

Abstract
It is estimated that one third of the human population is infected with Mycobacterium tuberculosis. Efforts to understand the molecular basis of its gene regulation have been focused on identification of protein encoding genes and regulons implicated in pathogenesis. Recently, a number of studies have described the identification of several non-coding RNAs that are likely to contribute significantly to the regulatory networks responsible for adaptation and virulence in M. tuberculosis. We have reviewed emerging information on the presence and abundance of different types of non-coding RNA in M. tuberculosis and consider their potential contribution to the adaptive responses that underlie disease pathogenesis.
Keywords: Mycobacterium tuberculosis, non-coding RNA, pathogenesis, persistence
Introduction
Mycobacterium tuberculosis is regarded as one of the most successful human pathogens. It is responsible for more deaths than any other microorganism, and moreover, one third of the global population is showing immunological evidence of past or current infection. This occurs predominantly in the form an asymptomatic “latent” infection, resembling carriage of a commensal organism, and only a minority of individuals develop the progressive lung disease that is necessary for maintenance of the transmission cycle. In contrast to well-characterized enteric pathogens that rely on the acquisition and expression of specialized “virulence factors,” pathogenesis of M. tuberculosis depends on an ability to adjust to changing environments within the host and, under appropriate circumstance, to transmit signals that subvert the immune response in order to cause localized immunopathology.1
Efforts to understand the molecular basis of mycobacterial pathogenesis, with the aim of identifying interventions that prevent or resolve disease, focus on understanding of the ability of the bacteria to modulate their metabolism and replication in response to different environmental signals. The 4.4 megabase genome of M. tuberculosis encodes 13 sigma factors and more than 100 annotated transcriptional regulators,2 and an extensive literature documents transcriptional changes in response to environmental stresses imposed in vitro, during macrophage infection, and in human disease (e.g., refs 3–5).
In this article, we will review emerging information on the presence of non-coding RNA in M. tuberculosis and consider its potential contribution to the adaptive responses that underlie disease pathogenesis and persistence.
Different types of non-coding RNA in bacteria
Application of high-density tiling arrays and RNA sequencing technologies (RNA-seq) have uncovered an extensive and previously unknown repertoire of non-coding RNA in bacteria, including 5′ and 3′ untranslated regions (UTRs), antisense transcripts, and intergenic small RNAs (sRNAs), reviewed in e.g., refs 6 and 7. This is also the case in M. tuberculosis.8
The 5′ UTR, refers to the region between the transcriptional and the translational start sites. This part of the transcript can in some cases change its conformation in response to external stimuli such as changes in temperature9 or the availability of certain metabolites, in which case the RNA element is referred to as a riboswitch.10 The change in conformation in turn leads to a change in the expression of the downstream gene either by blocking/unblocking of the ribosome binding site or by transcriptional termination/antitermination.11 An intriguing finding is that the riboswitch moiety of the SAM-IV riboswitch from Listeria monocytogenes can act in trans as an sRNA thereby adding further complexity to the regulation by riboswitches.12 Whether this applies to other riboswitches or perhaps attenuated transcripts in general remains to be determined.
sRNAs are, as the name indicates small transcripts, mostly in the range of 50 to 250 nucleotides, and they can be encoded opposite open reading frames (antisense or cis-encoded) or between open reading frames (intergenic or trans-encoded).
The majority of sRNAs regulate gene expression by base-pairing to one or more target mRNAs thereby modifying translation efficiency and/or mRNA stability, reviewed in refs 13 and 14. One widespread exception is 6S RNA, which binds to RNA polymerase thereby specifically downregulating the usage of σ70 promoters.15 Other examples of protein binding sRNAs include CsrB and CsrC, which both bind to the carbon storage regulatory protein, CsrA (reviewed in ref. 16).
Although sRNAs are generally referred to as regulatory and/or non-coding RNAs, a few have been found to encode small peptides as well as acting on RNA level.17 Other small intergenic transcripts appear to be acting purely as templates for the synthesis of small peptides.18,19
The RNA chaperone Hfq
In Gram negative bacteria, including many pathogens the RNA chaperone Hfq is required to facilitate the interaction between trans-encoded sRNAs and their targets.
Deletion of the hfq gene often leads to loss of virulence, which may in part be due to loss of proper sRNA function, reviewed in refs 20 and 21. However, the role of Hfq in the Gram positive Staphylococcus aureus is more controversial,22,23 and the protein is completely absent in several pathogens including M. tuberculosis.20 This lack of Hfq represents a limitation in extrapolation of experimental approaches as well as sRNA mechanisms from other bacterial systems both in terms of sRNA-mRNA interaction, but also in terms of the subsequent fate of transcripts via engagement with the degradosome, which is mediated by Hfq.24
The obvious question is, whether M. tuberculosis sRNAs can and do interact with their targets unaided or whether in M. tuberculosis there is one or more alternative, hitherto unidentified chaperones. The high GC content (67%) as well as the structure of intrinsic terminators in M. tuberculosis (see below), limits the frequency of AU-rich stretches implicated in conventional Hfq interactions.25 Nevertheless, an alternative chaperone could be considered an appropriate Hfq analog if it were to display a broad, albeit different sequence specificity toward the majority of sRNAs and their targets. One candidate for this type of chaperone is Rv2367. This protein is a homolog of the Sinorhizobium meliloti YbeY protein that has been shown to display certain Hfq functions,26 although the low level expression of Rv2367 contrasts with the abundance of Hfq in Escherichia coli.8,27
Alternatively (or in parallel) one could imagine a range of regulon-specific chaperones, each of which could interact with a subset of sRNAs and their targets in a manner similar to the FbpABC proteins in Bacillus subtilis.28 Finally there is the possibility that sRNA function in M. tuberculosis is independent of chaperones. But what are the pros and cons of riboregulation without RNA chaperones? On one hand the nucleotide composition of M. tuberculosis sRNAs will in all likelihood lead to structures that have a significantly higher degree of intra-molecular stability, which are more difficult to disrupt and these would therefore impair the interaction with target RNAs. On the other hand it is also possible that a higher proportion of G and C nucleotides in the seed sequence will facilitate the interaction between sRNA and target. Supporting this notion is the fact that some M. tuberculosis sRNAs are predicted to harbour unpaired C-rich stretches that would make for strong first interactions.29
Future investigations, in which tagged sRNAs are used as bait for the isolation of RNA-binding proteins and hence putative chaperones30 will hopefully shed more light on these questions in near future.
sRNAs and pathogenesis
While functional characterization of intergenic sRNAs has only recently been initiated in M. tuberculosis, numerous sRNAs have been characterized in other pathogens. Many of these sRNAs are induced by stress and hence associated with host adaptation and virulence, reviewed in refs 6, 21 and 31. In some cases sRNAs enhance their effect by regulating regulators such as the Qrr1–4 sRNAs, which regulate HapR in Vibrio cholera,32 or ArcZ, which regulates the expression of RpoS in both E. coli and Salmonella.33,34 Other sRNAs that are intimately related to pathogenesis and survival within the host are RybB and MicA, which upon acid stress promote the degradation of several outer membrane protein (OMP) mRNAs in Salmonella. This leads to a rapid rearrangement of the outer membrane, and hence the host-pathogen interface until a new homeostasis has been achieved.35 In spite of the close relationship between stress, host adaptation and pathogenesis, only a fraction of sRNAs has been shown to directly affect virulence in different infection models.21 These include the S. aureus Sprd,36 L. monocytogenes Rli38,37 Legionella pneumophilia ssrS (encoding 6S RNA),38 and the three Salmonella sRNAs IsrM,39 IstR and SroA;40 all of these sRNAs result in impaired virulence upon deletion. Deletion of L. monocytogenes RliB and V. cholerae VrrA lead to increased colonization of spleen and intestines, respectively.37,41
Coding vs non-coding RNA in M. tuberculosis
The identification of putative regulatory sRNAs in M. tuberculosis is so far limited to three studies.8,29,42Figure 1 presents an overview of some of the putative RNA-based regulatory networks in M. tuberculosis.
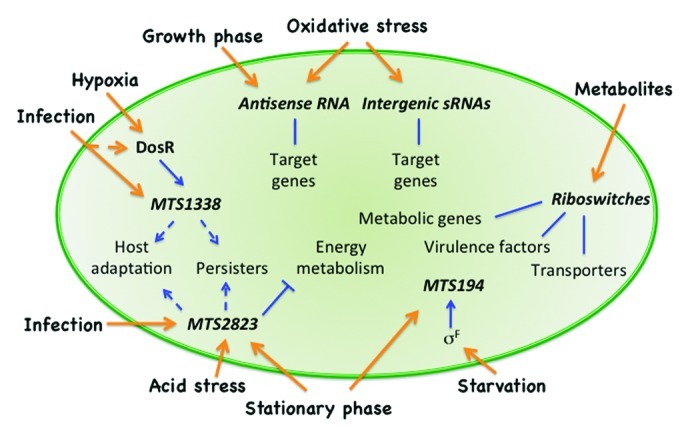
Figure 1. Overview of the potential roles of regulatory RNA in M. tuberculosis. The figure illustrates how different external signals, shown as orange arrows, can regulate either riboswitches, antisense or intergenic RNAs (italics). Known protein regulators, i.e., DosR and SigF as well as selected sRNAs have been indicated with names. Blue lines indicate that the resulting regulation can be either activation or inhibition; full lines indicate known activation/inhibition; dashed arrows indicate possible consequence.
The most recent study is a detailed quantitative analysis of M. tuberculosis transcriptomes from exponential and stationary phases using RNA-seq.8 After removal of signals from rRNAs, 17% of the reads identified from exponentially growing M. tuberculosis H37Rv mapped to intergenic regions, with a further 12% mapping in antisense orientation to coding sequences. Non-coding RNA increased to 58% of total (non-rRNA) reads in stationary phase cultures, largely due to accumulation of a single, highly abundant sRNA transcript, MTS2823 (see below). It is likely that these numbers represent an underestimate of the total non-coding transcriptome, since the protocol used in this study was not optimised for detection of shorter transcripts (< 200 nucleotides), which was reflected by the relatively low ratio of 5S to the larger 16S and 23S rRNA species.8
Not all intergenic transcripts will be non-coding and the distinction between coding and non-coding transcripts is contingent on the methods employed to predict protein-coding transcripts; the outcome varies according to different algorithms that have been used to generate multiple annotations of M. tuberculosis genomes. Identical short open reading frames are more frequently annotated as coding sequences in M. tuberculosis CDC155143,44 than in the closely related M. tuberculosis H37Rv.2 For example, transcription of the sequence annotated as an intergenic region between Rv2395 and Rv2396 in H37Rv has been described as an sRNA (mcr7),42 but it is annotated as two hypothetical proteins, MT2466 and MT2467 in the CDC1551 genome, and was recently reported to encode two small proteins involved in the mycobacterial response to low pH.45 Conversely, an abundant transcript encoded between Rv3661 and Rv3662c has been alternatively annotated as sRNA MTS2823,8 and as hypothetical protein MT3762.44 In this case, the inverse orientation of the transcript with respect to the predicted protein strongly favors the sRNA annotation. Moreover, it is likely that some transcripts will have dual function as both sRNA and mRNA as it has been found in other bacteria.17 Initial predictions of translation start sites are also likely to be subject to revision in light of experimental data, and this will result in reassignment of coding and non-coding portions of mRNA transcripts. Application of mass spectrometry technology for proteome analysis, e.g., refs 46 and 47, will play an important role in resolving these ambiguities.
Antisense RNAs
Bacterial gene regulation by short antisense transcripts that base-pair with 5′ regions of mRNAs has been well characterized for a subset of bacterial genes, including several involved in transposition of foreign genetic elements, reviewed in ref. 48. A surprising finding from several studies using RNA-seq is the extent and heterologous nature of antisense transcriptomes. Thus, an antisense component has been identified for up to 75% of all protein-coding genes, with transcripts ranging in size from less than 50 nucleotides to several kilobases and mapping to sequences throughout the length of cognate mRNAs.49 A similar pattern is observed for M. tuberculosis, where 65% of genes have an antisense component corresponding to ≥ 10% of the coding transcript during exponential growth; a number that increases to > 90% in stationary phase.8 Antisense transcripts have been shown to regulate transcription and translation of coding transcripts by a variety of mechanisms.48 Recently, RNase III-mediated digestion of double strand sequences has been proposed as an attractive model by which antisense transcription could play a pervasive genome-wide role in fine-tuning of expression patterns.49 During exponential growth in M. tuberculosis, all gene classes have some level of antisense RNA, but the gene classes with the lowest occurrence of antisense, are also the ones that are the most highly expressed, i.e., energy metabolism and synthesis of macromolecules.8
Several M. tuberculosis antisense transcripts have been identified by cloning and sequencing from low molecular weight RNA fractions or by RNA-seq, and their sizes as well as locations relative to their cognate open reading frames (ORFs) vary significantly; some are encoded at the 5′ end of the ORF, some in the center and some at the 3′ end, and a few cover an entire ORF or more.8,29,42 Most of the M. tuberculosis antisense RNAs appear to be independent transcripts, but a few are derived from long, overlapping 3′ UTRs (see below).8
PhoPR is a pathogenesis-associated two-component system that may be subject to riboregulation. There is a prominent internal antisense transcript toward the 5′ end of the phoP gene, and the 3′ UTR of Rv0795c generates an antisense transcript that extends over the 3′ end of the converging phoR8 (KBA, TC, DBY unpublished). Similarly, the ino1 gene (Rv0046c) is covered by a mid-ORF antisense transcript, which is significantly downregulated between exponential and stationary phase growth8 (Fig. 2). ino1 encodes an enzyme that catalyzes the first step in inositol synthesis and this gene has been shown to be essential for virulence.50 It is not unlikely that differential expression of either of these transcripts could affect the expression of their cognate mRNAs thereby playing a role in M. tuberculosis pathogenesis.
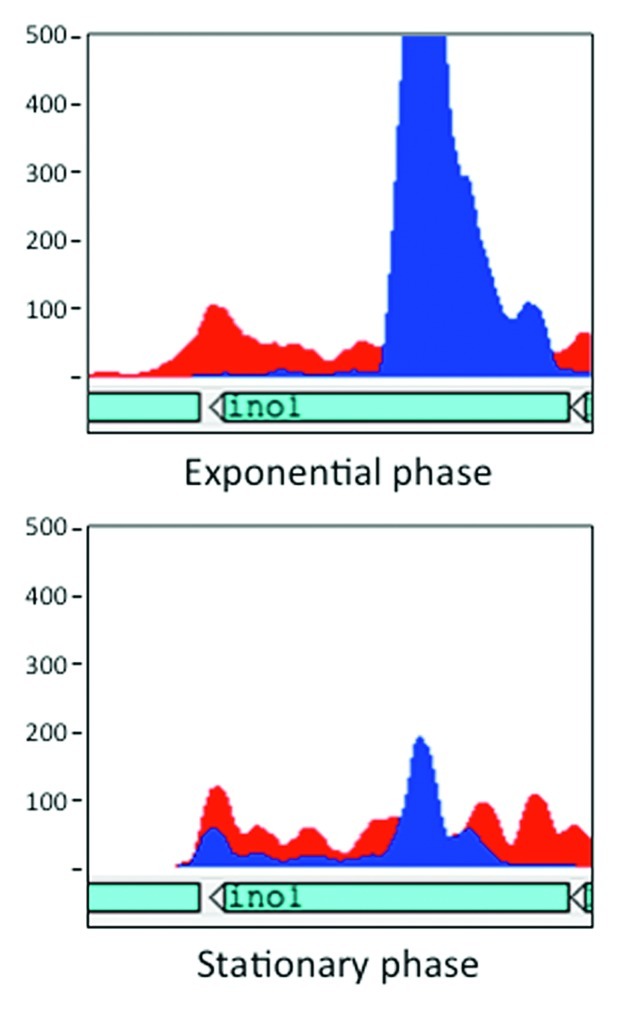
Figure 2. Expression of the ino1 antisense RNA is significantly downregulated in stationary phase. The figure illustrates RNA-seq data visualized with the Artemis genome browser; reads mapping to the plus strand, i.e., antisense to ino1, are shown in red, and reads mapping to the minus strand, i.e., ino1 itself, are shown in blue. Expression levels in the two phases have been adjusted for comparative reasons.
Two antisense RNAs associated with lipid metabolism have the interesting characteristic of the potential to base-pair with multiple mRNAs generated from duplicated gene sequences.29 The first such antisense RNA, ASdes, maps to Rv0842c, encoding an acyl-carrier protein desaturase (DesA1) and it has the potential for base-pairing with the mRNA of a second homologous target mRNA, DesA2 (Rv1094).29 The second example, ASpks, is encoded opposite of pks12 that encodes one of several polyketide synthases. These enzymes are required for biosynthesis of a series of complex lipid molecules that are involved in pathogenesis,51-53 and act as targets for host immune recognition.54 ASpks is a 75-nucleotide transcript that maps to a duplicated module in the 12.5kb pks12 gene (Rv2048). Moreover, it has significant sequence identity with the equivalent modules in pks7, 8 and 15 and hence it could potentially base-pair with these mRNAs as well. During oxidative stress, and to a lesser extent at low pH, an extended 200-nucleotide ASpks transcript is induced.29 Oxidative stress also results in a downregulation of pks12, pks8 and pks15 mRNAs;5 further analysis should clarify if this is due to the increased ASpks levels.
Thus, the extent of the antisense transcriptome suggests that it may represent a common component of gene regulation in M. tuberculosis, with the potential in some cases for cis-regulation of their cognate mRNA partner as well as coordinated trans-regulation of related genes.
3′ and 5′ untranslated regions (UTRs)
Sequences flanking either end of mRNA transcripts make an important contribution to the total complement of non-coding RNA. While it is relatively straightforward to identify the primary 5′ ends of transcripts by RLM-RACE,55 or more globally by exploiting their resistance to digestion by Terminator 5′-phosphate-dependent exonuclease,56 3′ ends are harder to define. A subset of M. tuberculosis mRNAs have well-defined 3′ ends associated with a recently identified mycobacterial terminator, TRIT,57 but the general paucity of L-shaped intrinsic terminators – i.e., a stem-loop followed by a stretch of U residues58 – frustrates bioinformatics approaches to the prediction of 3′ ends in M. tuberculosis. Visual inspection of RNA-seq profiles using a genome browser suggests that many mRNAs have 3′ UTRs extending over more than 50 nucleotides. Long 3′ UTRs stand out in the context of convergent gene pairs when they form a stretch of antisense transcript with potential base-pairing to the adjacent gene. The same phenomenon has been observed, albeit to a lesser extent, in e.g., L. monocytogenes.37 As in the case of the internal antisense transcripts discussed above, understanding of the functional consequence of overlapping 3′ UTRs will require further experimental analysis, but it is intriguing that the set of genes subject to this form of antisense coverage shows a non-random distribution that is skewed toward genes with a shared role in cell wall processes.8 It will be interesting to explore whether the presence of a long 3′ UTR has an influence on mRNA half-life and translation efficiency.
In contrast to 3′ UTRs, which are more commonly associated with mRNA processing in eukaryotic cells, the role of 5′ UTRs has been extensively characterized in bacteria. 5′ UTRs typically provide binding sites for regulatory proteins or small molecules that modulate the efficiency of transcription or translation of the downstream sequence. A well-characterized example is the inhibition of ribosomal protein expression by binding of free ribosomal protein to appropriate 5′ UTRs, and release of inhibition when no free protein is available, reviewed in ref. 59. Although no functional data are available, the presence of analogous 5′ UTRs associated with corresponding genes in M. tuberculosis suggests that similar circuits play an important role in post-transcriptional regulation.8
Riboswitches
A number of riboswitches have been identified in M. tuberculosis by sequence homology;60 one of these, the cobalamin riboswitch has been subject to further investigation.61 This type of riboswitch represses the expression of the downstream gene in the presence of cobalamin (vitamin B12) and is often found upstream of genes involved in the synthesis and/or transport of cobalamin.62 There are two copies of this motif in M. tuberculosis; one is upstream of the metE gene, encoding a B12-independent methionine synthase, and the second is upstream of an operon comprising PPE2, cobQ1 and cobU (Fig. 3). PPE2 belongs to an extensive family of proteins sharing proline-glutamate (PE) or proline-proline-glutamate (PPE) N-terminal motifs that were identified from the M. tuberculosis genome sequence but for the most part lack any obvious biological function, reviewed in ref. 63. The genomic location of PPE2, together with the presence of several transmembrane domains has led to the suggestion that it may be a cobalt transporter.62 The cobalamin riboswitches in M. tuberculosis are predicted to act on the level of translation by masking/unmasking the ribosome-binding site;64 on the other hand, a B12-dependent reduction in metE RNA has been interpreted as regulation on the level on transcription.61 This apparent conflict is possibly due to a destabilization of the metE transcript in the absence of translation.

Figure 3.M. tuberculosis riboswitches and PE-PPE genes; the figure illustrates how riboswitches in M. tuberculosis are sometimes separated from their predicted cognate mRNAs (shown in blue), by the insertion of one or more PE-PPE genes (shown in red). Due to the sequence conservation inherent in these riboswitches, the regions have been annotated as ‘conserved hypotheticals’ (shown in green). Other genes are shown in gray. The top panel shows how the B12 riboswitch is separated from cobQ1 and cobU genes by a PPE2 insertion. The bottom panel shows how the Mbox has been separated from mgtC by several PE-PPE genes.
Another riboswitch motif that is represented twice in the genome of M. tuberculosis but which has not been functionally characterized, is the ykok leader or Mbox, which is usually found in the context of magnesium transporters.65 The Bacillus subtilis Mbox has been shown to attenuate transcription upon binding Mg2+, thereby ensuring magnesium homeostasis.66 In M. tuberculosis one highly expressed Mbox is found upstream of Rv1535, a conserved hypothetical protein that is induced during Mg-starvation.8,67 Rv1535 is followed by and co-transcribed with a downstream Tbox (a riboswitch that binds tRNAs68) that forms the 5′ UTR of the essential isoleucine tRNA synthase mRNA (IleS, Rv1536)8 (KBA, DBY unpublished). The second Mbox is located upstream of a putative operon encoding four PE-PPE genes, a conserved hypothetical protein and a predicted magnesium transporter, MgtC (Fig. 3), all of which are induced by magnesium starvation.67 The fact that the genes downstream of the two Mboxes in M. tuberculosis are induced upon magnesium starvation, strongly suggest that these are in fact functional magnesium-responsive RNA elements, which are activated by the low concentration of magnesium found in macrophages.69
Inspection of M. tuberculosis RNA-seq profiles identifies numerous additional candidate riboswitches expressed as truncated 5′ transcripts8 (KBA, DBY unpublished). One of these is located in the 5′ UTR of Rv0282, the first gene in the ESX3 operon that is essential for virulence and regulated at the level of transcription initiation by iron and zinc.70 In addition to its transcriptional attenuation profile the 5′ UTR contains a highly conserved region, which could constitute a ligand-binding aptamer domain. It is intriguing to note that once again this putative riboswitch is located upstream of an operon containing a PE-PPE gene pair, extending a pattern of PE-PPE gene insertion between riboswitches and their predicted regulated targets. A YdaO riboswitch (candidate) is present upstream of rpfA (Rv0867c) in M. tuberculosis. No ligand has been identified for the YdaO riboswitch though it is often associated with genes involved in processes related to cell wall degradation.71 The rpfA gene product is a member of a family of “resuscitation-promoting factors” that share structural homology with lysozyme, and may play a particularly important role in re-initiation of cell division after periods of non-replication.72-74
These examples suggest that riboswitches have a widespread role in regulation of M. tuberculosis gene expression and growth.
M. tuberculosis intergenic sRNAs and stress
Around 20 M. tuberculosis intergenic sRNAs have been identified and verified by northern blotting.8,29,42 They display varying degrees of conservation; some are restricted to closely related members of the M. tuberculosis complex, others are present in multiple pathogenic mycobacteria with Mycobacterium leprae being the most distant relative, and finally some are conserved in all mycobacteria and even other actinomycetes.8,29,42 The M. tuberculosis sRNAs range in size from ~50 to > 300 nucleotides, they are often differentially expressed in response to changing environments, and a couple have been shown to be lethal upon overexpression. Selected examples are listed in Table 1 using an MTS (“Mycobacterium tuberculosis sRNA”) designation followed by a number corresponding to the intergenic region (i.e., the TIGR website annotation of intergenic regions [http://cmr.jcvi.org/tigr-scripts/CMR/CmrHomePage.cgi].
Table 1. Selected intergenic sRNAs, verified by northern blotting.
| sRNA | Alternative name | Strand | Left CDS | Right CDS | Size* | Regulation** | Ref. |
|---|---|---|---|---|---|---|---|
| MTS0194 |
F6 |
F |
Rv0243 |
Rv0244c |
55, 110 |
SigF, starvation, H2O2, low pH |
29 |
| MTS0479 |
B55 |
F |
Rv0609A |
Rv0610c |
60 |
H2O2 |
29 |
| MTS0823 |
mpr5 |
F |
Rv1051c |
Rv1052 |
120 |
- |
42 |
| MTS0858 |
|
R |
Rv1092c |
Rv1093 |
100 |
- |
8 |
| MTS0997 |
mcr11 |
R |
Rv1264 |
Rv1265 |
120 |
CRP, stationary phase, infection |
8,42 |
| MTS1082 |
|
F |
Rv1373 |
Rv1375 |
130 |
Exponential phase |
8 |
| MTS1310 |
G2 |
R |
Rv1689 |
Rv1690 |
65 |
- |
29 |
| MTS1338 |
|
F |
Rv1733c |
Rv1735c |
120 |
DosR, hypoxia, infection |
8 |
| MTS2774 |
mpr17 |
F |
Rv3596c |
RV3597c |
80, 110 |
- |
42 |
| MTS2822 |
B11 |
R |
Rv3660c |
Rv3661 |
95 |
H2O2 |
28 |
| MTS2823 |
mpr4 |
F |
Rv3661 |
Rv3662c |
305 |
stationary phase, infection, low pH |
8,42 |
| MTS2975 | F | Rv3843c | Rv3844 | 100 | Exponential phase | 8 |
Notes: *Approximate size (nucleotides) of the dominant transcripts as determined by northern blotting; **Regulation refers to relevant factors and conditions where increased expression is observed
The differential expression of sRNAs upon changing growth conditions may give some clues about function in terms of under what conditions a given sRNA may be required, although it will not reveal the entire story.
MTS194, MTS479, MTS2822
Oxidative stress is a potent inducer of stress responses in M. tuberculosis.75 Three of the sRNAs listed in Table 1, namely MTS194 (“F6”29), MTS479 (“B55”29) and MTS2822 (“B11”29) show increased expression in response to H2O2, which mimics the oxidative stress encountered inside the host macrophage, and hence these sRNAs could be associated with intracellular survival during the early stages of infection. MTS194 is encoded in the intergenic region between two genes involved in lipid degradation, Rv0243 and Rv0244c. It is transcribed by the starvation associated alternative sigma factor, SigF and the MTS194 promoter has recently been shown to represent the highest occupancy of this sigma factor suggesting that expression of this sRNA has high priority under certain conditions such as starvation.76 Overexpression of MTS194 leads to reduced growth in M. tuberculosis, but although an MTS194 homolog is present in the distantly related, non-pathogenic Mycobaterium smegmatis, overexpression of MTS194 in M. smegmatis has no obvious phenotype.29
MTS2822
is ~95 nucleotides in size and located in the intergenic region between Rv3660c and Rv3661, both predicted to be involved in cellular differentiation. Downstream of Rv3661 is another sRNA, MTS2823 (see below), and the entire locus is highly conserved among mycobacteria, with the notable exception of M. leprae. MTS2822 contains a so-called 6C motif, consisting of two C rich loops found to be widespread between Actinobacteria.77 The structure of MTS2822 has led to the suggestion that this sRNA may be a structural/protein binding RNA rather than a conventional regulatory sRNA.77 M. smegmatis harbours an sRNA that is > 90% identical to MTS2822 and while overexpression of MTS2822 in M. tuberculosis is lethal, overexpression of MTS2822 in M. smegmatis results in a dramatic phenotype; growth is very poor on solid and in liquid media, and the cells display aberrant morphology including irregular and elongated shape, suggesting association with cell wall synthesis and/or cell division.29
MTS997, MTS1338, MTS2823
Three other sRNAs, MTS997, MTS1338 and MTS2823 accumulate to high levels during the transition from exponential to stationary phase, (representing a mixture of stresses), and to even higher levels during infection, suggesting a role in pathogenesis.8,42 MTS997 and MTS2823 are both expressed during exponential growth while MTS1338 is almost undetectable under the same conditions. The number of MTS1338 molecules, relative to the number of ribosomes, estimated to be around 4000 per cell during fast growth,78 imply that less than 10% of exponentially growing cells express MTS1338. It therefore seems plausible that the few cells expressing this sRNA may be in a different metabolic state than the remaining population and therefore they could represent putative persister cells. Moreover, the induction of MTS1338 seen in stationary phase is almost eliminated by deletion of dormancy regulator, DosR.8 DosR interacts with DosS or DosT to form a phosphorylation-dependent two-component regulatory system activated during growth arrest induced by hypoxia or exposure to nitric oxide.79 The DosR regulon includes genes required for remodelling of protein, lipid and energy metabolism together with members of the universal stress protein family and multiple proteins of unknown function.5 The DosR genes are strongly expressed during infection in mice and in sputum samples from patients, and include several prominent antigens, and their expression has been linked to the generation of drug tolerant persister cells.3,80,81 It remains to be determined if MTS1338 is directly involved in the generation of persister cells or simply a marker for this subpopulation.
The most in-depth functional analysis has been performed on the relatively large (~300-nucleotide) MTS2823 sRNA, which is encoded on the plus strand between Rv3661 and Rv3662c. It is the most highly expressed non-rRNA in exponentially growing cultures, is induced a further 10-fold in stationary phase, and accumulates to a level approaching 1:1 stoichiometry with rRNA in tissues from infected mice.8 MTS2823 is conserved in a broad range of mycobacteria (and other actinomycetes) though not M. leprae.
Constitutive overexpression of MTS2823 in exponential phase cultures results in a slightly reduced growth rate and in a widespread downregulation of energy metabolism genes, analogous to that observed during transition from exponential growth to stationary phase.8 The most marked reduction is seen for genes associated with the methyl citrate cycle in particular prpC (methyl citrate synthase, > 100 fold) and prpD (methyl citrate dehydratase, > 60 fold). This metabolic pathway contributes to detoxification of metabolites generated during catabolism of odd-numbered fatty acids and cholesterol, which has been shown to be an essential carbon source during intracellular growth.82,83
Downregulation of active growth functions during stationary phase transition in bacterial cultures is commonly associated with increased expression of the structurally conserved 6S RNA molecule, that binds to RNA polymerase and inhibits transcription of genes controlled by the principal sigma factor.15,84,85 A bioinformatics search based on matching for energetically suboptimal structures recently identified the M. smegmatis homolog of MTS2823 as a potential 6S RNA candidate, but lack of binding to RNA polymerase led the authors to conclude that it is not a genuine 6S RNA,86 and so far no 6S RNA homolog has been identified in mycobacteria. It remains to be determined if MTS2823 is a conventional sRNA that acts via base-pairing with specific mRNA targets or with protein or whether its mode of action differs entirely from other sRNAs.
M. tuberculosis sRNA networks?
Generally, the roles of individual sRNAs within a cell’s regulatory networks vary substantially. Well-characterized sRNAs can be, and in the case of Salmonella and E. coli have been, assigned to different types of regulatory circuits depending on the interplay between regulators, sRNAs and targets.87 However, in the case of M. tuberculosis this requires a significant increase in the amount of data about individual sRNAs that we are only just beginning to collect. Once the different regulators and targets of M. tuberculosis sRNA have been identified, monitoring the fate of target mRNAs upon deletion or overexpression of sRNAs will help to distinguish between different mechanistic models.
CRISPR
The Clustered Regularly Interspaced Short Palindromic Repeat (CRISPR) locus provides a specialized source for generation of M. tuberculosis sRNAs. The CRISPR locus incorporates sequences from phage and other invading genetic elements and repackages them as sRNA defense molecules that confer resistance to re-infection, reviewed in ref. 88. CRISPRs are found in only a subset of mycobacteria, and the presence of the CRISPR in Mycobacterium canetti suggests acquisition by horizontal transfer prior to final branching of the M. tuberculosis complex. Sporadic deletion of integrated foreign sequence elements from the CRISPR domain has been exploited in a widely used system for differentiation of M. tuberculosis strains referred to as “spoligotyping.”89 Phylogenetically, the M. tuberculosis CRISPR system belongs to the Type III-A family,90 and is related to a well-characterized CRISPR system in Staphylococcus epidermis.91 The Type III-A system is characterized by the presence of Cas10 (Rv2823c) and Csm6 (Rv2818) in the CRISPR Associated Complex for Antiviral Defense (CASCADE), which in M. tuberculosis is likely to target foreign DNA rather than RNA.90 Inspection of RNA-seq profiles shows that the CRISPR domain is transcribed and processed into a series of 50–60 nucleotide sRNAs that are presumably incorporated into a CASCADE effector complex (KBA, TC, DBY unpublished). The transcription start site driving CRISPR sRNA expression overlaps with a divergent start site that generates a long antisense transcript covering Rv2816c and Rv2817c encoding Cas1 and Cas2 proteins involved in incorporation of novel sequences into the CRISPR locus8,92 (KBA, TC, DBY unpublished). This profile is consistent with silencing of the genes required to capture novel CRISPR sRNAs, but functional expression of Cas6 and Cas10 enzymes (i.e., Rv2824c and Rv2823c) required for processing of pre-existing sRNAs. An exception is the sub-lineage of M. tuberculosis known as the Beijing family, which is undergoing a current global expansion in marked association with multiple drug resistance.93 A genomic deletion has removed most of the CRISPR locus and many of the associated Cas genes, eliminating expression of CRISPR sRNAs in these strains.94 A similar deletion has been found in an unrelated sub-lineage referred to as “pseudo-Beijing.”95 It is open to speculation whether the presence of a functional CRISPR system in the majority of M. tuberculosis strains has current biological relevance or reflects fortuitous retention of a defense system appropriate to an ancestral lifestyle.
Implications for pathogenesis and disease control
Progression from documentation of the non-coding transcriptome to elucidation of its biological function will require an extensive program of experimental research. Given the limitations of working with a slow-growing pathogen under strict containment conditions – the timeframe of biologically relevant experiments using M. tuberculosis routinely extends from 6 to 12 mo – it may be important to explore fundamental mechanisms using more tractable organisms and to focus experimental work with M. tuberculosis on aspects of particular relevance to pathogenesis.
It can be envisaged that research on non-coding RNA in M. tuberculosis will contribute in three areas: improved understanding of the regulation of pathways already implicated in pathogenesis, characterization of M. tuberculosis phenotypes relevant to human infection, and hence, although more long-term, improved interventions for disease control.
Improved understanding
Over more than a decade since the sequencing of the M. tuberculosis genome, research in many laboratories has focused on identification of protein encoding genes and regulons implicated in pathogenesis; typically by generation of recombinant strains and subsequent testing of their phenotype in a mouse model of infection. Analysis by genome-wide transposon mutagenesis identified almost 200 protein encoding genes that are dispensable for growth in laboratory culture but are required for infection,52,53 and individual studies have characterized multiple genetic loci involved in control of defense against host antimicrobial systems, adaptation to changing nutritional and physicochemical constraints, and production of macromolecules that modulate interactions with host cells. RNA-seq profiling identifies non-coding RNA elements associated with many of these loci.8
Characterization of M. tuberculosis phenotypes
While mouse infection provides a convenient model to analyze M. tuberculosis growth dynamics in the presence of host innate and adaptive immune cells, it does not recapitulate the more complex biological processes underlying the disease transmission cycle. In humans and non-human primates, infection with M. tuberculosis induces formation of a heterogeneous and dynamic range of highly structured granulomatous lesions that provide a multiplicity of bacterial microenvironments.1 Successful pathogenesis involves an ability to tolerate the resulting unfavorable environments, and to exploit environments that are favorable for bacterial multiplication, access to airways and onward transmission. It is likely that the mouse model interrogates only a portion of the phenotypic repertoire involved in human disease, and characterization of phenotypic states relevant to different stages of human pathology presents a major research challenge. Accumulation of sRNAs linked to different stress regulons may act as sensitive markers of the physiological status of the small numbers of bacteria present in different parts of human and non-human lesions, providing crucial information to support rational targeting of improved therapies.1
Improved interventions
The success of treatment-based strategies for tuberculosis control is limited by the requirement for therapeutic regimens extending over 6 mo or more. Prolonged treatment is needed to clear a residual population of bacteria that persist in a phenotypic condition in which they are tolerant to antibacterial drugs. Identification of drugs that have a cidal effect against persister populations is a key research goal. Phenotypic tolerance can be reproduced in vitro by driving bacteria into a non-replicating state by limitation of oxygen or nutrients. In vitro “persister” models share the characteristic of a widespread downregulation of genes required for active growth,96-98 and it is likely that MTS2823 makes a contribution to this process. An alternative to the environment-induced model is that the persister phenotype is generated spontaneously in a few individuals within an apparently homogeneous bacterial population as a result of stochastic variations in gene expression.99 Mathematical modeling suggests that post-transcriptional regulation could provide an important source of such stochastic variation,100 and measurement of uninduced levels of an sRNA such as MTS1338 does indeed suggest occurrence at less than one copy per cell.
In summary the abundance of non-coding RNA in M. tuberculosis and its association with genes implicated in pathogenesis suggests that post-transcriptional regulation plays a significant role in the intracellular survival of this bacterium as it is the case for many other pathogens. Moreover, it is possible that riboregulation may also be involved in the generation of persister cells that are phenotypically tolerant to conventional antibacterial drugs. Future investigations will hopefully shed more light on this aspect of M. tuberculosis biology.
Acknowledgments
We would like to thank Teresa Cortes for sharing unpublished data. We would also like to thank Finn Werner and Paul Gardner for helpful comments on the manuscript.
Glossary
Abbreviations:
- sRNA
small RNA
- RNA-seq
RNA sequencing
- MTS
M. tuberculosis sRNA
- RLM-RACE
RNA ligase-mediated RACE
- PE
proline-glutamate
- PPE
proline-proline-glutamate
- UTR
untranslated region
- CRISPR
Clustered Regularly Interspaced Short Palindromic Repeat
Footnotes
Previously published online: www.landesbioscience.com/journals/rnabiology/article/20105
References
- 1.Barry CE, 3rd, Boshoff HI, Dartois V, Dick T, Ehrt S, Flynn J, et al. The spectrum of latent tuberculosis: rethinking the biology and intervention strategies. Nat Rev Microbiol. 2009;7:845–55. doi: 10.1038/nrmicro2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cole ST, Brosch R, Parkhill J, Garnier T, Churcher C, Harris D, et al. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature. 1998;393:537–44. doi: 10.1038/31159. [DOI] [PubMed] [Google Scholar]
- 3.Garton NJ, Waddell SJ, Sherratt AL, Lee SM, Smith RJ, Senner C, et al. Cytological and transcript analyses reveal fat and lazy persister-like bacilli in tuberculous sputum. PLoS Med. 2008;5:e75. doi: 10.1371/journal.pmed.0050075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rustad TR, Harrell MI, Liao R, Sherman DR. The enduring hypoxic response of Mycobacterium tuberculosis PLoS One 2008; 3:e1502. [DOI] [PMC free article] [PubMed]
- 5.Schnappinger D, Ehrt S, Voskuil MI, Liu Y, Mangan JA, Monahan IM, et al. Transcriptional Adaptation of Mycobacterium tuberculosis within Macrophages: Insights into the Phagosomal Environment. J Exp Med. 2003;198:693–704. doi: 10.1084/jem.20030846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gripenland J, Netterling S, Loh E, Tiensuu T, Toledo-Arana A, Johansson J. RNAs: regulators of bacterial virulence. Nat Rev Microbiol. 2010;8:857–66. doi: 10.1038/nrmicro2457. [DOI] [PubMed] [Google Scholar]
- 7.Sorek R, Cossart P. Prokaryotic transcriptomics: a new view on regulation, physiology and pathogenicity. Nat Rev Genet. 2010;11:9–16. doi: 10.1038/nrg2695. [DOI] [PubMed] [Google Scholar]
- 8.Arnvig KB, Comas I, Thomson NR, Houghton J, Boshoff HI, Croucher NJ, et al. Sequence-based analysis uncovers an abundance of non-coding RNA in the total transcriptome of Mycobacterium tuberculosis. PLoS Pathog. 2011;7:e1002342. doi: 10.1371/journal.ppat.1002342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Narberhaus F, Waldminghaus T, Chowdhury S. RNA thermometers. FEMS Microbiol Rev. 2006;30:3–16. doi: 10.1111/j.1574-6976.2005.004.x. [DOI] [PubMed] [Google Scholar]
- 10.Nahvi A, Sudarsan N, Ebert MS, Zou X, Brown KL, Breaker RR. Genetic control by a metabolite binding mRNA. Chem Biol. 2002;9:1043. doi: 10.1016/S1074-5521(02)00224-7. [DOI] [PubMed] [Google Scholar]
- 11.Breaker RR. Riboswitches and the RNA world. Cold Spring Harb Perspect Biol. 2012;4:4. doi: 10.1101/cshperspect.a003566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Loh E, Dussurget O, Gripenland J, Vaitkevicius K, Tiensuu T, Mandin P, et al. A trans-acting riboswitch controls expression of the virulence regulator PrfA in Listeria monocytogenes. Cell. 2009;139:770–9. doi: 10.1016/j.cell.2009.08.046. [DOI] [PubMed] [Google Scholar]
- 13.Storz G, Vogel J, Wassarman KM. Regulation by small RNAs in bacteria: expanding frontiers. Mol Cell. 2011;43:880–91. doi: 10.1016/j.molcel.2011.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Waters LS, Storz G. Regulatory RNAs in bacteria. Cell. 2009;136:615–28. doi: 10.1016/j.cell.2009.01.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wassarman KM. 6S RNA: a small RNA regulator of transcription. Curr Opin Microbiol. 2007;10:164–8. doi: 10.1016/j.mib.2007.03.008. [DOI] [PubMed] [Google Scholar]
- 16.Babitzke P, Romeo T. CsrB sRNA family: sequestration of RNA-binding regulatory proteins. Curr Opin Microbiol. 2007;10:156–63. doi: 10.1016/j.mib.2007.03.007. [DOI] [PubMed] [Google Scholar]
- 17.Vanderpool CK, Balasubramanian D, Lloyd CR. Dual-function RNA regulators in bacteria. Biochimie. 2011;93:1943–9. doi: 10.1016/j.biochi.2011.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hemm MR, Paul BJ, Schneider TD, Storz G, Rudd KE. Small membrane proteins found by comparative genomics and ribosome binding site models. Mol Microbiol. 2008;70:1487–501. doi: 10.1111/j.1365-2958.2008.06495.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hobbs EC, Astarita JL, Storz G. Small RNAs and small proteins involved in resistance to cell envelope stress and acid shock in Escherichia coli: analysis of a bar-coded mutant collection. J Bacteriol. 2010;192:59–67. doi: 10.1128/JB.00873-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chao Y, Vogel J. The role of Hfq in bacterial pathogens. Curr Opin Microbiol. 2010;13:24–33. doi: 10.1016/j.mib.2010.01.001. [DOI] [PubMed] [Google Scholar]
- 21.Papenfort K, Vogel J. Regulatory RNA in bacterial pathogens. Cell Host Microbe. 2010;8:116–27. doi: 10.1016/j.chom.2010.06.008. [DOI] [PubMed] [Google Scholar]
- 22.Bohn C, Rigoulay C, Bouloc P. No detectable effect of RNA-binding protein Hfq absence in Staphylococcus aureus. BMC Microbiol. 2007;7:10. doi: 10.1186/1471-2180-7-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu Y, Wu N, Dong J, Gao Y, Zhang X, Mu C, et al. Hfq is a global regulator that controls the pathogenicity of Staphylococcus aureus. PLoS One. 2010;5:5. doi: 10.1371/journal.pone.0013069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jousselin A, Metzinger L, Felden B. On the facultative requirement of the bacterial RNA chaperone, Hfq. Trends Microbiol. 2009;17:399–405. doi: 10.1016/j.tim.2009.06.003. [DOI] [PubMed] [Google Scholar]
- 25.Vogel J, Luisi BF. Hfq and its constellation of RNA. Nat Rev Microbiol. 2011;9:578–89. doi: 10.1038/nrmicro2615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pandey SP, Minesinger BK, Kumar J, Walker GC. A highly conserved protein of unknown function in Sinorhizobium meliloti affects sRNA regulation similar to Hfq. Nucleic Acids Res. 2011;39:4691–708. doi: 10.1093/nar/gkr060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kajitani M, Kato A, Wada A, Inokuchi Y, Ishihama A. Regulation of the Escherichia coli hfq gene encoding the host factor for phage Q beta. J Bacteriol. 1994;176:531–4. doi: 10.1128/jb.176.2.531-534.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gaballa A, Antelmann H, Aguilar C, Khakh SK, Song KB, Smaldone GT, et al. The Bacillus subtilis iron-sparing response is mediated by a Fur-regulated small RNA and three small, basic proteins. Proc Natl Acad Sci U S A. 2008;105:11927–32. doi: 10.1073/pnas.0711752105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Arnvig KB, Young DB. Identification of small RNAs in Mycobacterium tuberculosis. Mol Microbiol. 2009;73:397–408. doi: 10.1111/j.1365-2958.2009.06777.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Said N, Rieder R, Hurwitz R, Deckert J, Urlaub H, Vogel J. In vivo expression and purification of aptamer-tagged small RNA regulators. Nucleic Acids Res. 2009;37:e133. doi: 10.1093/nar/gkp719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Arnvig KB, Young DB. Regulation of pathogen metabolism by small RNA. Drug Discov Today Dis Mech. 2010;7:e19–24. doi: 10.1016/j.ddmec.2010.09.001. [DOI] [Google Scholar]
- 32.Svenningsen SL, Tu KC, Bassler BL. Gene dosage compensation calibrates four regulatory RNAs to control Vibrio cholerae quorum sensing. EMBO J. 2009;28:429–39. doi: 10.1038/emboj.2008.300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mandin P, Gottesman S. Integrating anaerobic/aerobic sensing and the general stress response through the ArcZ small RNA. EMBO J. 2010;29:3094–107. doi: 10.1038/emboj.2010.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Monteiro C, Papenfort K, Hentrich K, Ahmad I, Le Guyon S, Reimann R, et al. Hfq and Hfq-dependent small RNAs are major contributors to multicellular development in Salmonella enterica serovar Typhimurium. RNA Biol. 2012;9:9. doi: 10.4161/rna.19682. [DOI] [PubMed] [Google Scholar]
- 35.Papenfort K, Pfeiffer V, Mika F, Lucchini S, Hinton JC, Vogel J. SigmaE-dependent small RNAs of Salmonella respond to membrane stress by accelerating global omp mRNA decay. Mol Microbiol. 2006;62:1674–88. doi: 10.1111/j.1365-2958.2006.05524.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chabelskaya S, Gaillot O, Felden B. A Staphylococcus aureus small RNA is required for bacterial virulence and regulates the expression of an immune-evasion molecule. PLoS Pathog. 2010;6:e1000927. doi: 10.1371/journal.ppat.1000927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Toledo-Arana A, Dussurget O, Nikitas G, Sesto N, Guet-Revillet H, Balestrino D, et al. The Listeria transcriptional landscape from saprophytism to virulence. Nature. 2009;459:950–6. doi: 10.1038/nature08080. [DOI] [PubMed] [Google Scholar]
- 38.Faucher SP, Friedlander G, Livny J, Margalit H, Shuman HA. Legionella pneumophila 6S RNA optimizes intracellular multiplication. Proc Natl Acad Sci U S A. 2010;107:7533–8. doi: 10.1073/pnas.0911764107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gong H, Vu GP, Bai Y, Chan E, Wu R, Yang E, et al. A Salmonella small non-coding RNA facilitates bacterial invasion and intracellular replication by modulating the expression of virulence factors. PLoS Pathog. 2011;7:e1002120. doi: 10.1371/journal.ppat.1002120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Santiviago CA, Reynolds MM, Porwollik S, Choi SH, Long F, Andrews-Polymenis HL, et al. Analysis of pools of targeted Salmonella deletion mutants identifies novel genes affecting fitness during competitive infection in mice. PLoS Pathog. 2009;5:e1000477. doi: 10.1371/journal.ppat.1000477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Song T, Mika F, Lindmark B, Liu Z, Schild S, Bishop A, et al. A new Vibrio cholerae sRNA modulates colonization and affects release of outer membrane vesicles. Mol Microbiol. 2008;70:100–11. doi: 10.1111/j.1365-2958.2008.06392.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.DiChiara JM, Contreras-Martinez LM, Livny J, Smith D, McDonough KA, Belfort M. Multiple small RNAs identified in Mycobacterium bovis BCG are also expressed in Mycobacterium tuberculosis and Mycobacterium smegmatis. Nucleic Acids Res. 2010;38:4067–78. doi: 10.1093/nar/gkq101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chaudhuri RR, Loman NJ, Snyder LA, Bailey CM, Stekel DJ, Pallen MJ. xBASE2: a comprehensive resource for comparative bacterial genomics. Nucleic Acids Res. 2008;36(Database issue):D543–6. doi: 10.1093/nar/gkm928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Reddy TB, Riley R, Wymore F, Montgomery P, DeCaprio D, Engels R, et al. TB database: an integrated platform for tuberculosis research. Nucleic Acids Res. 2009;37(Database issue):D499–508. doi: 10.1093/nar/gkn652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Abramovitch RB, Rohde KH, Hsu FF, Russell DG. aprABC: a Mycobacterium tuberculosis complex-specific locus that modulates pH-driven adaptation to the macrophage phagosome. Mol Microbiol. 2011;80:678–94. doi: 10.1111/j.1365-2958.2011.07601.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bell C, Smith GT, Sweredoski MJ, Hess S. Characterization of the Mycobacterium tuberculosis proteome by liquid chromatography mass spectrometry-based proteomics techniques: a comprehensive resource for tuberculosis research. J Proteome Res. 2012;11:119–30. doi: 10.1021/pr2007939. [DOI] [PubMed] [Google Scholar]
- 47.Kruh NA, Troudt J, Izzo A, Prenni J, Dobos KM. Portrait of a pathogen: the Mycobacterium tuberculosis proteome in vivo. PLoS One. 2010;5:e13938. doi: 10.1371/journal.pone.0013938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Thomason MK, Storz G. Bacterial antisense RNAs: how many are there, and what are they doing? Annu Rev Genet. 2010;44:167–88. doi: 10.1146/annurev-genet-102209-163523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lasa I, Toledo-Arana A, Dobin A, Villanueva M, de los Mozos IR, Vergara-Irigaray M, et al. Genome-wide antisense transcription drives mRNA processing in bacteria. Proc Natl Acad Sci U S A. 2011;108:20172–7. doi: 10.1073/pnas.1113521108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Movahedzadeh F, Smith DA, Norman RA, Dinadayala P, Murray-Rust J, Russell DG, et al. The Mycobacterium tuberculosis ino1 gene is essential for growth and virulence. Mol Microbiol. 2004;51:1003–14. doi: 10.1046/j.1365-2958.2003.03900.x. [DOI] [PubMed] [Google Scholar]
- 51.Hotter GS, Collins DM. Mycobacterium bovis lipids: virulence and vaccines. Vet Microbiol. 2011;151:91–8. doi: 10.1016/j.vetmic.2011.02.030. [DOI] [PubMed] [Google Scholar]
- 52.Sassetti CM, Boyd DH, Rubin EJ. Genes required for mycobacterial growth defined by high density mutagenesis. Mol Microbiol. 2003;48:77–84. doi: 10.1046/j.1365-2958.2003.03425.x. [DOI] [PubMed] [Google Scholar]
- 53.Sassetti CM, Rubin EJ. Genetic requirements for mycobacterial survival during infection. Proc Natl Acad Sci U S A. 2003;100:12989–94. doi: 10.1073/pnas.2134250100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Matsunaga I, Bhatt A, Young DC, Cheng TY, Eyles SJ, Besra GS, et al. Mycobacterium tuberculosis pks12 produces a novel polyketide presented by CD1c to T cells. J Exp Med. 2004;200:1559–69. doi: 10.1084/jem.20041429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Argaman L, Hershberg R, Vogel J, Bejerano G, Wagner EG, Margalit H, et al. Novel small RNA-encoding genes in the intergenic regions of Escherichia coli. Curr Biol. 2001;11:941–50. doi: 10.1016/S0960-9822(01)00270-6. [DOI] [PubMed] [Google Scholar]
- 56.Sharma CM, Hoffmann S, Darfeuille F, Reignier J, Findeiss S, Sittka A, et al. The primary transcriptome of the major human pathogen Helicobacter pylori. Nature. 2010;464:250–5. doi: 10.1038/nature08756. [DOI] [PubMed] [Google Scholar]
- 57.Gardner PP, Barquist L, Bateman A, Nawrocki EP, Weinberg Z. RNIE: genome-wide prediction of bacterial intrinsic terminators. Nucleic Acids Res. 2011;39:5845–52. doi: 10.1093/nar/gkr168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Unniraman S, Prakash R, Nagaraja V. Alternate paradigm for intrinsic transcription termination in eubacteria. J Biol Chem. 2001;276:41850–5. doi: 10.1074/jbc.M106252200. [DOI] [PubMed] [Google Scholar]
- 59.Lindahl L, Zengel JM. Ribosomal genes in Escherichia coli. Annu Rev Genet. 1986;20:297–326. doi: 10.1146/annurev.ge.20.120186.001501. [DOI] [PubMed] [Google Scholar]
- 60.Gardner PP, Daub J, Tate J, Moore BL, Osuch IH, Griffiths-Jones S, et al. Rfam: Wikipedia, clans and the “decimal” release. Nucleic Acids Res. 2011;39(Database issue):D141–5. doi: 10.1093/nar/gkq1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Warner DF, Savvi S, Mizrahi V, Dawes SS. A riboswitch regulates expression of the coenzyme B12-independent methionine synthase in Mycobacterium tuberculosis: implications for differential methionine synthase function in strains H37Rv and CDC1551. J Bacteriol. 2007;189:3655–9. doi: 10.1128/JB.00040-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rodionov DA, Vitreschak AG, Mironov AA, Gelfand MS. Comparative genomics of the vitamin B12 metabolism and regulation in prokaryotes. J Biol Chem. 2003;278:41148–59. doi: 10.1074/jbc.M305837200. [DOI] [PubMed] [Google Scholar]
- 63.Akhter Y, Ehebauer MT, Mukhopadhyay S, Hasnain SE. The PE/PPE multigene family codes for virulence factors and is a possible source of mycobacterial antigenic variation: perhaps more? Biochimie. 2012;94:110–6. doi: 10.1016/j.biochi.2011.09.026. [DOI] [PubMed] [Google Scholar]
- 64.Vitreschak AG, Rodionov DA, Mironov AA, Gelfand MS. Regulation of the vitamin B12 metabolism and transport in bacteria by a conserved RNA structural element. RNA. 2003;9:1084–97. doi: 10.1261/rna.5710303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Dann CE, 3rd, Wakeman CA, Sieling CL, Baker SC, Irnov I, Winkler WC. Structure and mechanism of a metal-sensing regulatory RNA. Cell. 2007;130:878–92. doi: 10.1016/j.cell.2007.06.051. [DOI] [PubMed] [Google Scholar]
- 66.Barrick JE, Corbino KA, Winkler WC, Nahvi A, Mandal M, Collins J, et al. New RNA motifs suggest an expanded scope for riboswitches in bacterial genetic control. Proc Natl Acad Sci U S A. 2004;101:6421–6. doi: 10.1073/pnas.0308014101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Walters SB, Dubnau E, Kolesnikova I, Laval F, Daffe M, Smith I. The Mycobacterium tuberculosis PhoPR two-component system regulates genes essential for virulence and complex lipid biosynthesis. Mol Microbiol. 2006;60:312–30. doi: 10.1111/j.1365-2958.2006.05102.x. [DOI] [PubMed] [Google Scholar]
- 68.Vitreschak AG, Mironov AA, Lyubetsky VA, Gelfand MS. Comparative genomic analysis of T-box regulatory systems in bacteria. RNA. 2008;14:717–35. doi: 10.1261/rna.819308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Alix E, Blanc-Potard AB. MgtC: a key player in intramacrophage survival. Trends Microbiol. 2007;15:252–6. doi: 10.1016/j.tim.2007.03.007. [DOI] [PubMed] [Google Scholar]
- 70.Serafini A, Boldrin F, Palù G, Manganelli R. Characterization of a Mycobacterium tuberculosis ESX-3 conditional mutant: essentiality and rescue by iron and zinc. J Bacteriol. 2009;191:6340–4. doi: 10.1128/JB.00756-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Block KF, Hammond MC, Breaker RR. Evidence for widespread gene control function by the ydaO riboswitch candidate. J Bacteriol. 2010;192:3983–9. doi: 10.1128/JB.00450-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Biketov S, Potapov V, Ganina E, Downing K, Kana BD, Kaprelyants A. The role of resuscitation promoting factors in pathogenesis and reactivation of Mycobacterium tuberculosis during intra-peritoneal infection in mice. BMC Infect Dis. 2007;7:146. doi: 10.1186/1471-2334-7-146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Keep NH, Ward JM, Cohen-Gonsaud M, Henderson B. Wake up! Peptidoglycan lysis and bacterial non-growth states. Trends Microbiol. 2006;14:271–6. doi: 10.1016/j.tim.2006.04.003. [DOI] [PubMed] [Google Scholar]
- 74.Telkov MV, Demina GR, Voloshin SA, Salina EG, Dudik TV, Stekhanova TN, et al. Proteins of the Rpf (resuscitation promoting factor) family are peptidoglycan hydrolases. Biochemistry (Mosc) 2006;71:414–22. doi: 10.1134/S0006297906040092. [DOI] [PubMed] [Google Scholar]
- 75.Ehrt S, Schnappinger D. Mycobacterial survival strategies in the phagosome: defence against host stresses. Cell Microbiol. 2009;11:1170–8. doi: 10.1111/j.1462-5822.2009.01335.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hartkoorn RC, Sala C, Uplekar S, Busso P, Rougemont J, Cole ST. Genome-wide Definition of the SigF Regulon in Mycobacterium tuberculosis. J Bacteriol. 2012 doi: 10.1128/JB.06692-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Weinberg Z, Barrick JE, Yao Z, Roth A, Kim JN, Gore J, et al. Identification of 22 candidate structured RNAs in bacteria using the CMfinder comparative genomics pipeline. Nucleic Acids Res. 2007;35:4809–19. doi: 10.1093/nar/gkm487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Beste DJ, Peters J, Hooper T, Avignone-Rossa C, Bushell ME, McFadden J. Compiling a molecular inventory for Mycobacterium bovis BCG at two growth rates: evidence for growth rate-mediated regulation of ribosome biosynthesis and lipid metabolism. J Bacteriol. 2005;187:1677–84. doi: 10.1128/JB.187.5.1677-1684.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kumar A, Toledo JC, Patel RP, Lancaster JR, Jr., Steyn AJ. Mycobacterium tuberculosis DosS is a redox sensor and DosT is a hypoxia sensor. Proc Natl Acad Sci U S A. 2007;104:11568–73. doi: 10.1073/pnas.0705054104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Shi L, North R, Gennaro ML. Effect of growth state on transcription levels of genes encoding major secreted antigens of Mycobacterium tuberculosis in the mouse lung. Infect Immun. 2004;72:2420–4. doi: 10.1128/IAI.72.4.2420-2424.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Voskuil MI, Schnappinger D, Visconti KC, Harrell MI, Dolganov GM, Sherman DR, et al. Inhibition of respiration by nitric oxide induces a Mycobacterium tuberculosis dormancy program. J Exp Med. 2003;198:705–13. doi: 10.1084/jem.20030205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Chang JC, Miner MD, Pandey AK, Gill WP, Harik NS, Sassetti CM, et al. igr Genes and Mycobacterium tuberculosis cholesterol metabolism. J Bacteriol. 2009;191:5232–9. doi: 10.1128/JB.00452-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Griffin JE, Pandey AK, Gilmore SA, Mizrahi V, McKinney JD, Bertozzi CR, et al. Cholesterol Catabolism by Mycobacterium tuberculosis Requires Transcriptional and Metabolic Adaptations. Chem Biol. 2012;19:218–27. doi: 10.1016/j.chembiol.2011.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Barrick JE, Sudarsan N, Weinberg Z, Ruzzo WL, Breaker RR. 6S RNA is a widespread regulator of eubacterial RNA polymerase that resembles an open promoter. RNA. 2005;11:774–84. doi: 10.1261/rna.7286705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wassarman KM. 6S RNA: a regulator of transcription. Mol Microbiol. 2007;65:1425–31. doi: 10.1111/j.1365-2958.2007.05894.x. [DOI] [PubMed] [Google Scholar]
- 86.Pánek J, Krásny L, Bobek J, Jezková E, Korelusová J, Vohradsky J. The suboptimal structures find the optimal RNAs: homology search for bacterial non-coding RNAs using suboptimal RNA structures. Nucleic Acids Res. 2011;39:3418–26. doi: 10.1093/nar/gkq1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Beisel CL, Storz G. Base pairing small RNAs and their roles in global regulatory networks. FEMS Microbiol Rev. 2010;34:866–82. doi: 10.1111/j.1574-6976.2010.00241.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Deveau H, Garneau JE, Moineau S. CRISPR/Cas system and its role in phage-bacteria interactions. Annu Rev Microbiol. 2010;64:475–93. doi: 10.1146/annurev.micro.112408.134123. [DOI] [PubMed] [Google Scholar]
- 89.Driscoll JR. Spoligotyping for molecular epidemiology of the Mycobacterium tuberculosis complex. Methods Mol Biol. 2009;551:117–28. doi: 10.1007/978-1-60327-999-4_10. [DOI] [PubMed] [Google Scholar]
- 90.Makarova KS, Aravind L, Wolf YI, Koonin EV. Unification of Cas protein families and a simple scenario for the origin and evolution of CRISPR-Cas systems. Biol Direct. 2011;6:38. doi: 10.1186/1745-6150-6-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Marraffini LA, Sontheimer EJ. CRISPR interference limits horizontal gene transfer in staphylococci by targeting DNA. Science. 2008;322:1843–5. doi: 10.1126/science.1165771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Makarova KS, Haft DH, Barrangou R, Brouns SJ, Charpentier E, Horvath P, et al. Evolution and classification of the CRISPR-Cas systems. Nat Rev Microbiol. 2011;9:467–77. doi: 10.1038/nrmicro2577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Hanekom M, Gey van Pittius NC, McEvoy C, Victor TC, Van Helden PD, Warren RM. Mycobacterium tuberculosis Beijing genotype: a template for success. Tuberculosis (Edinb) 2011;91:510–23. doi: 10.1016/j.tube.2011.07.005. [DOI] [PubMed] [Google Scholar]
- 94.Tsolaki AG, Hirsh AE, DeRiemer K, Enciso JA, Wong MZ, Hannan M, et al. Functional and evolutionary genomics of Mycobacterium tuberculosis: insights from genomic deletions in 100 strains. Proc Natl Acad Sci U S A. 2004;101:4865–70. doi: 10.1073/pnas.0305634101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Fenner L, Malla B, Ninet B, Dubuis O, Stucki D, Borrell S, et al. “Pseudo-Beijing”: evidence for convergent evolution in the direct repeat region of Mycobacterium tuberculosis. PLoS One. 2011;6:e24737. doi: 10.1371/journal.pone.0024737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Betts JC, Lukey PT, Robb LC, McAdam RA, Duncan K. Evaluation of a nutrient starvation model of Mycobacterium tuberculosis persistence by gene and protein expression profiling. Mol Microbiol. 2002;43:717–31. doi: 10.1046/j.1365-2958.2002.02779.x. [DOI] [PubMed] [Google Scholar]
- 97.Keren I, Minami S, Rubin E, Lewis K. Characterization and transcriptome analysis of Mycobacterium tuberculosis persisters. MBio. 2011;2:e00100–11. doi: 10.1128/mBio.00100-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Sherman DR, Voskuil M, Schnappinger D, Liao R, Harrell MI, Schoolnik GK. Regulation of the Mycobacterium tuberculosis hypoxic response gene encoding alpha -crystallin. Proc Natl Acad Sci U S A. 2001;98:7534–9. doi: 10.1073/pnas.121172498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Dhar N, McKinney JD. Mycobacterium tuberculosis persistence mutants identified by screening in isoniazid-treated mice. Proc Natl Acad Sci U S A. 2010;107:12275–80. doi: 10.1073/pnas.1003219107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Komorowski M, Miekisz J, Kierzek AM. Translational repression contributes greater noise to gene expression than transcriptional repression. Biophys J. 2009;96:372–84. doi: 10.1016/j.bpj.2008.09.052. [DOI] [PMC free article] [PubMed] [Google Scholar]