermylation of the resulting dienylgermanium intermediates.11,xxiii,xxiv However, the overall efficiency of this process was limited by the low yielding preparation of key building block (Z)-I-1. With the development of the efficient preparation of (Z)-Br-1, we have been able to prepare the dienylgermanium intermediates 21 with substantially improved overall efficiency (Scheme 5). Specifically, the Stille coupling between (Z)-Br-1 and (E)-20 yields (E,Z)-21 as a single stereoisomer and coupling between (Z)-Br-1 and (Z)-20 yields (Z,Z)-21. In a similar fashion, the coupling of (E)-Br-1 with (E)-20 and (Z)-20 yields (E,E)-21 and (Z,E)-21, respectively. We have previously reported that the iododegermylation of 21 proceeds smoothly with I2 in MeOH to complete the synthesis of all possible stereoisomeric forms of the iododienyl MIDA boronate building blocks 22.11
Scheme 5.
Thus, (Z)-Br-1 and (E)-Br-1 represent a very powerful pair of bifunctional haloalkenyl MIDA boronate building blocks. In addition to being versatile coupling partners themselves, they provide ready access to a collection of more complex bifunctional building blocks that have the potential to enable the synthesis of a wide range of polyene motifs via ICC. For example, iterative cross-coupling of building blocks (E,E)-22 and (Z,Z)-22 was recently shown to provide synthetic access to the highly complex (E,E,E,Z,Z,E,E)-heptaene portion of vacidin A (Scheme 6).11,xxv
Scheme 6.
(ref. 11).
3. Summary and conclusions
Access to a wide range of bifunctional building blocks representing motifs that commonly appear in natural products and pharmaceuticals stands to greatly increase the efficiency and flexibility of small molecule synthesis. The cis-olefin represents a very important substructure that appears in a wide range of polyenyl natural product motifs. We herein describe a very practical and scalable synthesis of the bifunctional building block (Z)-Br-1 and demonstrate its utility for the stereocontrolled synthesis of a wide range of cis-alkenes. Collectively, these findings expand the utility of ICC with MIDA boronates as a simple and flexible platform for the efficient synthesis of a wide range of functional small molecules.
4. Experimental
4.1. Materials
Commercial reagents were purchased from Sigma-Aldrich, Fisher Scientific, Alfa Aesar, TCI America, Strem Chemicals Inc., or Frontier Scientific and were used without further purification unless otherwise noted. Solvents were purified via passage through packed columns as described by Pangborn and coworkersxxvi (THF, Et2O, CH3CN, CH2Cl2: dry neutral alumina; hexane, benzene, and toluene: dry neutral alumina and Q5 reactant; DMSO, DMF: activated molecular sieves). All water was deionized prior to use. The following compounds were prepared according to known literature procedures: ethynyl MIDA boronate 3,11 pinacol ester 2,9 (E)-(2-bromovinyl)-MIDA boronate (E)-Br-1,5,7 vinyl boronic acid 14h,11 vinyl stannane (E)-20,11 vinyl stannane (Z)-20.11
4.2. General experimental procedures
Unless noted, all reactions were performed in flame-dried round bottom or modified Schlenk flasks fitted with rubber septa under a positive pressure of argon or nitrogen. Organic solutions were concentrated via rotary evaporation under reduced pressure with a bath temperature of 35 – 40 °C. Reactions were monitored by analytical thin layer chromatography (TLC) performed using the indicated solvent on E. Merck silica gel 60 F254 plates (0.25mm). Compounds were visualized by exposure to a UV lamp (λ = 254 nm) and/or a solution of basic KMnO4 followed by brief heating using a Varitemp heat gun. MIDA boronates are compatible with standard silica gel chromatography, including standard loading techniques. Column chromatography was performed using standard methodsxxvii or on a Teledyne-Isco CombiFlash Rf purification system using Merck silica gel grade 9385 (60Å, 230-400 mesh). For loading, compounds were adsorbed onto non acid-washed Celite in vacuo from an acetone solution. Specifically, for a 1 g mixture of crude material the sample is dissolved in reagent grade acetone (25 to 50 mL) and to the flask is added Celite 545 Filter Aid (5 to 15 g). The mixture is then concentrated in vacuo to afford a powder, which is then loaded on top of a silica gel column. The procedure is typically repeated with a small amount of acetone (5 mL) and Celite (2 g) to ensure quantitative transfer.
4.3. Structural analysis
1H NMR spectra were recorded at 23 °C on one of the following instruments: Varian Unity 400, Varian Unity 500, Varian Unity Inova 500NB. Chemical shifts (δ) are reported in parts per million (ppm) downfield from tetramethylsilane and referenced to residual protium in the NMR solvent (CHCl3, δ = 7.26; CD2HCN, δ = 1.94, center line; d6-acetone, δ = 2.05, center line) or to added tetramethylsilane (δ = 0.00). Data are reported as follows: chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, quint = quintet, sext = sextet, sept = septet, m = multiplet, b = broad, app = apparent), coupling constant (J) in Hertz (Hz), and integration. 13C NMR spectra were recorded at 23 °C on a Varian Unity 400 or Varian Unity 500. Chemical shifts (δ) are reported in ppm downfield from tetramethylsilane and referenced to carbon resonances in the NMR solvent (CDCl3, δ = 77.0, center line; CD3CN, δ = 1.30, center line; d6-acetone, δ = 29.80, center line) or to added tetramethylsilane (δ = 0.00). Carbons bearing boron substituents were not observed (quadrupolar relaxation). High resolution mass spectra (HRMS) were performed by Furong Sun, Haijun Yao, and Beth Eves at the University of Illinois School of Chemical Sciences Mass Spectrometry Laboratory. X-ray crystallographic analyses were carried out by Dr. Danielle Gray and Amy Fuller at the University of Illinois George L. Clark X-Ray facility.
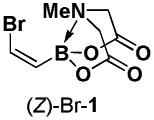
4.4. MIDA boronate (Z)-Br-1
Bromination/elimination procedure
A 300 mL round bottom flask equipped with a stir bar was charged with MIDA boronate 2 (3.07g, 9.9 mmol) and CH2Cl2 (100 mL). To this solution was added dropwise neat bromine (0.75 mL, 14.6 mmol). The resulting solution was stirred at 23 °C for 1 hr and then concentrated in vacuo to afford a pale yellow solid. Residual bromine was removed by azeotroping with CH2Cl2 (3 × 50 mL). To the resulting pale yellow solid was added finely ground K3PO4 (20.02 g, 94.3 mmol) and MeCN (100 mL). The resulting suspension was stirred at 23 °C for 3.5 hr. The resulting suspension was poured into 200 mL EtOAc, 100 mL pH 7 phosphate buffer (0.5 M), and 100 mL DI H2O. The mixture was shaken and the aqueous layer was removed. The organic layer was washed with pH 7 phosphate buffer (0.5 M, 1 × 100 mL). The combined aqueous layers were back extracted with 9:1 EtOAc:Acetone (1 × 200 mL). The combined organic layers were dried over MgSO4, filtered, and concentrated in vacuo. The resulting residue was azeotroped with CH2Cl2 (2 × 50 mL) and then suspended in Et2O (100 mL). This suspension was placed in a sonicator bath for 1 hr. The resulting solid was collected by vacuum filtration and rinsed with Et2O (15 mL) to yield MIDA boronate (Z)-Br-1 as a colorless solid (1.91g, 73%).
One-pot CuBr2 procedure
A 500-mL round bottom flask equipped with a stir bar was charged with MIDA boronate 2 (2.00 g, 6.47 mmol), CuBr2 (7.23 g, 32.4 mmol) and CH3CN:H2O (19:1, 100 mL). The resulting solution was stirred at 23 °C for 1.75 h. The reaction was poured into a separatory funnel containing 1 M aq. HCl (150 mL). The aqueous layer was extracted with EtOAc (1 × 250 mL, 2 × 150 mL). The combined organic layers were sequentially washed with sat. aq. Na2S2O3 (100 mL), half-saturated brine (100 mL), and brine (100 mL). The organic solution was vigorously stirred with an aq. solution of 2Na+ EDTA2- (0.05 M, 150 mL) for 45 min at 23 °C. The biphasic mixture was transferred to a separatory funnel and the aqueous layer removed. The organic layer was washed with brine (100 mL) and then dried over MgSO4 and decolorized with charcoal. Filtration and concentration of the filtrate in vacuo afforded crude (Z)-Br-1 as a white solid. This solid was suspended in Et2O:Acetone (25:1, ∼260 mL) and placed in a sonicator bath for 1 hr. The resulting solid was collected by vacuum filtration and rinsed with Et2O to yield MIDA boronate (Z)-Br-1 as a white powder (0.96 g, crop 1). The filtrate was concentrated in vacuo and the sonication process was repeated to afford (Z)-Br-1 as a white powder (0.13 g, crop 2; 1.09 g total, 64%).
1H NMR (500 MHz, d6-acetone) δ 7.00 (br d, J = 7.5 Hz, 1H), 6.43 (d, J = 9 Hz, 1H), 4.31 (d, J = 17 Hz, 2H), 4.10 (d, J = 17 Hz, 2H), 3.11 (s, 3H). 13C NMR (125 MHz, d6-acetone) δ 168.7, 120.6, 63.7, 47.9. HRMS (ESI+) calculated for C7H10BBrNO4 (M+H)+: 261.9886. Found: 261.9884.
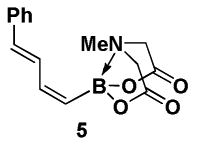
4.5. MIDA boronate 5
Preparation of catalyst solution
In a glovebox, to a 7 mL vial equipped with a stir bar and charged with XPhosxxviii (9.6 mg, 0.02 mmol) and Pd(OAc)2 (2.2 mg, 0.01 mmol) was added THF (2 mL). The solution was stirred at 23 °C for 10 min.
The freshly prepared catalyst solution was used in the following reaction
In a glovebox, to a 7 mL vial equipped with a stir bar and charged with MIDA boronate (Z)-Br-1 (52.1 mg, 0.20 mmol) and boronic acid 4 (45.4 mg, 0.31 mmol) was added Cs2CO3 (200.2 mg, 0.61 mmol). The prepared catalyst solution was added in one portion. The vial was sealed with a cap and removed from the glovebox. The solution was stirred in a subdued light environment at 23 °C for 23.5 hr. The reaction mixture was filtered through a pad of Celite, concentrated in vacuo, dry loaded onto Celite from an acetone solution, and purified via flash chromatography on silica gel (Et2O:acetone 100:0 → 3:1) to afford MIDA boronate 5 as a pale yellow solid (40.2 mg, 71%).
1H NMR (400 MHz, d6-acetone) δ 7.46 (m, 3H), 7.34 (t, J = 7.5 Hz, 2H), 7.25 (m, 1H), 6.87 (app t, J = 12 Hz, 1H), 6.62 (d, J = 15.5 Hz, 1H), 5.52 (d, J = 14 Hz, 1H), 4.29 (d, J = 17 Hz, 2H), 4.09 (d, J = 17 Hz, 2H), 3.07 (s, 3H). 13C NMR (100 MHz, d6-acetone) δ 169.0, 144.3, 138.2, 135.6, 129.4, 128.7, 128.5, 127.3, 62.3, 42.3. HRMS (ESI+) calculated for C15H17BNO4 (M+H)+: 286.1251. Found: 286.1253.
General procedure: Stille coupling
In a glovebox, to a 7 mL vial equipped with a stir bar and charged with (Z)-Br-1 or (E)-Br-1 (0.2 mmol) and vinyl stannane 6 or 1-triethylgermanium-2-tributyltin ethylene (Z)-20 or (E)-20 (0.22 mmol) was added Pd2(dba)3 (0.01 mmol) and Ph3As (0.02 mmol). The vial was sealed with a PTFE-lined septum screw-cap and removed from the glovebox. At 0 °C, under a positive pressure of Ar, THF (0.5 mL) and DMF (1.5 mL) were added sequentially via syringe. The resulting mixture was stirred in a subdued light environment at 0 °C for 2 hr and then slowly warmed to 23 °C and stirred for an additional 8-18 hr at 23 °C. The reaction mixture was poured into brine (5.0 mL) and extracted with EtOAc (2 × 15 mL). The combined organic phases were dried over MgSO4, concentrated in vacuo, dry loaded onto Celite from an acetone solution, and purified via flash chromatography on silica gel or Florisil to afford the desired compound.
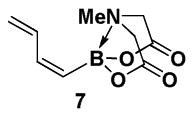
4.6. MIDA boronate 7
The general Stille coupling procedure was followed using (Z)-Br-1 (39 mg, 0.15 mmol), vinyl stannane 6 (53 mg, 0.17 mmol), Pd2(dba)3 (7.0 mg, 0.01 mmol), Ph3As (4.6 mg, 0.02 mmol), THF (0.5 mL) and DMF (1.5 mL). The resulting mixture was stirred at 0 °C for 2 hr and then slowly warmed to 23 °C and stirred for an additional 18 hr at 23 °C. Purification via flash chromatography on Florisil (EtOAc:petroleum ether 1:1 → EtOAc → EtOAc:MeCN 9:1) afforded 7 as a foam (25 mg, 79%).
1H NMR (500 MHz, CD3CN) δ 6.85 (ddd, J = 17, 10, 1 Hz, 1H), 6.70 (app t, J = 11 Hz, 1H), 5.39 (d, J = 13.5 Hz, 1H), 5.27 (dd, J = 17, 2 Hz, 1H), 5.23 (ddd, J = 10, 2, 1 Hz, 1H), 3.95 (d, J = 17 Hz, 2H), 3.79 (d, J = 17 Hz, 2H), 2.79 (s, 3H). 13C NMR (125 MHz, CD3CN) δ 169.1, 145.2, 136.7, 120.6, 62.4, 47.5. HRMS (ESI+) calculated for C9H13BNO4 (M+H)+: 210.0938. Found: 210.0940.
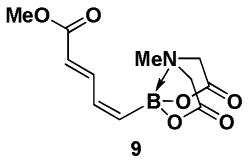
4.7. MIDA boronate 9
In a glovebox, to a 7 mL vial equipped with a stir bar and charged with MIDA boronate (Z)-Br-1 (48.3 mg, 0.18 mmol) was added Pd(OAc)2 (3.4 mg, 0.05 mmol), a solution of methyl acrylate 8 (0.4 M in DMF, 1.0 mL), and a solution of freshly distilled Et3N (0.4 M in DMF, 1.0 mL). The vial was sealed with a cap and removed from the glovebox. The solution was stirred in a subdued light environment at 23 °C for 46.5 hr. The reaction mixture was filtered through a pad of Celite, concentrated in vacuo, dry loaded onto Celite from an acetone solution, and purified via flash chromatography on Florisil (Et2O:acetone 100:0 → 2:1) to afford MIDA boronate 9 as a pale yellow solid (42.4 mg, 86%).
1H NMR (500 MHz, CD3CN) δ 7.78 (dd, J = 11.5, 15 Hz, 1H), 6.81 (app t, J = 12.5 Hz, 1H), 5.94 (d, J = 15 Hz, 1H), 5.85 (d, J = 14 Hz, 1H), 3.98 (d, J = 17 Hz, 2H), 3.83 (d, J = 17 Hz, 2H), 3.69 (s, 3H), 2.82 (s, 3H). 13C NMR (125 MHz, CD3CN) δ 169.0, 168.0, 143.5, 141.4, 124.5, 62.6, 52.1, 47.6. HRMS (ESI+) calculated for C11H15BNO6 (M+H)+: 268.0992. Found: 268.0991.
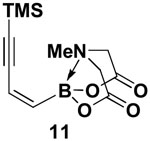
4.8. MIDA boronate 11
In a glovebox, to a 7 mL vial equipped with a stir bar and charged with (Z)-Br-1 (62.8 mg, 0.24 mmol) was added CuI (2.6 mg, 0.014 mmol) and PdCl2(PPh3)2 (19.5 mg, 0.028 mmol). The vial was sealed with a PTFE-lined septum screw-cap and removed from the glovebox. Under a positive pressure of Ar, THF (1.2 mL), TMS-acetylene 10 (0.050 mL, 0.36 mmol), and Et3N (0.10 mL, 0.72 mmol) were sequentially added via syringe. The reaction was stirred at 23 °C for 4 hr. The crude reaction was transferred to a separatory funnel containing brine (10 mL) and extracted with EtOAc (2 × 15 mL). The combined organic extracts were dried over Na2SO4, filtered, dry loaded onto Celite and purified via flash chromatography on Florisil (Et2O:EtOAc 3:1 → 2:1 → 1:1) to afford MIDA boronate 11 as a yellow solid (54 mg, 80 %). Colorless crystals could be obtained by recrystallization from Et2O (42 mg, 63 %).
1H NMR (500 MHz, CDCl3) δ 6.27 (d, J = 14.5 Hz, 1H), 6.05 (d, J = 14.5 Hz, 1H), 4.00 (d, J = 16.5 Hz, 2H), 3.87 (d, J = 16.5 Hz, 2H), 2.91 (s, 3H), 0.17 (s, 9H). 13C NMR (125 MHz, CDCl3) δ 167.7, 124.2, 103.8, 101.1, 63.0, 47.4, −0.38. HRMS (ESI+) Calculated for C12H19BNO4Si (M+H)+: 280.1176, Found: 280.1170.
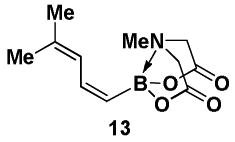
4.9. MIDA boronate 13
(2-methyl-1-propenyl)zinc bromide solution 12 was prepared as follows
A 4 mL vial equipped with a magnetic stir bar was charged with zinc bromide (68 mg, 0.31 mmol), flushed with Ar and sealed with a PTFE-lined septum screw-cap. THF (1.0 mL) was added and the solution was stirred at 0 °C for 10 min. 2-methyl-1-propenyl magnesium bromide (0.6 mL, 0.30 mmol, 0.5 M in THF) and was added and the solution was stirred at 0 °C for 30 min.
The freshly prepared solution of 12 was used in the following reaction
In a glovebox, to a 7 mL vial equipped with a magnetic stir bar and charged with (Z)-Br-1 (39 mg, 0.15 mmol) was added Pd(OAc)2 (2 mg, 0.01 mmol) and SPhos (6 mg, 0.02 mmol). The vial was sealed with a PTFE-lined septum screw-cap and removed from the glovebox. The resulting slurry was stirred for 30 min. at 23 °C and was then cooled to 0 °C. The freshly prepared solution of 12 was added dropwise to the reaction vial. The resulting mixture was stirred in a subdued light environment at 0 °C for 2 hr. The reaction mixture was poured into brine (5 mL) and extracted with EtOAc (2 × 15 mL). The combined organic phases were dried over MgSO4, concentrated in vacuo, dry loaded onto Celite from an acetone solution, and purified via flash chromatography on Florisil (EtOAc:petroleum ether 1:1 → EtOAc → EtOAc:MeCN 9:1) to afford MIDA boronate 13 as a pale yellow solid (33 mg, 92 %).
1H NMR (500 MHz, CD3CN) δ 6.94 (app t, J = 14 Hz, 1H), 6.25 (d, J = 12 Hz, 1H), 5.18 (d, J = 14 Hz, 1H), 3.94 (d, J = 17 Hz, 2H), 3.77 (d, J = 17 Hz, 2H), 2.78 (s, 3H), 1.78 (s, 3H), 1.75 (s, 3H). 13C NMR (125 MHz, CD3CN) δ 169.2, 140.8, 139.1, 124.7, 62.4, 47.4, 26.5, 17.7. HRMS (ESI+) calculated for C11H17BNO4 (M+H)+: 238.1251. Found: 238.1250.
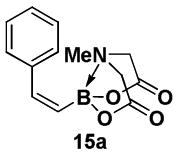
4.10. MIDA boronate 15a
Preparation of catalyst solution
In a glovebox, to a 7 mL vial equipped with a stir bar and charged with Pd2(dba)3 (9.7 mg, 0.01 mmol) and XPhos (19.2 mg, 0.04 mmol) was added THF (2 mL). The solution was stirred at 23 °C for 10 min.
The freshly prepared catalyst solution was used in the following reaction
In a glovebox, to a 7 mL vial equipped with a stir bar and charged with MIDA boronate (Z)-Br-1 (50.5 mg, 0.19 mmol) and boronic acid 14a (37.9 mg, 0.31 mmol) was added finely ground K3PO4 (129.4 mg, 0.61 mmol). The prepared catalyst solution was added in one portion. The vial was sealed with a cap and removed from the glovebox. The solution was stirred in a subdued light environment at 35 °C for 47.5 hr. The reaction mixture was filtered through a pad of Celite, concentrated in vacuo, dry loaded onto Celite from an acetone solution, and purified via flash chromatography on silica gel (Et2O:acetone 100:0 → 3:1) to afford MIDA boronate 15a as a pale yellow solid. This solid was triturated with 0.10 mL of acetone to provide a white solid (27.5 mg, 55%).
1H NMR (500 MHz, CD3CN) δ 7.36 (d, J = 7.5 Hz, 2H), 7.30 (t, J = 7.5 Hz, 2H), 7.25 (d, J = 7.5 Hz, 1H), 7.18 (br d, J = 15 Hz, 1H), 5.66 (d, J = 15 Hz, 1H), 3.88 (d, J = 17 Hz, 2H), 3.66 (d, J = 17 Hz, 2H), 2.86 (s, 3H). 13C NMR (125 MHz, CD3CN) δ 168.9, 145.0, 140.1, 129.4, 128.9, 128.1, 62.6, 47.4. HRMS (ESI+) calculated for C13H15BNO4 (M+H)+: 260.1094. Found: 260.1088.
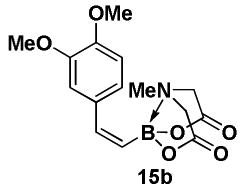
4.11. MIDA boronate 15b
Preparation of catalyst solution
In a glovebox, to a 7 mL vial equipped with a stir bar and charged with Pd2(dba)3 (10.8 mg, 0.01 mmol) and XPhos (20.9 mg, 0.04 mmol) was added THF (2 mL). The solution was stirred at 23 °C for 10 min.
The freshly prepared catalyst solution was used in the following reaction
In a glovebox, to a 7 mL vial equipped with a stir bar and charged with MIDA boronate (Z)-Br-1 (50.8 mg, 0.19 mmol) and boronic acid 14b (72.0 mg, 0.40 mmol) was added finely ground K3PO4 (137.7 mg, 0.65 mmol). The prepared catalyst solution was added in one portion. The vial was sealed with a cap and removed from the glovebox. The solution was stirred in a subdued light environment at 35 °C for 48 hr. The reaction mixture was filtered through a pad of Celite, concentrated in vacuo, dry loaded onto Celite from an acetone solution, and purified via flash chromatography on silica gel (Et2O:acetone 100:0 → 3:1) to afford MIDA boronate 15b as a pale yellow solid (52.6 mg, 85%).
1H NMR (500 MHz, d6-acetone) δ 7.29 (d, J = 2 Hz, 1H), 7.07 (d, J = 15 Hz, 1H), 6.92 (dd, J = 2, 8 Hz, 1H), 6.86 (d, J = 8 Hz, 1H), 5.56 (d, J = 15 Hz, 1H), 4.18 (d, J = 17 Hz, 2H), 3.93 (d, J = 17 Hz, 2H), 3.83 (s, 3H), 3.79 (s, 3H), 3.04 (s, 3H). 13C NMR (125 MHz, d6-acetone) δ 168.8, 149.7, 149.6, 144.7, 132.8, 122.6, 113.0, 112.0, 62.4, 55.9, 55.8, 46.9. HRMS (ESI+) calculated for C15H19BNO6 (M+H)+: 320.1305. Found: 320.1302.
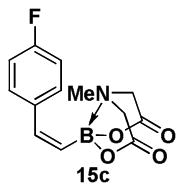
4.12. MIDA boronate 15c
Preparation of catalyst solution
In a glovebox, to a 7 mL vial equipped with a stir bar and charged with Pd2(dba)3 (10.6 mg, 0.01 mmol) and XPhos (19.5 mg, 0.04 mmol) was added THF (2 mL). The solution was stirred at 23 °C for 10 min.
The freshly prepared catalyst solution was used in the following reaction
In a glovebox, to a 7 mL vial equipped with a stir bar and charged with MIDA boronate (Z)-Br-1 (43.7 mg, 0.17 mmol) and boronic acid 14c (63.8 mg, 0.46 mmol) was added finely ground K3PO4 (136.8 mg, 0.64 mmol). The prepared catalyst solution was added in one portion. The vial was sealed with a cap and removed from the glovebox. The solution was stirred in a subdued light environment at 35 °C for 48.5 hr. The reaction mixture was filtered through a pad of Celite, concentrated in vacuo, dry loaded onto Celite from an acetone solution, and purified via flash chromatography on silica gel (Et2O:acetone 100:0 → 3:1) to afford MIDA boronate 15c as a pale yellow solid (29.6 mg, 64%).
1H NMR (400 MHz, d6-acetone) δ 7.47 (m, 2H), 7.27 (br d, J = 15 Hz, 1H), 7.09 (m, 2H), 5.71 (d, J = 15 Hz, 1H), 4.17 (d, J = 17 Hz, 2H), 3.94 (d, J = 17 Hz, 2H), 3.11 (s, 3H). 13C NMR (125 MHz, d6-acetone) δ 168.6, 143.3, 137.4, 131.4, 131.3, 115.4, 62.4, 47.0. HRMS (ESI+) calculated for C13H14BFNO4 (M+H)+: 278.1000. Found: 278.1000.
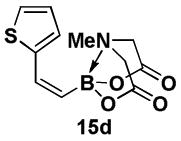
4.13. MIDA boronate 15d
Preparation of catalyst solution
In a glovebox, to a 7 mL vial equipped with a stir bar and charged with P(o-tolyl)3 (12.4 mg, 0.04 mmol) and Pd2(dba)3 (9.8 mg, 0.01 mmol) was added THF (2 mL). The solution was stirred at 23 °C for 10 min.
The freshly prepared catalyst solution was used in the following reaction
In a glovebox, to a 7 mL vial equipped with a stir bar and charged with MIDA boronate (Z)-Br-1 (49.8 mg, 0.19 mmol), boronic acid 14d (36.8 mg, 0.29 mmol), and Ag2O (145.0 mg, 0.63 mmol) was added the prepared catalyst solution in one portion. The vial was sealed with a cap and removed from the glovebox. The solution was stirred in a subdued light environment at 23 °C for 25 hr. The reaction mixture was filtered through a pad of Celite, concentrated in vacuo, dry loaded onto Celite from an acetone solution, and purified via flash chromatography on silica gel (Et2O:acetone 100:0 → 3:1) to afford MIDA boronate 15d as a pale yellow solid (36.0 mg, 72%).
1H NMR (500 MHz, d6-acetone) δ 7.40 (m, 1H), 7.24 (m, 1H), 7.19 (br d, J = 15.5 Hz, 1H), 7.00 (dd, J = 3.5, 5 Hz, 1H), 5.54 (d, J = 15.5 Hz, 1H), 4.25 (d, J = 17 Hz, 2H), 4.05 (d, J = 17 Hz, 2H), 3.08 (s, 3H). 13C NMR (125 MHz, d6-acetone) δ 168.8, 142.7, 136.3, 129.8, 127.8, 127.3, 62.7, 47.1. HRMS (ESI+) calculated for C11H13BNO4S (M+H)+: 266.0658. Found: 266.0648.
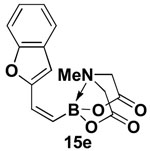
4.14. MIDA boronate 15e
Preparation of catalyst solution
In a glovebox, to a 7 mL vial equipped with a stir bar and charged with P(o-tolyl)3 (11.7 mg, 0.04 mmol) and Pd2(dba)3 (9.3 mg, 0.01 mmol) was added THF (2 mL). The solution was stirred at 23 °C for 10 min.
The freshly prepared catalyst solution was used in the following reaction
In a glovebox, to a 7 mL vial equipped with a stir bar and charged with MIDA boronate (Z)-Br-1 (46.6 mg, 0.18 mmol), boronic acid 14e (66.1 mg, 0.41 mmol), and Ag2O (143.5 mg, 0.62 mmol) was added the prepared catalyst solution in one portion. The vial was sealed with a cap and removed from the glovebox. The solution was stirred in a subdued light environment at 23 °C for 24 hr. The reaction mixture was filtered through a pad of Celite, concentrated in vacuo, dry loaded onto Celite from an acetone solution, and purified via flash chromatography on silica gel (Et2O:acetone 100:0 → 3:1) to afford MIDA boronate 15e as a pale yellow solid (49.1 mg, 92%).
1H NMR (500 MHz, d6-acetone) δ 7.58 (d, J = 8 Hz, 1H), 7.42 (d, J = 8.5 Hz, 1H), 7.29 (t, J = 7.5 Hz, 1H), 7.21 (t, J = 7.5 Hz, 1H), 7.04 (br d, J = 15.5 Hz, 1H), 6.88 (s, 1H), 5.75 (d, J = 15.5 Hz, 1H), 4.38 (d, J = 17 Hz, 2H), 4.24 (d, J = 17 Hz, 2H), 3.11 (s, 3H). 13C NMR (125 MHz, d6-acetone) δ 169.5, 156.1, 155.7, 131.2, 129.4, 125.6, 123.9, 122.0, 112.0, 108.6, 64.4, 48.9. HRMS (ESI+) calculated for C15H15BNO5 (M+H)+: 300.1043. Found: 300.1045.
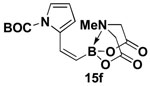
4.15. MIDA boronate 15f
Preparation of catalyst solution
In a glovebox, to a 7 mL vial equipped with a stir bar and charged with Pd(OAc)2 (2.3 mg, 0.01 mmol) and SPhos (9.2 mg, 0.02 mmol) was added THF (2 mL). The solution was stirred at 23 °C for 10 min.
The freshly prepared catalyst solution was used in the following reaction
In a glovebox, to a 7 mL vial equipped with a stir bar and charged with MIDA boronate (Z)-Br-1 (43.0 mg, 0.16 mmol) and boronic acid 14f (91.6 mg, 0.40 mmol) was added finely ground K3PO4 (136.7 mg, 0.64 mmol). The prepared catalyst solution was added in one portion. The vial was sealed with a cap and removed from the glovebox. The solution was stirred in a subdued light environment at 23 °C for 24 hr. The reaction mixture was filtered through a pad of Celite, concentrated in vacuo, dry loaded onto Celite from an acetone solution, and purified via flash chromatography on silica gel (Et2O:acetone 100:0 → 3:1) to afford MIDA boronate 15f as a pale yellow solid (39.7 mg, 70%).
1H NMR (500 MHz, d6-acetone) δ 7.24 (br d, J = 15.5 Hz, 1H), 7.23 (dd, J = 2, 3.5 Hz, 1H), 6.44 (m, 1H), 6.11 (t, J = 3.5 Hz, 1H), 5.55 (d, J = 15.5 Hz, 1H), 4.13 (d, J = 17 Hz, 2H), 3.84 (d, J = 17 Hz, 2H), 3.02 (s, 3H), 1.59 (s, 9H). 13C NMR (125 MHz, d6-acetone) δ 168.8, 150.0, 136.2, 133.7, 122.3, 115.4, 111.3, 84.4, 62.7, 47.2, 28.0. HRMS (ESI+) calculated for C16H22BN2O6 (M+H)+: 349.1571. Found: 349.1575.
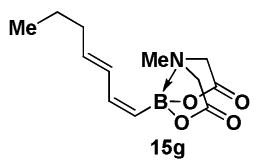
4.16. MIDA boronate 15g
Preparation of catalyst solution
In a glovebox, to a 7 mL vial equipped with a stir bar and charged with XPhos (8.6 mg, 0.02 mmol) and Pd(OAc)2 (2.1 mg, 0.01 mmol) was added THF (2 mL). The solution was stirred at 23 °C for 10 min.
The freshly prepared catalyst solution was used in the following reaction
In a glovebox, to a 7 mL vial equipped with a stir bar and charged with MIDA boronate (Z)-Br-1 (46.5 mg, 0.18 mmol) and boronic acid 14g (53.4 mg, 0.47 mmol) was added Cs2CO3 (209.4 mg, 0.64 mmol). The prepared catalyst solution was added in one portion. The vial was sealed with a cap and removed from the glovebox. The solution was stirred in a subdued light environment at 23 °C for 24 hr. The reaction mixture was filtered through a pad of Celite, concentrated in vacuo, dry loaded onto Celite from an acetone solution, and purified via flash chromatography on Florisil (Et2O:acetone 100:0 → 3:1) to afford MIDA boronate 15g as a pale yellow solid (28.5 mg, 64%). For characterization of 15g see ref. 11.
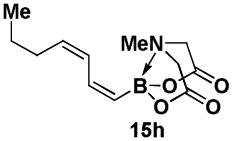
4.17. MIDA boronate 15h
Preparation of catalyst solution
In a glovebox, to a 7 mL vial equipped with a stir bar and charged with XPhos (8.6 mg, 0.02 mmol) and Pd(OAc)2 (2.1 mg, 0.01 mmol) was added THF (2 mL). The solution was stirred at 23 °C for 10 min.
The freshly prepared catalyst solution was used in the following reaction
In a glovebox, to a 7 mL vial equipped with a stir bar and charged with MIDA boronate (Z)-Br-1 (42.8 mg, 0.16 mmol) and boronic acid 14h (57.5 mg, 0.50 mmol) was added Cs2CO3 (195.8 mg, 0.60 mmol). The prepared catalyst solution was added in one portion. The vial was sealed with a cap and removed from the glovebox. The solution was stirred in a subdued light environment at 23 °C for 24 hr. The reaction mixture was filtered through a pad of Celite, concentrated in vacuo, dry loaded onto Celite from an acetone solution, and purified via flash chromatography on Florisil (Et2O:acetone 100:0 → 3:1) to afford MIDA boronate 15h as a pale yellow solid (24.6 mg, 60%). For characterization of 15h see ref. 11.
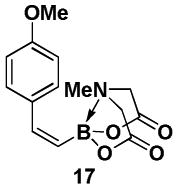
4.19. MIDA boronate 17
Preparation of catalyst solution
In a glovebox, to a 7 mL vial equipped with a stir bar and charged with Pd2(dba)3 (9.7 mg, 0.01 mmol) and XPhos (19.2 mg, 0.04 mmol) was added THF (2 mL). The solution was stirred at 23 °C for 10 min.
The freshly prepared catalyst solution was used in the following reaction
In a glovebox, to a 7 mL vial equipped with a stir bar and charged with MIDA boronate (Z)-Br-1 (50.0 mg, 0.19 mmol) and boronic acid 16 (47.2 mg, 0.31 mmol) was added finely ground K3PO4 (131.0 mg, 0.62 mmol). The prepared catalyst solution was added in one portion. The vial was sealed with a cap and removed from the glovebox. The solution was stirred in a subdued light environment at 35 °C for 47.5 hr. The reaction mixture was filtered through a pad of Celite, concentrated in vacuo, dry loaded onto Celite from an acetone solution, and purified via flash chromatography on silica gel (Et2O:acetone 100:0 → 3:1) to afford MIDA boronate 17 as a pale yellow solid (52.4 mg, 95%).
1H NMR (500 MHz, CD3CN) δ 7.34 (d, J = 8.5 Hz, 2H), 7.08 (br d, J = 15 Hz, 1H), 6.85 (d, J = 8.5 Hz, 2H), 5.53 (d, J = 15 Hz, 1H), 3.89 (d, J = 17 Hz, 2H), 3.77 (s, 3H), 3.69 (d, J = 17 Hz, 2H), 2.86 (s, 3H). 13C NMR (125 MHz, CD3CN) δ 169.0, 159.9, 144.5, 132.5, 130.9, 114.2, 62.5, 55.8, 47.3. HRMS (ESI+) calculated for C14H17BNO5 (M+H)+: 290.1200. Found: 290.1201.
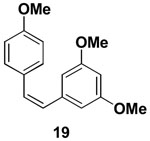
4.19. (Z)-3,5,4′-trimethoxystilbene 19
Preparation of catalyst solution
In a glovebox, to a 7mL vial charged with SPhos (6.5 mg, 0.02 mmol) and Pd(OAc)2 (2.1 mg, 0.009 mmol) was added THF (1.4 mL). The solution was stirred at 23 °C for 10 minutes.
The freshly prepared catalyst solution was used in the following reaction
In a glovebox, to a 7 mL vial equipped with a stir bar and charged with MIDA boronate 17 (39.2 mg, 0.14 mmol) and 1-bromo-3,5-dimethoxybenzene 18 (26.1 mg, 0.12) was added the prepared catalyst solution in one portion. The vial was sealed with a PTFE-lined septum screw-cap and removed from the glovebox. Under a positive pressure of Ar, 1 M aq. NaOH (0.40 mL) was added via syringe. The solution was stirred in a subdued light environment at 23 °C for 6 hr. The reaction mixture was poured into 1 M aq. phosphate buffer pH 7 (2 mL) and extracted with Et2O (3 × 2 mL). The combined organic phases were dried over MgSO4, filtered, and concentrated in vacuo. The crude material was dry loaded onto Celite from an acetone solution and purified via flash chromatography on Florisil (petroleum ether:CH2Cl2 100:0 → 80:20) to afford the 19 as a pale yellow oil (28.3 mg, 76%). Spectral data for 19 were consistent with those previously reported.xxix
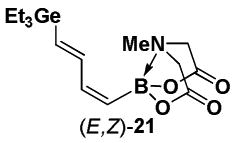
4.20. MIDA boronate (E,Z)-21
The general Stille coupling procedure was followed using (Z)-Br-1 (52 mg, 0.20 mmol), (E)-18 (105 mg, 0.22 mmol), Pd2(dba)3 (9.2 mg, 0.01 mmol), Ph3As (6.1 mg, 0.02 mmol), THF (0.5 mL), and DMF (1.5 mL). The resulting mixture was stirred at 0 °C for 2 hr and then slowly warmed to 23 °C and stirred for an additional 12 hr at 23 °C. Purification via flash chromatography on Florisil (EtOAc:petroleum ether 1:1 → EtOAc → EtOAc:MeCN 9:1) afforded (E,Z)-19 as a white solid (59 mg, 81%). Spectral data for (E,Z)-19 were consistent with those previously reported from our laboratories.11
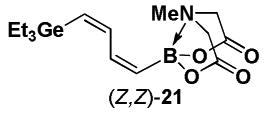
4.21. MIDA boronate (Z,Z)-21
The general Stille coupling procedure was followed using (Z)-Br-1 (52 mg, 0.20 mmol), (Z)-18 (105 mg, 0.22 mmol), Pd2(dba)3 (9.2 mg, 0.01 mmol), Ph3As (6.1 mg, 0.02 mmol), THF (0.5 mL), and DMF (1.5 mL). The resulting mixture was stirred at 0 °C for 2 hr and then slowly warmed to 23 °C and stirred for an additional 18 hr at 23 °C. Purification via flash chromatography on Florisil (EtOAc:petroleum ether 1:1 → EtOAc → EtOAc:MeCN 9:1) afforded (Z,Z)-19 as a pale yellow solid (51.8 mg, 71%). The 1H NMR of the crude product indicated 10% (Z,E)-19 as a byproduct which was inseparable by normal phase Florisil chromatography. Spectral data for (Z,Z)-19 were consistent with those previously reported from our laboratories.11
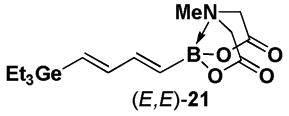
4.22. MIDA boronate (E,E)-21
The general Stille coupling procedure was followed using (E)-Br-1 (52 mg, 0.20 mmol), (E)-18 (105 mg, 0.22 mmol), Pd2(dba)3 (9.2 mg, 0.01 mmol), Ph3As (6.1 mg, 0.02 mmol), THF (0.5 mL), and DMF (1.5 mL). The resulting mixture was stirred at 0 °C for 2 hr and then slowly warmed to 23 °C and stirred for an additional 8 hr at 23 °C. Purification via flash chromatography on silica gel (EtOAc:petroleum ether 1:1 → EtOAc → EtOAc:MeCN 9:1) afforded (E,E)-19 as a white solid (59.5 mg, 81 %). Spectral data for (E,E)-19 were consistent with those previously reported from our laboratories.11
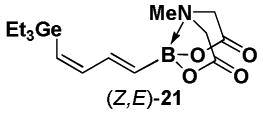
4.23. MIDA boronate (Z,E)-21
The general Stille coupling procedure was followed using (E)-Br-1 (39 mg, 0.15 mmol), (Z)-18 (79 mg, 0.17 mmol), Pd2(dba)3 (7.0 mg, 0.01 mmol), Ph3As (4.6 mg, 0.02 mmol), THF (0.5 mL), and DMF (1.5 mL). The resulting mixture was stirred at 0 °C for 2 hr and then slowly warmed to 23 °C and stirred for an additional 12 hr at 23 °C. Purification via flash chromatography on Florisil (EtOAc:petroleum ether 1:1 → EtOAc → EtOAc:MeCN 9:1) afforded (Z,E)-19 as a pale yellow solid (43 mg, 78%). Spectral data for (Z,E)-19 were consistent with those previously reported from our laboratories.11
Acknowledgments
We gratefully acknowledge the NIH for funding (R01 GM080436). E.M.W. is an NSF Graduate Fellow and M.D.B. is an Early Career Scientist of the Howard Hughes Medical Institute.
Footnotes
Dedicated to Professor Dean Toste on his receipt of the Tetrahedron Young Investigator Award.
References
- i.a) Merrifield RB. Angew Chem, Int Ed Engl. 1985;24:799–810. [Google Scholar]; b) Caruthers MH. Science. 1985;230:281–285. doi: 10.1126/science.3863253. [DOI] [PubMed] [Google Scholar]; c) Plante OJ, Palmacci MR, Seeberger PH. Science. 2001;291:1523–-527. doi: 10.1126/science.1057324. [DOI] [PubMed] [Google Scholar]; d) For the iterative cross-coupling based synthesis of oligophenylacetylenes, see: Zhang J, Moore JS, Xu Z, Aguirre RA. J Am Chem Soc. 1992;114:2273.
- ii.a) Fischbach MA, Walsh CT. Chem Rev. 2006;106:3468–3496. doi: 10.1021/cr0503097. [DOI] [PubMed] [Google Scholar]; b) Garret, Grisham . Biochemistry. Saunders College Publishing; 1995. [Google Scholar]
- iii.Gillis EP, Burke MD. Aldrichimica Acta. 2009;42:17–27. [PMC free article] [PubMed] [Google Scholar]
- iv.Gillis EP, Burke MD. J Am Chem Soc. 2007;129:6716–6717. doi: 10.1021/ja0716204. [DOI] [PubMed] [Google Scholar]
- v.Lee SJ, Gray KC, Paek JS, Burke MD. J Am Chem Soc. 2008;130:466–468. doi: 10.1021/ja078129x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- vi.Gillis EP, Burke MD. J Am Chem Soc. 2008;130:14084–14085. doi: 10.1021/ja8063759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- vii.Uno BE, Gillis EP, Burke MD. Tetrahedron. 2009;65:3130–3138. [Google Scholar]
- viii.Knapp DM, Gillis EP, Burke MD. J Am Chem Soc. 2009;131:6961–6963. doi: 10.1021/ja901416p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ix.Struble JR, Lee SJ, Burke MD. Tetrahedron. 2010;66:4710–4718. [Google Scholar]
- x.Woerly EM, Cherney AH, Davis EK, Burke MD. J Am Chem Soc. 2010;132:6941–6943. doi: 10.1021/ja102721p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- xi.Lee SJ, Anderson TM, Burke MD. Angew Chem Int Ed. 2010;49:8860–8863. doi: 10.1002/anie.201004911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- xii.Ballmer SG, Gillis EP, Burke MD. Org Syn. 2009;86:344–359. [Google Scholar]
- xiii.Dick GR, Knapp DM, Gillis EP, Burke MD. Org Lett. 2010;12:2314–2317. doi: 10.1021/ol100671v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- xiv.Warren CB, Malec EJ. Science. 1972;176:277–279. doi: 10.1126/science.176.4032.277. [DOI] [PubMed] [Google Scholar]
- xv.Mancilla T, Contreras R, Wrackmeyer B. J Organomet Chem. 1986;307:1–6. [Google Scholar]
- xvi.www.aldrich.com/mida
- xvii.Dictionary of Natural Products. Vol. 7. Chapman & Hall; London: 1994. [Google Scholar]
- xviii.Takeda T. Modern Carbonyl Olefination: Methods and Applications. Wiley-VCH; Weinheim: 2004. [Google Scholar]
- xix.a) Brown HC, Subrahmanyam C, Hamaoka T, Ravindran N, Bowman DH, Misumi S, Unni MK, Somayaji V, Bhat NG. J Org Chem. 1989;54:6068–6075. [Google Scholar]; b) Morrill C, Grubbs RH. J Org Chem. 2003;68:6031–6034. doi: 10.1021/jo0345345. [DOI] [PubMed] [Google Scholar]
- xx.Likhtenshtein G. Stilbenes: Applications in Chemistry, Life Sciences and Material Science. Wiley-VCH; Weinheim: 2010. [Google Scholar]
- xxi.Cushman M, Nagarathnam D, Gopal D, Chakraborti AK, Lin CM, Hamel E. J Med Chem. 1991;34:2579–2588. doi: 10.1021/jm00112a036. [DOI] [PubMed] [Google Scholar]
- xxii.Schneider Y, Chabert P, Stutzmann J, Coelho D, Fougerousse A, Gosse F, Launay JF, Brouillard R, Raul F. Int J Cancer. 2003;107:189–196. doi: 10.1002/ijc.11344. [DOI] [PubMed] [Google Scholar]
- xxiii.David-Quillot F, Thibonnet J, Marsacq D, Abarbri M, Duchêne A. Tetrahedron Lett. 2000;41:9981–9984. [Google Scholar]
- xxiv.a) Oda H, Morizawa Y, Oshima K, Nozaki H. Tetrahedron Lett. 1984;25:3221–3224. [Google Scholar]; b) Schwier T, Gevorgyan V. Org Lett. 2005;7:5191–5194. doi: 10.1021/ol0521026. [DOI] [PubMed] [Google Scholar]
- xxv.a) Igarashi S, Ogata K, Miyake A. Jpn J Antibiotics, Ser B. 1956;9:79–80. [Google Scholar]; b) Sowiński P, Gariboldi P, Czerwiński A, Borowski E. J Antibiot. 1989;42:1631–1638. doi: 10.7164/antibiotics.42.1631. [DOI] [PubMed] [Google Scholar]
- xxvi.Pangborn AB, Giardello MA, Grubbs RH, Rosen RK, Timmers FJ. Organometallics. 1996;15:1518. [Google Scholar]
- xxvii.Still WC, Kahn M, Mitra A. J Org Chem. 1978;43:2923. [Google Scholar]
- xxviii.Martin R, Buchwald SL. Acc Chem Res. 2008;41:1461–1473. doi: 10.1021/ar800036s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- xxix.Sore HF, Blackwell DT, MacDonald SJF, Spring DR. Org Lett. 2010;12:2806–2809. doi: 10.1021/ol100895d. [DOI] [PubMed] [Google Scholar]