

Abstract
Allergic asthma is a complex disease that has been modeled extensively in small rodents. Airway eosinophilia and changes in lung function have been documented using a variety of protocols. However, recent efforts have improved these models by trying to replicate the structural remodeling that occurs in the lung as a consequence of chronic allergen-driven inflammation. This review documents the recent developments in protocols and systems designed to examine pathways leading to allergen-induced airway remodeling.
Introduction
Animal models of the allergic response to inhaled antigens have been widely studied to investigate the mechanisms leading to the development of pulmonary inflammation and airway hyperreactivity (AHR) during asthma. In particular, many different protocols have been employed to render rodents, generally mice, sensitive to protein antigens such as ovalbumin (OVA) [1] (Fig. 1). After a peripheral sensitization, by peritoneal, dermal, or subcutaneous routes, and subsequent challenge via the airways, intransally, or by inhalation, mice exhibit AHR to stimuli such as methacholine. Changes in lung function are accompanied by recruitment of inflammatory leukocytes, including eosinophils, to the airway lumen and parenchyma. In addition, the majority of protocols elicit a helper T cell (Th) 2 profile of disease, with production of classic Th2 cytokines such as interleukin (IL)-4, IL-5, and IL-13 and increased serum levels of antigen-specific immunoglobulin (Ig) E and IgG1. However, the clinical relevance of these models has been questioned because most involve relatively short-term exposure to antigen via the airways (usually 1 to 2 weeks), and many do not show the chronic inflammatory and epithelial changes or mucosal inflammation that typify human asthma. Moreover, the inflammation and hyperreactivity that are observed generally resolve after cessation of the allergen challenge. However, more recently, investigators have tried to model the more chronic features of the human disease in rodents, namely airway remodeling. Airway remodeling incorporates deposition of extracellular matrix, changes in epithelial cell hyperplasia, smooth muscle cell proliferation, production of fibrogenic growth factors, mucus production, and matrix dysregulation. Newer models have tried to replicate these features in mice (Fig. 2).
Figure 1.

Summary of different models used to replicate features of allergic airways disease. Arrows represent challenges with allergens. Alum—aluminum hydroxide; HDM—house dust mite; OVA—ovalbumin.
Figure 2.
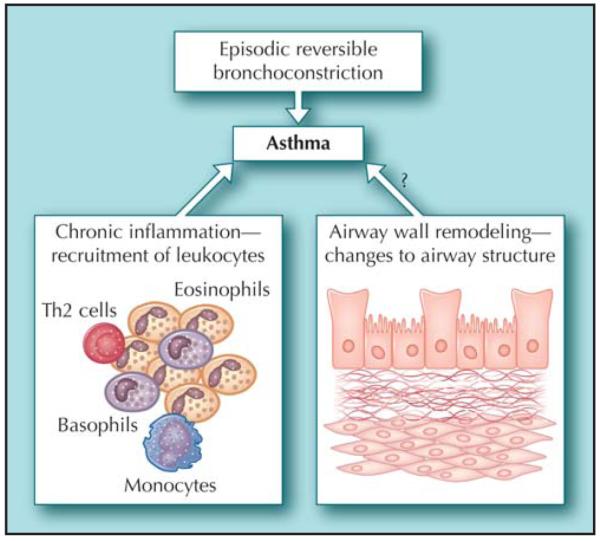
The pathophysiological features of asthma that investigators try to model in mice. Th2—helper T cell 2.
Transgenic Models
Transgenic technology has been used to generate models where the features of asthma, including airway remodeling, develop spontaneously [2]. Expression of individual cytokines or growth factors under the control of a lung-specific promoter has defined roles for these molecules in some of the pathophysiological features of asthma. The Th2-type cytokines in particular have exhibited features of remodeling, suggesting that at least some component of the remodeling response is dependent on Th2 cytokines. Pulmonary expression of IL-5 results in spontaneous eosinophilia, AHR, and some remodeling [3]. Lung-specific expression of IL-13 in particular results in spontaneous airway remodeling characterized by excessive mucus production and increased matrix deposition in the airways, in addition to a mononuclear and eosinophilic inflammatory response, eotaxin production, airways obstruction, and nonspecific AHR [4]. Similarly, pulmonary overexpression of IL-9 resulted in eosinophil- and lymphocyte-rich pulmonary inflammation, a striking lung mast cell hyperplasia, epithelial cell hypertrophy, and subepithelial collagen deposition [5]. Although mice showed normal baseline airway resistance, their response to inhaled methacholine was markedly increased. IL-11 expression in lung epithelium manifested as nodular peribronchiolar mononuclear infiltrates, with B cells present in larger numbers than T-cells, and substantial airway remodeling with subepithelial fibrosis [6]. A further refinement of the model, using a tissue-specific inducible transgenic system, was used to show that IL-11 caused abnormalities dependent (large alveoli) and independent (airway remodeling, peribronchiolar nodules) of lung growth and development [7].
Interestingly, direct targeting of classic profibrotic growth factors, such as transforming growth factor (TGF)-β and vascular endothelial growth factor (VEGF), show phenotypic similarities to asthmatic patients. Lung-targeted VEGF resulted in an asthmalike phenotype characterized by inflammation, parenchymal and vascular remodeling, edema, mucus metaplasia, myocyte hyperplasia, and AHR [8•]. Interestingly, these features were shown to develop via IL-13–dependent and independent pathways. In contrast, lung-specific expression of TGF-β has been more difficult to achieve because the transgene induces fetal lethality. However, development of a conditional transgenic expression system using a tetracycline-controlled transcriptional suppressor and reverse tetracycline transactivator has circumvented these problems [9]. Analysis of 6-week-old transgene-positive and transgene-negative mice following activation of expression by intake of doxycycline revealed a complex phenotype [10]. Overexpression of TGF-β in the epithelium induced an early wave of epithelial apoptosis that declines with continued expression of TGF-β. Prominent inflammation was also noted, as well as an airway and parenchymal fibrotic response characterized by increased collagen deposition. Moreover, there was a significant increase in accumulation of myofibroblasts and myocytes.
Human asthma is generally considered to be associated with classic Th2-type pathology, so it is interesting that targeted deletion of the Th1 transcription factor T-bet resulted in spontaneous development of the pathophysiologic features of asthma. In addition to AHR and pulmonary eosinophilia, T-bet deficiency was associated with subepithelial collagen deposition, an increase in bronchial myofibroblasts, and increased production of TGF-β [11].
Although these transgenic models allow us to determine the particular contribution of individual mediators, one could argue that they may overemphasize the contribution of that molecule, when it is likely that pathological changes occur as a consequence of a tightly controlled network of mediators. In addition, they do not involve allergen challenge, which likely elicits a distinct array of immunological pathways resulting in chronic inflammation that impacts lung function.
Allergen Challenge Models
In an attempt to model the structural changes exhibited in the lungs of patients with chronic asthma, investigators have tried to manipulate acute allergen challenge protocols by using prolonged challenge regimens. In comparison to the shorter acute models (generally several days of allergen challenge), the chronic models incorporate weeks or months of allergen challenge in sensitized mice. These prolonged challenge models generally elicit less inflammation in the lungs than in the acute protocols, but eosinophilia is still prominent. Moreover, inflammation is accompanied by features of airway remodeling, including increased matrix deposition in the airways, an increase in smooth muscle cells, and goblet cell hyperplasia [12–16]. In addition, these pathological changes are generally accompanied by changes in lung function [13–16]. These models have now been used to establish relationships between individual cell types and specific mediators with the development of remodeling events. For example, IL-13 is thought to be a critical mediator in airway remodeling because absence of IL-13, either by blockage with neutralizing antibody or in genetically deficient mice, ameliorates many of the symptoms associated with chronic allergen challenge [17–19]. Similarly, targeting IL-5, either by anti–IL-5 antibody or in IL-5–deficient mice, is also beneficial in reducing airway remodeling [16, 20], perhaps due to effects on eosinophils, which are critical contributors to parameters of remodeling [21•]. Others have determined that mast cells are important contributors to the remodeling reaction [14,22].
One of the criticisms of models of airway inflammation in mice has been the fact that the pathophysiological symptoms disappear with the cessation of allergen challenge. However, in some of the more recent chronic challenge models, investigators have established that the features of airway remodeling have persisted even in the absence of further allergen challenge [13,15,23,24]. In contrast, there is disagreement between different models as to the persistence of inflammation in the absence of allergen challenge. In some models, sustained mononuclear and eosinophilic inflammation is reported several weeks after the cessation of allergen challenge [13,24], while other protocols result in a return to baseline eosinophilia without continued allergen challenge [15,23]. Similarly, there are differences in the persistence of lung dysfunction. In some models this is maintained after allergen challenge is stopped [15], but in other models it resolves and resistance returns to baseline levels [13]. Thus, different aspects of sustained airway remodeling, and in some cases dysfunction, existed beyond the resolution of the acute inflammatory events.
The majority of mouse models report some change in lung function after prolonged challenge [15,16, 25,26,27••] that correlates with airway inflammation and airway remodeling. However, the relationship between airway inflammation, airway remodeling, and changes in airway function is still ambiguous. Several studies argue that airway structural changes can be uncoupled from airway hyperresponsiveness. One elegant study compared brief or chronic exposure of mice to allergen and determined that airway dysfunction and remodeling persisted beyond the resolution of immune-mediated inflammatory events [15]. Airway responses to methacholine were measured, and increases in the maximal inducible bronchoconstriction observed after either brief or chronic allergen challenge were maintained for at least 8 weeks after cessation of allergen challenge. These functional changes were seen in conjunction with increases in contractile tissue in the airway wall. In contrast, observed increases in airway reactivity (rate of increase in respiratory resistance for a given dose of methacholine) only persisted beyond resolution of allergen-induced inflammation in chronically challenged mice. The authors concluded that sustained AHR was not associated with ongoing Th2 inflammatory markers such as eosinophilia or IL-13, but that sustained dysfunction occurred as a consequence of airway remodeling rather than these immune-mediated events. One can speculate that the initial development of airway dysfunction is dependent on acute inflammatory responses to allergen challenge, such as recruitment of eosinophils and Th2 cells and production of Th2 cytokines, but that sustained dysfunction is dependent on structural changes to the airways, such as changes in the smooth muscle layer and increased deposition of extracellular matrix. Subsequent studies using knockout mice revealed that IL-4 and IL-13, but not IL-5, are critical for the development of sustained airway remodeling and AHR [28]. However, one might argue that because these mice are completely devoid of the individual cytokines for the duration of the entire protocol, the observed effect may reflect differences in the initial development of remodeling or AHR rather than in the persistence. Studies with neutralizing antibodies during different phases of the protocol would determine this.
Other studies have also revealed interesting correlations between airway inflammation, development of airway dysfunction, and airway remodeling. Several models have shown that it is possible to uncouple airway function from either inflammation or airway remodeling. We have previously shown that prolonged allergen challenge leads to persistent airway remodeling, but that inflammation and AHR are dependent upon continued allergen challenge [13]. Moreover, using the same challenge protocol, mice genetically deficient in eosinophils were protected from development of remodeling but, surprisingly, not from changes in airway function [21•]. These mice develop robust AHR to acute and chronic allergen challenge but failed to develop airway fibrosis or airway smooth muscle hypertrophy after chronic challenge. Another study has shown that airway inflammation and AHR are preserved in mice that lack mast cells, but that allergen-induced subepithelial fibrosis was partially attenuated [29]. Therapeutic administration of anti–TGF-β antibody after the onset of eosinophilic inflammation and AHR had no effect on either airway eosinophilia or AHR parameters, but did prevent the development of airway remodeling [30•].
Several studies have attempted to determine the role of inflammatory cytokines in the development of airway inflammation and remodeling. Most of these have used antibodies or knockout mice to target Th2 cytokines [16, 20,28]. In contrast, a transgenic approach has been used to determine whether genes expressed in the airway epithelium contribute to allergen-induced airway epithelium [31]. Multiple genes that are potentially involved in airway inflammation and remodeling are controlled by the transcription factor NF-κB, which in turn requires IκB kinase gene for activation. Chronic allergen challenge of mice with inducible, tissue-specific inactivation of the IκB kinase gene led to significantly less peribronchiolar fibrosis as measured by lung collagen content as well as peribronchiolar collagen staining. Airway mucus, eosinophilia, and peribronchiolar CD4 cells were also reduced after IκκB ablation. Moreover, the Th2-type chemokines CCL11 and CCL17 were reduced, as were levels of TGF-β.
Collectively, these studies show that it is possible to affect one parameter of chronic allergen challenge without affecting others, and will permit the testing of potential therapeutic treatment regimens on outcomes of the various aspects of chronic allergen challenge. In the future, it will be important to consider the success of these therapeutic regimens on facets of chronic allergen challenge, such as airway remodeling rather than just eosinophil recruitment. In addition, it will be important to test potential therapeutics using a therapeutic rather than a prophylactic regimen. We have shown that treatment of mice after onset of allergen challenge is successful in reducing airway inflammation, AHR, and remodeling [32].
Valid criticism of all of these models has been their reliance on systemic sensitization of mice with OVA, often in the presence of adjuvant such as aluminum hydroxide/alum. This has been necessary because repeated airway challenge of mice with OVA, in the absence of prior sensitization, has been shown to lead to tolerance [33–36]. Moreover, even in sensitized mice eosinophilia declines despite continued aerosol exposure to allergen [13,37]. In contrast, intranasal administration of OVA to A/J mice resulted in airway remodeling, inflammation, and persistent AHR [37]. This was observed only in A/J mice and not in three other strains studied, highlighting the importance of genetics in the development of allergen-induced pathophysiology. In addition, the results were obtained only if the allergen was given intranasally and not by inhalation, thus the protocol for induction of disease is critical.
A similar study has also tried to eliminate the need for systemic sensitization, using an environmentally relevant antigen. Johnson et al. [27••] determined that prolonged intranasal administration of house dust mite (HDM) antigen resulted in severe and persistent eosinophilia, AHR, and Th2-type pathology. Moreover, mice showed evidence of airway remodeling, with goblet cell hyperplasia, collagen deposition, and increased contractile elements. Interestingly, although airway inflammation had fully resolved by 9 weeks after cessation of HDM exposure, the AHR had resolved only partially, but there was no resolution in the remodeling changes. Further analysis revealed that the airway pathology elicited by intranasal HDM exposure is mediated by granulocyte-macrophage colony-stimulating factor (GM-CSF) [38]. Moreover, exposure to HDM facilitates a lung microenvironment that allows induction of inflammation in response to an otherwise innocuous antigen, such as OVA [39].
Conclusions
Mouse models of asthma have proved a valuable tool in elucidating the cells and molecules responsible for the development of the pathophysiological features of the allergic pulmonary response. However, the information that they yield is valuable only if the models are valid. Efforts are constantly being made to refine these models; recent advances have been the development of models that recapitulate the structural changes observed in lungs of patients with chronic asthma. These improved models should provide important information regarding the relationships between airway inflammation and airway dysfunction and remodeling, as well as the contributions of different cells and mediators to these processes. They will be critical to test novel therapeutics because lessons from previous drug development in asthma have shown that an integrated approach is critical.
References and Recommended Reading
Papers of particular interest, published recently, have been highlighted as:
• Of importance
•• Of major importance
- 1.Lloyd CM, Gonzalo JA, Coyle AJ, Gutierrez-Ramos JC. Mouse models of allergic airway disease. Adv Immunol. 2001;77:263–295. doi: 10.1016/s0065-2776(01)77019-8. [DOI] [PubMed] [Google Scholar]
- 2.Elias J. The relationship between asthma and COPD: lessons from transgenic mice. Chest. 2004;126:111S–116S. doi: 10.1378/chest.126.2_suppl_1.111S. [DOI] [PubMed] [Google Scholar]
- 3.Lee JJ, McGarry MP, Farmer SC, et al. Interleukin-5 expression in the lung epithelium of transgenic mice leads to pulmonary changes pathognomonic of asthma. J Exp Med. 1997;185:2143–2156. doi: 10.1084/jem.185.12.2143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhu Z, Homer RJ, Wang Z, et al. Pulmonary expression of interleukin-13 causes inflammation, mucus hypersecretion, subepithelial fibrosis, physiologic abnormalities, and eotaxin production. J Clin Invest. 1999;103:779–788. doi: 10.1172/JCI5909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Temann UA, Geba GP, Rankin JA, Flavell RA. Expression of interleukin 9 in the lungs of transgenic mice causes airway inflammation, mast cell hyperplasia, and bronchial hyperresponsiveness. J Exp Med. 1998;188:1307–1320. doi: 10.1084/jem.188.7.1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tang W, Geba GP, Zheng T, et al. Targeted expression of IL-11 in the murine airway causes lymphocytic inflammation, bronchial remodeling, and airways obstruction. J Clin Invest. 1996;98:2845–2853. doi: 10.1172/JCI119113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ray P, Tang W, Wang P, et al. Regulated overexpression of interleukin 11 in the lung. Use to dissociate development-dependent and -independent phenotypes. J Clin Invest. 1997;100:2501–2511. doi: 10.1172/JCI119792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8•.Lee CG, Link H, Baluk P, et al. Vascular endothelial growth factor (VEGF) induces remodeling and enhances TH2-mediated sensitization and inflammation in the lung. Nat Med. 2004;10:1095–1103. doi: 10.1038/nm1105. [This work defines a role for VEGF in Th2-mediated allergic lung disease.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhu Z, Zheng T, Lee CG, et al. Tetracycline-controlled transcriptional regulation systems: advances and application in transgenic animal modeling. Semin Cell Dev Biol. 2002;13:121–128. doi: 10.1016/s1084-9521(02)00018-6. [DOI] [PubMed] [Google Scholar]
- 10.Lee CG, Kang HR, Homer RJ, et al. Transgenic modeling of transforming growth factor-1: role of apoptosis in fibrosis and alveolar remodeling. Proc Am Thorac Soc. 2006;3:418–423. doi: 10.1513/pats.200602-017AW. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Finotto S, Neurath MF, Glickman JN, et al. Development of spontaneous airway changes consistent with human asthma in mice lacking T-bet. Science. 2002;295:336–338. doi: 10.1126/science.1065544. [DOI] [PubMed] [Google Scholar]
- 12.Temelkovski J, Hogan SP, Shepherd DP, et al. An improved murine model of asthma: selective airway inflammation, epithelial lesions and increased methacholine responsiveness following chronic exposure to aerosolised allergen. Thorax. 1998;53:849–856. doi: 10.1136/thx.53.10.849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McMillan S, Lloyd C. Prolonged allergen challenge in mice leads to persistent airway remodeling. Clin Exp Allergy. 2004;34:497–507. doi: 10.1111/j.1365-2222.2004.01895.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Henderson WRJ, Tang LO, Chu SJ, et al. A role for cysteinyl leukotrienes in airway remodeling in a mouse asthma model. Am J Respir Crit Care Med. 2002;165:108–116. doi: 10.1164/ajrccm.165.1.2105051. [DOI] [PubMed] [Google Scholar]
- 15.Leigh R, Ellis R, Wattie J, et al. Dysfunction and remodeling of the mouse airway persist after resolution of acute allergen-induced airway inflammation. Am J Respir Cell Mol Biol. 2002;27:526–535. doi: 10.1165/rcmb.2002-0048OC. [DOI] [PubMed] [Google Scholar]
- 16.Cho JY, Miller M, Baek KJ, et al. Inhibition of airway remodeling in IL-5–deficient mice. J Clin Invest. 2004;113:551–560. doi: 10.1172/JCI19133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kumar RK, Herbert C, Yang M, et al. Role of interleukin-13 in eosinophil accumulation and airway remodeling in a mouse model of chronic asthma. Clin Exp Allergy. 2002;32:1104–1111. doi: 10.1046/j.1365-2222.2002.01420.x. [DOI] [PubMed] [Google Scholar]
- 18.Foster PS, Webb DC, Yang M, et al. Dissociation of T helper type 2 cytokine-dependent airway lesions from signal transducer and activator of transcription 6 signalling in experimental chronic asthma. Clin Exp Allergy. 2003;33:688–695. doi: 10.1046/j.1365-2222.2003.01647.x. [DOI] [PubMed] [Google Scholar]
- 19.Yang G, Li L, Volk A, et al. Therapeutic dosing with anti-interleukin-13 monoclonal antibody inhibits asthma progression in mice. J Pharmacol Exp Ther. 2005;313:8–15. doi: 10.1124/jpet.104.076133. [DOI] [PubMed] [Google Scholar]
- 20.Kumar RK, Herbert C, Webb DC, et al. Effects of anticytokine therapy in a mouse model of chronic asthma. Am J Respir Crit Care Med. 2004;170:1043–1048. doi: 10.1164/rccm.200405-681OC. [DOI] [PubMed] [Google Scholar]
- 21•.Humbles AA, Lloyd CM, McMillan SJ, et al. A critical role for eosinophils in allergic airways remodeling. Science. 2004;305:1776–1779. doi: 10.1126/science.1100283. [This study provides for the first time direct evidence that eosinophils are involved in allergen-induced airway remodeling.] [DOI] [PubMed] [Google Scholar]
- 22.Ikeda RK, Miller M, Nayar J, et al. Accumulation of peribronchial mast cells in a mouse model of ovalbumin allergen induced chronic airway inflammation: modulation by immunostimulatory DNA sequences. J Immunol. 2003;171:4860–4867. doi: 10.4049/jimmunol.171.9.4860. [DOI] [PubMed] [Google Scholar]
- 23.Kumar RK, Herbert C, Kasper M. Reversibility of airway inflammation and remodeling following cessation of antigenic challenge in a model of chronic asthma. Clin Exp Allergy. 2004;34:1796–1802. doi: 10.1111/j.1365-2222.2004.02097.x. [DOI] [PubMed] [Google Scholar]
- 24.Wegmann M, Fehrenbach H, Fehrenbach A, et al. Involvement of distal airways in a chronic model of experimental asthma. Clin Expl Allergy. 2005;35:1263–1274. doi: 10.1111/j.1365-2222.2005.02306.x. [DOI] [PubMed] [Google Scholar]
- 25.Henderson WR, Jr, Lewis DD, Albert RK, et al. The importance of leukotrienes in airway inflammation in a mouse model of asthma. J Exp Med. 1996;184:1483–1494. doi: 10.1084/jem.184.4.1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tanaka H, Masuda T, Tokuoka S, et al. The effect of allergen-induced airway inflammation on airway remodeling in a murine model of allergic asthma. Inflamm Res. 2001;50:616–624. doi: 10.1007/PL00000243. [DOI] [PubMed] [Google Scholar]
- 27••.Johnson JR, Wiley RE, Fattouh R, et al. Continuous exposure to house dust mite elicits chronic airway inflammation and structural remodeling. Am J Respir Crit Care Med. 2004;169:378–385. doi: 10.1164/rccm.200308-1094OC. [This study describes a model of allergen-induced airway inflammation mediated by an environmentally relevant allergen without the need for prior sensitization of the immune system.] [DOI] [PubMed] [Google Scholar]
- 28.Leigh R, Ellis R, Wattie JN, et al. Type 2 cytokines in the pathogenesis of sustained airway dysfunction and airway remodeling in mice. Am J Respir Crit Care Med. 2004;169:860–867. doi: 10.1164/rccm.200305-706OC. [DOI] [PubMed] [Google Scholar]
- 29.Masuda T, Tanaka H, Komai M, et al. Mast cells play a partial role in allergen-induced subepithelial fibrosis in a murine model of allergic asthma. Clin Exp Allergy. 2003;33:705–713. doi: 10.1046/j.1365-2222.2003.01588.x. [DOI] [PubMed] [Google Scholar]
- 30•.McMillan SJ, Xanthou G, Lloyd CM. Manipulation of allergen-induced airway remodeling by treatment with anti–TGF-β antibody: effect on the SMAD signaling pathway. J Immunol. 2005;174:5774–5780. doi: 10.4049/jimmunol.174.9.5774. [This study describes for the fi rst time that treatment during a model of asthma can be successful when given after the onset of allergic symptoms.] [DOI] [PubMed] [Google Scholar]
- 31.Broide DH, Lawrence T, Doherty T, et al. Allergen-induced peribronchial fibrosis and mucus production mediated by I B kinase beta-dependent genes in airway epithelium. Proc Natl Acad Sci U S A. 2005;102:17723–17728. doi: 10.1073/pnas.0509235102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McMillan SJ, Xanthou G, Lloyd CM. Therapeutic administration of Budesonide ameliorates allergen-induced airway remodeling. Clin Exp Allergy. 2005;35:388–396. doi: 10.1111/j.1365-2222.02193.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McMenamin C, Holt PG. The natural immune response to inhaled soluble protein antigens involves major histo-compatibility complex (MHC) class I-restricted CD8+ T cell-mediated but MHC class II-restricted CD4+ T cell-dependent immune deviation resulting in selective suppression of immunoglobulin E production. J Exp Med. 1993;178:889–899. doi: 10.1084/jem.178.3.889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McMenamin C, Pimm C, McKersey M, Holt PG. Regulation of IgE responses to inhaled antigen in mice by antigen-specific gamma delta T cells. Science. 1994;265:1869–1871. doi: 10.1126/science.7916481. [DOI] [PubMed] [Google Scholar]
- 35.van Halteren AG, van der Cammen MJ, Cooper D, et al. Regulation of antigen-specific IgE, IgG1, and mast cell responses to ingested allergen by mucosal tolerance induction. J Immunol. 1997;159:3009–3015. [PubMed] [Google Scholar]
- 36.Stampfli MR, Wiley RE, Neigh GS, et al. GM-CSF transgene expression in the airway allows aerosolized ovalbumin to induce allergic sensitization in mice. J Clin Invest. 1998;102:1704–1714. doi: 10.1172/JCI4160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shinagawa K, Kojima M. Mouse model of airway remodeling: strain differences. Am J Respir Crit Care Med. 2003;168:959–967. doi: 10.1164/rccm.200210-1188OC. [DOI] [PubMed] [Google Scholar]
- 38.Cates EC, Fattouh R, Wattie J, et al. Intranasal exposure of mice to house dust mite elicits allergic airway inflammation via a GM-CSF–mediated mechanism. J Immunol. 2004;173:6384–6392. doi: 10.4049/jimmunol.173.10.6384. [DOI] [PubMed] [Google Scholar]
- 39.Fattouh R, Pouladi MA, Alvarez D, et al. House dust mite facilitates ovalbumin-specific allergic sensitization and airway inflammation. Am J Respir Crit Care Med. 2005;172:314–321. doi: 10.1164/rccm.200502-198OC. [DOI] [PubMed] [Google Scholar]