

Pentannulation of aromatic rings has emerged as an important reaction in the arsenal of the synthetic organic chemist. Several methods have been developed to achieve this transformation, most notably, the Nazarov and Karpf–Drieding reactions, which form 1-indanones.1,2 These processes commonly employ either strong protic or Lewis acids that eschew acid-labile functionality or require prohibitively high temperatures. Alternatively, 2-indanones may be prepared via C–H insertion from α-diazo ketone precursors, which are not ideal substrates because of their acid-, photo-, and thermal-lability.3 Due to these limitations, there remains a need for catalytic processes that effect pentannulations from readily available and robust starting materials.4
We are interested in the synthesis of highly functionalized indenes as intermediates in natural product synthesis and envisioned a strategy that employs readily obtained propargylic esters for this purpose (Scheme 1). The sequence would involve an initial, potentially reversible metal-catalyzed 5-exo-dig cyclization of an ester carbonyl (see 1) onto the alkyne to provide 2, followed by C–O bond cleavage to afford the metallo–carbenoids 3a/b.5 At this stage, preferential irreversible C–H insertion into the aryl moiety of 3b would then yield 4.6
Scheme 1.

Over the past decade, increasing attention has been paid to the activation of alkynes using transition metal complexes to generate metallo–carbenoid intermediates.7,8 The reactivity of the resulting metallo–carbenoid in the majority of these systems is found to be heavily dependent on the identity of the metal. In this communication, we report the development of a pentannulation of propargylic esters, which we believe proceeds via Pt–carbenoid intermediates.9 This methodology displays excellent generality and provides efficient access to important substructures for complex molecule synthesis.
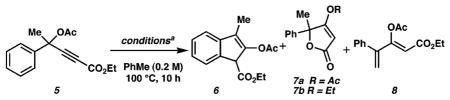
Our studies began with propargylic acetate 5, easily prepared from acetophenone in two steps (see Supporting Information). On the basis of ample literature precedent6 for the generation of metallo–carbenoids from alkynyl precursors, we examined the reaction of several transition metal complexes with this substrate. Various temperature (60–120 °C), solvent (1,2-dichloroethane, MeCN, PhH, PhMe), and catalyst (IrCl(CO)(PPh3)2, RhCl(PPh3)3, Pd(OAc)2, Rh2(OAc)4, Rh2(TFA)4, [RhCl(CO)2]2) combinations resulted in either no discernible reaction, decomposition, or complex mixtures of products, of which only trace amounts could be attributed to 6,10 7a/b, and 8 (Table 1).
Table 1.
Optimization Studies
| |||
|---|---|---|---|
| entry | catalyst | additives | product ratio (6:7:8)b |
| 1 | [RuCl2(CO)3]2 | 1:7:0 | |
| 2 | PtCl2 | 3:0:2 | |
| 3 | PtCl2(PPh3)2 | no reaction | |
| 4c | PtCl2(PPh3)2 | air | >9:0:1 |
| 5 | PtCl2(PPh3)2 | p-benzoquinone | trace |
| 6 | PtCl2(PPh3)2 | PhI(OAc)2 | 4:0:1 |
| 7 | PtCl2(PPh3)2 | PhIO | >9:0:1 |
All reactions were run with 10 mol % catalyst loading and 20 mol % additive.
Product ratios are based on integration of 1H NMR resonances.
Reaction was allowed to cool to 23 °C every 3 h and analyzed by TLC, which resulted in some exposure to air.
Interestingly, the use of [RuCl2(CO)3]2 as catalyst led to the formation of 7a and 7b as major products (entry 1).11 It is important to note that this catalyst, which was employed in a single, isolated example of indene formation from a propargylic carboxylate by Uemura et al.,9 proved to be unproductive for our desired transformation. On the other hand, PtCl2 led to a significant increase in the amount of the desired indene 6 along with diene 8 as the major byproduct (entry 2).12 Despite this initial success, solvent, temperature, and additive variations did not provide any noticeable improvements. We reasoned that the introduction of ligands on the Pt metal center might modulate the reactivity of the catalyst and perhaps improve selectivity. This notion led us to use commercially available PtCl2(PPh3)2. Disappointingly, this catalyst led to a complete recovery of starting material under the reaction conditions (PhMe, 100 °C, 24 h, entry 3) when the reaction was left undisturbed during this period. To our surprise, repeated exposure of the reaction mixture to air (during TLC analysis) led to almost exclusive formation of the desired indene product. On the basis of a hypothesis that oxidation, presumably of the phosphine ligands or metal, plays a key role in the generation of an active catalyst, we studied the addition of several common oxidants (entries 5–7) to the reaction mixture, from which iodosobenzene emerged as the optimal reagent.13 As far as we know, this constitutes the first report of the use of PtCl2(PPh3)2 to activate alkynes. As shown in Figure 1, the desired pentannulation products are easily obtained utilizing our optimized reaction conditions (10 mol % of PtCl2(PPh3)2, 20 mol % of PhIO, 0.2 M in PhMe, 100 °C, 8 h). Methyl and ethyl esters as well as acetate and benzoate propargylic esters readily participate in this reaction. Substitution of other groups (e.g., alkyl, phenyl, or silyl) for esters at the terminus of the alkyne did not lead to productive reactivity, pointing to the necessity of an electron-deficient alkyne for this transformation. Several substitution patterns and aromatic and heteroaromatic nuclei are tolerated as substrates and provide good yields of the desired products.
Figure 1.

Pt-catalyzed cyclization of propargylic esters. For a full description of reaction details, including the identity of propargylic ester substrates, see Supporting Information. Products were obtained as a mixture of olefin regioisomers (≥17:3 ratio). In each case, the major product is shown.
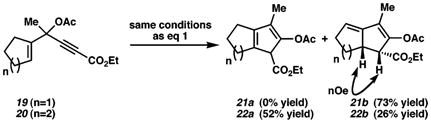
We have also explored nonaromatic-based substrates; in one case, a cyclopentadiene (18) is formed as the sole product from propargylic acetate 17 in good yield (eq 1). In contrast, the related substrate 19 yields 21b14 exclusively via a presumed allylic C–H insertion, while 20 provides isomeric products 22a and 22b via competitive formal allylic and vinylic insertion (eq 2).15
 |
(1) |
 |
(2) |
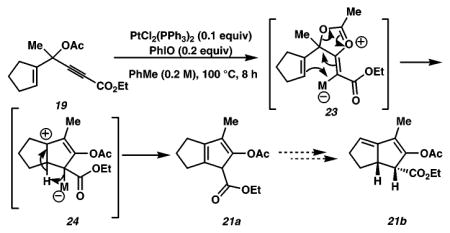
In a preliminary study to identify the active catalyst in these reactions, we have undertaken a series of 31P NMR and preparative experiments. NMR spectroscopic studies were performed in 1,2-dichloroethane-d4 (1,2-DCE-d4),16 in which PtCl2(PPh3)2 was found to be completely soluble. The starting complex displays an apparent triplet centered at δ 15.7 (J = 1836 Hz). Upon treatment with PhIO at 100 °C, a signal that corresponds to OPPh3 (δ 28.4) as well as other downfield signals (δ 37.9, 42.8, 43.9, and 72.2) was observed, suggestive of further oxidation of the Pt–metal center. On the basis of these findings, we were prompted to consider Pt(IV) catalysts and additive combinations that might mirror our in situ generated catalytic species.17 Thus, treatment of 5 with 10% PtCl4 as a catalyst (0.2 M in PhMe, 100 °C, 5 h) results in a conversion to the desired indene product 6 in 35% yield, along with several unidentified byproducts. Interestingly, the addition of PPh3 (20 mol %) completely inactivates the catalyst, resulting in no reaction.18 On the other hand, introduction of 20 mol % of OPPh3 does not impart any deleterious effects to the reaction and provides the desired indene products in 42% isolated yield. These preliminary investigations suggest the possible intermediacy of a Pt(IV) species as the active catalyst.19 Furthermore, our studies point to a clear advantage in utilizing the moisture tolerant and robust PtCl2(PPh3)2 as a precatalyst (compared to PtCl4) to effect a high-yielding conversion of propargylic esters (e.g., 5) to highly functionalized pentannulated compounds.
In conclusion, we have developed an efficient method for pentannulation using in situ generated Pt–carbenoid intermediates that arise from readily available propargylic esters. In addition, we have demonstrated that PtCl2(PPh3)2 is a practical precatalyst to effect these transformations. Further studies to probe the mechanism of these transformations, unambiguously identify the catalytically active species, and broaden the scope of these reactions are currently ongoing and will be reported in due course.
Acknowledgments
The authors are grateful to UC Berkeley, GlaxoSmithKline, and Eli Lilly for financial support, and Dr. H. van Halbeek for extensive NMR assistance. We also thank the Bartlett, Toste, Trauner, Bergman, Francis, and Ellman labs for chemicals and pertinent discussions.
Footnotes
Supporting Information Available: Experimental details and characterization data for all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.For reviews, see: Habermas KL, Denmark SE, Jones KL. Org React. 1994;45:1–158. (Nazarov)Karpf M, Dreiding AS. Helv Chim Acta. 1979;62:852–865. (Karpf–Dreiding)
- 2.For recent examples of 1-indanone synthesis, see: Shintani R, Okamoto K, Hayashi T. J Am Chem Soc. 2005;127:2872–2873. doi: 10.1021/ja042582g.Yamabe H, Mizuno A, Kusama H, Iwasawa N. J Am Chem Soc. 2005;127:3248–3249. doi: 10.1021/ja042581o.
- 3.For an example, see: Nakatani K. Tetrahedron Lett. 1987;28:165–166.
- 4.For recent examples, see: Nakamura I, Bajracharya GB, Wu H, Oishi K, Mizushima Y, Gridnev ID, Yamamoto Y. J Am Chem Soc. 2004;126:15423–15430. doi: 10.1021/ja044603c.
- 5.The C–O bond cleavage may not be a required step as the product may be envisioned to arise directly from 2.
- 6.Propargylic esters as substrates to access carbenes and metallo–carbenoids have been previously proposed. See: Frey LF, Tillyer RD, Ouellet SG, Reamer RA, Grabowski EJJ, Reider PJ. J Am Chem Soc. 2000;122:1215–1216.Mamane V, Gress T, Krause H, Fürstner A. J Am Chem Soc. 2004;126:8654–8655. doi: 10.1021/ja048094q.Harrak Y, Bi-aszykowski C, Benard M, Carlou K, Manetti E, Mouriés V, Dhimane A-L, Fensterbank L, Malacria M. J Am Chem Soc. 2004;126:8656–8657. doi: 10.1021/ja0474695.
- 7.For a recent review, see: Nevado C, Echavarren AM. Synthesis. 2005:167–182.
- 8.For seminal contributions, see: Rautenstrauch V. J Org Chem. 1984;49:950.Mainetti E, Mouriés V, Fensterbank L, Malacria M, Marco-Contelles J. Angew Chem, Int Ed. 2002;41:2132–2135.
- 9.A single isolated example of indene synthesis from a propargylic carboxylate via a Ru–carbenoid has been reported. See: Miki K, Ohe K, Uemura S. J Org Chem. 2003;68:8505–8513. doi: 10.1021/jo034841a.
- 10.Obtained as a 17:3 ratio of olefin isomers, major is shown. This mixture was saponified to give the β-ketoester exclusively (see Supporting Information).
- 11.Optimization and mechanistic studies of this transformation are underway.
- 12.For related precedent, see: Cariou K, Mainetti E, Fensterbank L, Malacria M. Tetrahedron. 2004;60:9745–9755.
- 13.Attempts to prepare a complex of PtCl2 and Ph3PO in situ in various solvents did not lead to any improvements in yield as compared to PtCl2 alone. Other oxidants that were tried include NaIO4, I2, and NMO.
- 14.A single diastereomer, established by nOe to be that shown (see Supporting Information), was obtained.
-
15.Recently, an elegant study describing the synthesis of nonaromatic compounds related to eqs 1 and 2 was reported. See: Shi X, Gorin DJ, Toste FD. J Am Chem Soc. 2005;127:5802–5803. doi: 10.1021/ja051689g.The insightful mechanistic hypothesis put forth in this account cannot be ruled out for the formation of 18, 21, or 22 and may also explain the formation of the other pentannulated products. However, the formation of 21b (via 23 and 24), for example, would require a seemingly unlikely isomerization of cyclopentadiene 21a to the diene 21b.
- 16.Propargylic ester substrates are readily converted under our reaction conditions in 1,2-DCE to the desired products, albeit in lower isolated yields compared to reactions performed in PhMe.
- 17.PtCl4 exhibits catalytic activity similar to that of PtCl2 and may even be more efficient. See: Fürstner A, Stelzer F, Szillat H. J Am Chem Soc. 2001;123:11863–11864. doi: 10.1021/ja0109343.Kobayashi S, Kakumoto K, Sugiura M. Org Lett. 2002;4:1319–1322. doi: 10.1021/ol0256163.Pastine SJ, Youn SW, Sames D. Org Lett. 2003;5:1055–1058. doi: 10.1021/ol034177k. and references therein.
- 18.Presumably, PPh3 outcompetes the alkyne substrate for open coordination sites on the metal center.
- 19.The oxidation state of the active Pt species is still under investigation.