

Abstract
Steroid hormones exhibit diverse biological activities. Despite intensive studies on steroid function at the genomic level, their non-genomic action remains an enigma. In this study, we investigated the role of reactive oxygen species (ROS) in androgen-stimulated prostate cancer (PCa) cell proliferation. In androgen-treated PCa cells, increased cell growth and ROS production correlated with elevated p66Shc protein, an authentic oxidase. This growth stimulation was blocked by anti-oxidants. Further, elevated expression of p66Shc protein by cDNA transfection encoding wild type (WT) protein, but not redox-deficient (W134F) mutant, was associated with increased PCa cell proliferation. Conversely, knockdown p66Shc expression by shRNA resulted in diminished cell growth. Increased p66Shc expression in PCa cells enhanced their tumorigenicity in xenograft animals. Importantly, p66Shc protein level is higher in clinical prostate adenocarcinomas than in adjacent non-cancerous cells. Expression of redox-deficient p66Shc mutant protein abolished androgen-stimulated cell growth. In androgen-treated, H2O2-treated and p66Shc cDNA-transfected PCa cells, cellular prostatic acid phosphatase (cPAcP), an authentic tyrosine phosphatase, was inactivated by reversible oxidation; subsequently, ErbB-2 was activated by phosphorylation at tyrosine1221/2. These results together support the notion that androgens induce ROS production through the elevation of p66Shc protein, which inactivates tyrosine phosphatase activity for the activation of interacting tyrosine kinase, leading to increased cell proliferation and enhanced tumorigenicity. Our results thus suggest that p66Shc protein functions at the critical junction point between androgens and tyrosine phosphorylation signaling in human PCa cells.
Keywords: Androgen, p66Shc, Reactive oxygen species, tyrosine phosphorylation
Introduction
Steroid hormones including androgen DHT are involved in regulating diverse physiological functions. While the pro-growth role of steroids via gene regulation in cells has been intensively studied, their non-genomic action has received much less attention and requires further investigation.
ROS, depending on its intracellular level, is proposed to mediate diverse cellular activities [1, 2]. For example, ROS-induced protein and DNA adducts are proposed to be involved in carcinogenesis [1, 3]. Higher intracellular ROS levels are found in various carcinomas than corresponding non-cancerous tissues and proposed to be involved in different stages of cancer progression, characterized by oxidative nuclear damage [2, 4-6]. Evidently, elevated levels of intracellular H2O2 and oxidant enzymes, such as NAD(P)H oxidase 1 (Nox1), correlate with cancerous and metastatic ability of PCa cells [4]. However, the molecular mechanism by which ROS increase tumorigenicity is yet to be elucidated.
ROS also plays a role in growth regulation. Growth factors stimulate cell growth at least in part by increasing ROS levels that inhibit growth-inhibitory PTPs through oxidation of its active site cysteine residues, leading to activation of corresponding receptor tyrosine kinases [7-10]. Recent advances reveal the existence of the histidine-dependent PTPs, a sub-group of PTP superfamily [11]. In parallel, histidine can also be oxidized in cells forming oxo-histidine [12]. However, the biological significance of histidine oxidation in proteins is much less understood. While, androgenic stimulation of AS PCa cells also increases ROS production [13]; the molecular mechanism by which ROS mediate androgen action remains an enigma.
Human PAcP exists as cellular (cPAcP) and secretory (sPAcP) forms in differentiated prostate epithelia [14, 15]. Different post-translational modifications attribute to the differences in their biochemical properties [15]. A trans-membrane form of PAcP was recently reported and could function as analgesic in mice [16]. Nevertheless, the role of this transmembrane protein in human remains unknown. Several lines of evidence support the notion that cPAcP functions as a tumor suppressor [14-17]. Evidently, PAcP-knockout mice developed prostate carcinomas spontaneously [16]. Intratumoral injection of plasmid encoding WT PAcP protein, but not inactive mutant, into xenograft prostate tumors led to tumor suppression [17], supporting the notion that tumor suppression is dependent on its phosphatase activity. In PCa cells, cPAcP functions as a neutral, histidine-dependent PTP with ErbB-2 as its primary substrate [14, 15, 17-19]. Interestingly, the PTP and tumor suppressor activities of PAcP are dependent on its active site His12, but not cysteine residues near the active domain [17-19]. In these cells, cPAcP dephosphorylates Tyr-P of ErbB-2 and attenuates its down-stream signaling and cell growth. Upon growth stimulation, e.g., DHT, cPAcP activity is inhibited by a yet unknown mechanism, which leads to ErbB-2 activation by Tyr-P and cell proliferation [14, 15, 17-19]. The mechanism of androgen-induced cPAcP inactivation remains to be investigated.
p66Shc is predominantly expressed in epithelia and can mediate apoptotic stress signals [20-25]. p66Shc, an oxidase, increases ROS levels by oxidizing Cyt C in mitochondria or through SOS-mediated Rac1 activation at cell membrane [22, 26, 27]. Despite p66Shc being a life span determinant in murine, its association with human longevity remains under investigation [20, 28]. Rather, data from human thyroid, prostate, ovarian and colon cancer tissues and cell lines show elevated p66Shc protein levels, implying its tumorigenic role in human [29-32]. In parallel, p66Shc protein level is increased in breast cancer cell lines with high metastatic ability [32], although inconsistent results were observed in primary breast tumors [33, 34]. Collectively, p66Shc plays a critical role in steroid hormone-related carcinogenesis, in part, by promoting cell proliferation [13, 30, 35, 36]. In sex steroid-treated prostate, testicular and breast cancer cells, p66Shc protein levels are elevated and cell growth is increased [30]. Accelerated PCa cell proliferation by p66Shc cDNA transfection is preceded by increased mitochondrial ROS production [13]. However, it is not known how steroid and p66Shc-ROS signaling promote PCa cell proliferation.
In this study, we report a novel molecular mechanism of non-genomic androgen action on PCa cell growth stimulation. Our data, to the best of our knowledge, for the first time clearly reveal a novel crosstalk between androgen and Tyr-P signaling connected by p66Shc via ROS production that results in PTP inhibition and subsequently ErbB-2 activation, leading to the enhanced tumorigenicity of PCa cells.
Materials and Methods
Materials
RPMI 1640 medium, gentamicin and L-glutamine were obtained from Invitrogen (Carlsbad, CA, USA). FBS and charcoal/dextran-treated FBS were from Atlanta Biologicals (Lawrenceville, GA, USA). Polyclonal Abs recognizing ErbB-2 and GPx1, and horseradish peroxidase-conjugated anti-mouse and anti-rabbit IgG Abs were all from Santa Cruz Biotechnology Ltd (Santa Cruz, CA, USA). Anti-pTyr (clone 4G10) was obtained from Millipore (USA). Anti-pY1221/2 of ErbB-2 Ab was obtained from Cell Signaling (USA). Polyclonal Abs recognizing all three isoforms of Shc protein, the Ab specifically recognizing p66Shc protein and anti-pY 1248 of ErbB-2 Ab were obtained from Upstate (Upstate, USA). Anti-β-actin Ab, mouse anti-human PAcP Ab, DHT, NAC and Vitamin E succinate (VES) were from Sigma (St. Louis, MO). Rabbit anti-human PAcP Ab (ATM-3) has been described previously [14, 37-39]. Plasmids encoding Myc-tagged wild-type, W134F mutant and S36A mutant of p66Shc cDNAs are described in previous publications [13]. The p66Shc cDNA plasmid was originally provided by Dr. Pier Giuseppe Pelicci at European Institute of Oncology (Milan, Italy) and Dr. A Raymond Frackelton Jr at Brown University (Providence, RI, USA) [36]. Full-length GPx1 cDNA was from OriGene Technologies (Rockville, MD, USA). All other reagents were as described previously [13, 36-38].
Cell lines, hormone treatment and plasmid transfection
Source and DHT treatment of AS LNCaP-FGC, MDA PCa2b and VCaP cells and AI PC-3 human PCa cell lines were described previously [14, 17, 37]. Briefly, LNCaP-FGC, MDA PCa2b, VCaP and PC-3 were originally purchased from the American Type Culture Collection (ATCC, Rockville, MD) and routinely maintained in the respective regular medium [18, 40]. LNCaP C-33 cells are androgen sensitive; while C-81 cells exhibit the androgen independent phenotype [14, 17, 18].
For DHT treatment, cells were seeded in regular medium for 3 days, and then steroid-starved for 48 hr in a SR medium, i.e., phenol red-free RPMI 1640 medium containing 5% charcoal/dextran-treated FBS (v/v), 2 mM glutamine and 50 μg/ml gentamicin. After fed with fresh SR medium, experimental PCa cells were exposed to 10 nM DHT, and controls cells received the solvent ethanol alone for various periods of time as indicated in each experiment.
For cell growth experiments, cells were counted in a Cellometer (Nexcelom, MA, USA). cDNA and shRNA transfection were done as described previously [13, 14, 36]. Stable subclones S-31, S-32 & S-36 of LNCaP C-33 cells overexpressing p66Shc were established after p66Shc cDNA transfection [13, 36].
AcP Assay
AcP assay was performed as described previously with pNPP as the substrate in citrate buffer, pH 5.5 [37, 38]. In LNCaP C-33 cells, L(+)-tartrate-sensitive AcP (TSP) activity is routinely used to represent PAcP activity because over 90% of TSP activity in these cells is represented by cPAcP [37, 38]. PAcP-specific AcP activity was analyzed in the immunocomplex by anti-PAcP Ab [37, 38]. Reversible oxidation of PAcP was analyzed by incubating the total lysate or immunoprecipitated PAcP with or without 10 mM DTT for 10 min at room temperature and then analyzed for AcP activity.
Immunoprecipitation and immunoblotting
For immunoblotting, subconfluent cells were harvested and the immunoblotting was performed as described in previous reports [13, 18, 39]. For rehybridization, the membranes were stripped as described previously [13, 18, 39], blocked and re-probed with specific Abs. β-actin protein level was used as a loading control. All western blot experiments were repeated at least in two sets of independent experiments. The protein level was semiquantified by densitometric analyses of autoradiograms using ImageJ.exe program (http://rsb.info.nih.gov/). The relative protein level was then normalized to the corresponding loading control protein level.
For immunoprecipitation, cells were harvested and lysed in ice-cold cell lysis buffer [18, 30, 39]. An aliquot of 250 μg of lysate protein was incubated with primary Ab at 4°C overnight, then added Protein A agarose beads (20 μl of 50% bead slurry) for 3 hr at 4°C. Normal corresponding animal serum was used as a negative control. The immunocomplexes were spun at 700 × g for 1 min, washed three times with ice-cold lysis buffer, and analyzed by immunoblotting.
Xenograft animal experiments
Animal maintenance and tumor development and measurement were done according to NIH guidelines and the specific guidelines at our medical center. The protocol was approved by the University of Nebraska Medical Center Institutional Animal Care and Use Committee prior to the initiation of experiments. To compare the tumorigenicity of p66Shc cDNA-transfected stable sublines, 1×106 and 2×106 cells at the exponential growth phase were suspended in 0.1 ml medium, mixed with 0.1 ml Matrigel (BD Biosciences, Bedford, Massachusetts) and injected subcutaneously in the hind flank of male and female nude mice, respectively (National Institutes of Health) [14, 17, 18]. The animals were monitored daily and the tumor size was measured weekly with a caliper. The comparison of tumor volume induced by p66Shc-transfected stable subclones vs. vector alone subclone was analyzed by the two-tailed Student’s t-test. p< 0.05 was considered statistically significant.
Immunohistochemistry
The protocol for the usage of human prostate archival specimens was approved by the Institutional Review Board at UNMC. According to tissue availability, we obtained 78 human prostate cancer archival specimens for analyzing p66Shc expression. Immunohistochemical staining was carried out as described previously [14, 30]. Due to the heterogenicity of prostate carcinomas, each sample was given a composite score based on the intensity and the extent of tissue staining, which were evaluated by a clinical pathologist (S.M.L.). The comparison of expression level of p66Shc protein in paired samples (benign and malignant) on the same tissue section was analyzed using the one population Student’s t-test
Statistical analyses
Each set of experiments was performed in duplicates or triplicates, specified in each figure legend or experimental designs, repeated at least 2-3 times of independent experiments and the mean and standard error values of experimental results were calculated. In general, paired two-tailed Student’s t-tests were used for comparison between each group. p <0.05 was considered statistically significant [14].
RESULTS
Androgens up regulate ROS, p66Shc protein and PCa cell proliferation
To investigate a molecular mechanism of non-genomic androgen action on upregulating cell proliferation, we analyzed androgen effect on ROS production in AS PCa cells. In 10 nM DHT-treated AS LNCaP C-33 cells, ROS production was increased (Fig. 1A), as seen in EGF-treated cells (Fig. 1A) [7-9], correlating with cell growth induction (data not shown). In DHT and EGF-treated C-33 cells, ROS production was associated with elevated p66Shc protein levels (Fig. 1A, right panel). Further, the cell growth and ROS production in DHT-treated cells were abolished by NAC (Fig. 1B, left panel). In NAC-treated slow growing cells, p66Shc protein was reduced (right panel). Additionally, NAC abolished the growth of DHT-stimulated VCaP and MDA PCa2b cells, another two AS PCa cells (data not shown). Similarly, VES could block DHT-stimulated proliferation of MDA PCa2b cells (Fig. 1C) and LNCaP C-33 cells (data not shown) [13]. The direct ROS effect on C-33 cell growth was shown by H2O2 treatment (Fig. 1D). These data together showed a positive association between androgen-stimulated proliferation, ROS production and p66Shc protein elevation in AS PCa cells.
Fig. 1.
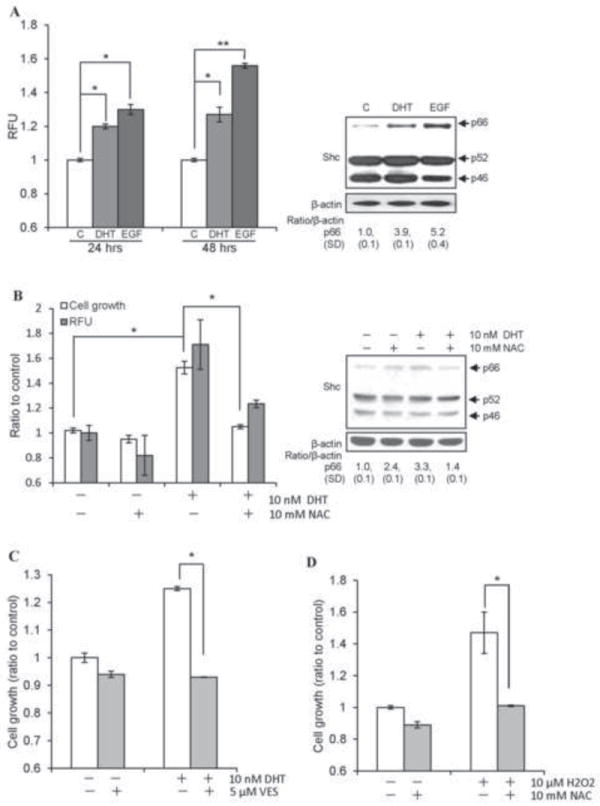
Androgens upregulate ROS, p66Shc protein and PCa cell proliferation. (A) DHT and EGF increased ROS production in LNCaP C-33 cells. Cells were plated at a density of 5×103 cells/cm2 in duplicates for 3 days in regular RPMI medium. Cells were steroid starved for 48 h in steroid-reduced (SR) medium, and then treated with 10 nM DHT or 10 ng/ml EGF for the indicated time periods. Cells were then incubated with 20 μM dichlorofluorescein diacetate (DCF-DA) for 15 min and ROS levels were measured by flow cytometry. Median fluorescence was calculated and normalized to the control values and expressed as Relative fluorescence units (RFU). (left panel, *p<0.05; **p<0.01). Total cell lysate proteins from 48 h DHT or EGF treatments were analyzed for Shc protein. β-actin protein level was used as a loading control (right panel). (B) LNCaP C-33 cells were seeded and followed by steroid starvation as described above. Cells were then treated with 10 nM DHT in the presence or absence of 10 mM NAC for 48 h. The cell growth was determined by cell counting and ROS levels were measured as described above. The data shown is an average of three sets of independent experiments in duplicates (left panel, n=2×3, *p<0.01). Total cell lysate protein was analyzed for Shc protein. β-actin protein level was used as a loading control (right panel). (C) MDA PCa2b cells were plated at a density of 1×104 cells/cm2 in duplicates for 3 days in regular medium. Cells were steroid starved for 48 h in SR medium and then treated with 10 nM DHT in the presence or absence of 5 μM VES for 48 h. The cell growth was determined by cell counting. The data shown is one set of representative results from two sets of independent experiments (*p<0.05). (D) LNCaP C-33 cells were seeded and followed by steroid starvation as described above. Cells were then treated with 10 μM H2O2 in the presence or absence of 10 mM NAC for 48 h. The cell growth was determined by cell counting (*p<0.05).
We first determined the role of p66Shc protein in growth regulation since it is associated with DHT-stimulated PCa cell growth (Fig. 1). cDNA transfection experiments revealed that elevated expression of WT p66Shc, but not the redox-defective p66Shc W134F mutant [26], correlated with increased cell growth, about 30% increase in cell number of the transiently transfected cell population in SR medium after 48 hr (Fig. 2A) [13]. Similarly, WT p66Shc cDNA transfection increased the growth of MDA PCa2b cells in SR condition (Fig. 2B). Thus, the ROS-producing ability of p66Shc protein is mitogenic activity to PCa cell growth.
Fig. 2.
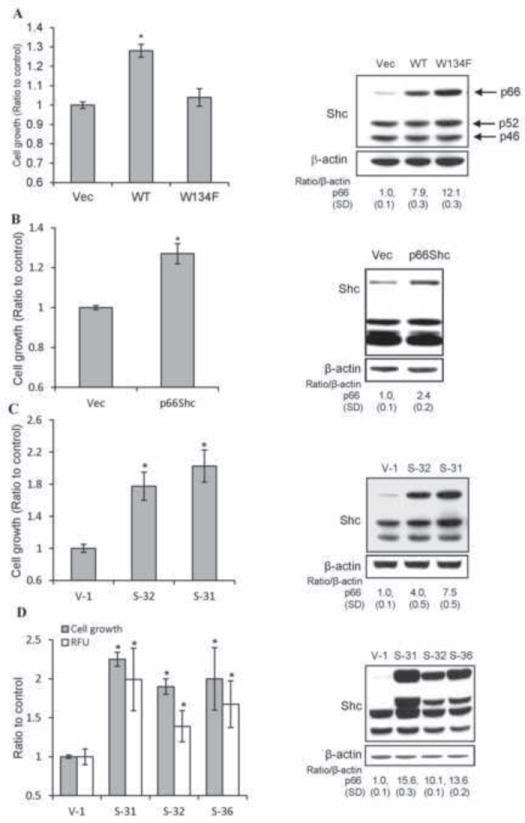
Effect of elevated p66Shc protein expression on PCa cell proliferation. (A) LNCaP C-33 cells were plated in duplicates for 48 h and then transfected with WT p66Shc cDNA (WT). Control cells were transfected with empty vector (Vec) or the redox-defective mutant W134F. After transfection, cells were fed with regular medium for overnight, then steroid-starved for 48 h, and cell growth was analyzed by cell counting (n=2×3, *p<0.01). The data shown is one set of representative results. Immunoblot analysis was performed for p66Shc level in respective cells (right panel). β-actin protein level was used as a loading control. (B) MDA PCa2b cells were transfected with WT p66Shc cDNA (WT) or empty vector (Vec). Cells were maintained in regular medium for overnight, steroid-starved for 48 h, and cell growth was analyzed by cell counting (n=2×3, *p<0.01). Immunoblot analysis was performed for p66Shc level in respective MDA PCa2b cells (right panel). β-actin protein level was used as a loading control. (C) LNCaP C-33 stable subclones of WT p66Shc cDNA (S-31 and S-32) and vector-alone (V-1) control cells were plated at a density of 8×103 cells/cm2 in regular medium for 3 days, replenished with fresh regular medium, and cell growth was analyzed by cell counting after 2 days (n=2×3, *p<0.01). The total cell lysates were analyzed for p66Shc protein levels by immunoblotting (right panel). β-actin protein level was used as a loading control. (D) As described above in (C), WT p66Shc cDNA stable subclones (S-31, S-32 and S-36) and control vector-alone (V-1) cells were plated in regular medium for 3 days. Cells were steroid-starved for 48 h, replenished with fresh SR medium for 3 days. Cells were then incubated with 20 μM DCF-DA for 30 min, and trypsinized for counting cell number or measuring ROS levels by flow cytometry (n=2×3; *p<0.01). Total cell lysates were analyzed for p66Shc protein levels by immunoblotting (right panel). β-actin protein level was used as a loading control.
We established p66Shc WT cDNA stable transfectants in LNCaP C-33 cells that express a low level of endogenous p66Shc protein (Fig. 2A) [30, 36]. Elevated WT p66Shc protein levels in S-31 and S-32 stable subclone cells were associated with increased cell growth, significantly higher than V-1 cells transfected with the control vector alone in regular culture medium (Fig. 2C). The increased cell growth was validated by cell cycle analyses on these stable subclone cells that in average, approximately 23% of p66Shc stable subclone cells were in the S-phase of cell cycle, significantly higher than C-33 parental cells (17%) and V-1 control cells (18%) (p<0.05, data not shown, and [36]). Interestingly, p66Shc effect on cell growth was much distinctly seen in a SR condition where p66Shc subclones exhibited a growth rate about 2-fold of that of V-1 control cells (Fig. 2D). We quantified ROS level in p66Shc stable subclone cells vs. V-1 control cells in a SR condition. Elevated p66Shc protein levels correlated with increased ROS production in these subclone cells, which is significantly higher than that of V-1 control cells (Fig. 2D). Further, NAC treatment effectively reduced the growth of p66Shc subclone cells by approximately 35%, much more potent than on V-1 control cells (6%) in SR condition (data not shown).
To validate p66Shc role in PCa cell growth, we performed knockdown experiments in LNCaP C-81 and PC-3 cells because these cells express higher levels of p66Shc protein and ROS production than that of LNCaP C-33 cells, which correlates with rapid growth rates with reduced doubling time [36] and ERK activation (Fig. 3A). Conversely, NAC treatment decreased C-81 cell growth by 32% vs. C-33 cells by 4% (data not shown). Further, reduced expression of p66Shc protein by shRNA was associated with decreased growth rates of LNCaP C-81 (Fig. 3B) and PC-3 cells (Fig. 3C) [36]. In p66Shc shRNA-transfected cells, ERK1/2 activation was also decreased, lower than that in vector alone-transfected control cells (Fig. 3B & 3C). These data together suggest that WT p66Shc via ROS production is involved in regulating PCa cell growth which is in part via ERK pathway.
Fig. 3.
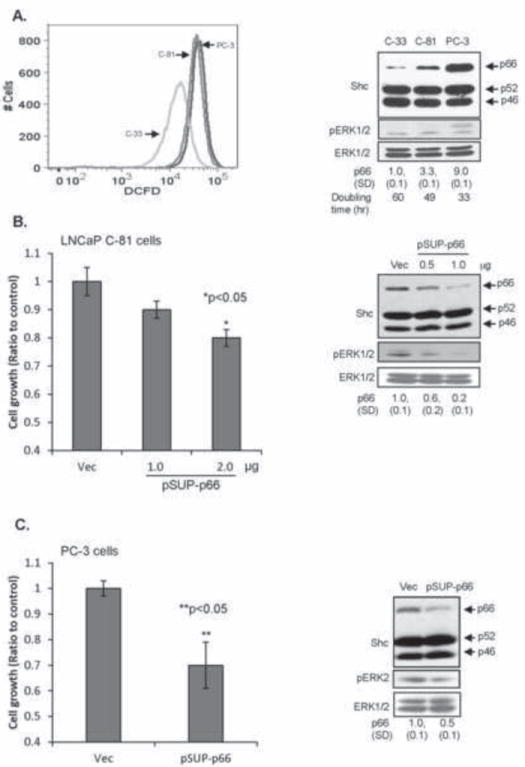
Effect of p66Shc expression on PCa cell growth and ROS production. (A) LNCaP C-33, C-81 and PC-3 cells were labeled with 10 μM DCF-DA for 30 min. The labeled cells were subjected to flow cytometric analysis for quantitative estimation of ROS production (left panel). Cell lysates were analyzed for p66Shc protein and ERK1/2 phosphorylation (right panel). The data shown is a representative of four sets of independent experiments. (B) Cell growth and western blot analyses of p66Shc knock-down LNCaP C-81 cells. LNCaP C-81 cells were plated at a density of 1×105 cells/well of six-well plates in duplicates for 48 h. The cells were transiently transfected with shRNA to p66Shc (pSUP-p66). Control cells were transfected with pSUPER vector alone (Vec). 3 days after transfection, cells were harvested and counted (left panel). Cell lysates from other set of cells transfected with different amounts of pSUP-p66 were analyzed for p66Shc protein and ERK1/2 phosphorylation (right panel). (C) As described in (B), PC-3 cells were transfected with pSUP-p66. Cell growth, p66Shc protein and ERK2 phosphorylation were analyzed 3 days post-transfection. The data shown is the average of two sets of independent experiments.
Elevated p66Shc expression enhances the tumorigenicity of PCa cells
We explored the biological significance of elevated p66Shc proteins in prostate carcinogenesis utilizing xenograft animal models. As shown in Fig. 4A, in male athymic mice, three p66Shc stable subclone cells developed measurable xenograft tumors two weeks after inoculation; while control V-1 cells developed measurable tumors after 4 weeks. Furthermore, over 80% of animals (13/15) inoculated with p66Shc subclone cells developed xenograft tumors by week 6, and 60% of animals (3/5) inoculated with control V-1 cells developed xenograft tumors through the entire experiment, similar to parental C-33 cells [17]. The average size of xenograft tumors by p66Shc subclones was about 4-fold of that by control V-1 cells (data not shown). Similarly, these p66Shc subclone cells developed tumors more rapidly with higher incidence than V-1 control cells in low androgenic female animals (data not shown). At 7, 8 and 9 week time points, the tumor size of p66Shc S-31 and S-36 subclone cells was significantly different from that of corresponding V-1 subclone cells (p<0.05). Further, at 9-week time point, the average tumor size of p66Shc subclone cells was approximately 10-fold of that from V-1 subclone cells (Fig. 4B), supporting that p66Shc subclone cells obtain proliferation ability under androgen-reduced condition (Fig. 2D) with enhanced tumorigenicity (Fig. 4A & 4B). To corroborate clinical relevance, we conducted immunohistochemistry staining of p66Shc protein in PCa archival specimens that contain both cancerous and non-cancerous cells. As shown in Fig. 4C, immunohistochemical staining showed that the prostatic adenocarcinoma (bottom) had increased p66Shc protein level as compared to benign prostatic acini (top). In a total of 78 clinical specimens, p66Shc protein levels in cancerous cells were in general elevated with an average of about 50% higher level of staining index than that in non-cancerous acini cells (p<0.05). Nevertheless, there was no correlation between p66Shc protein level and the Gleason grade or the Pathological stage (data not shown). These data collectively supports the notion that elevated p66Shc protein can increase the tumorigenicity of PCa cells.
Fig. 4.
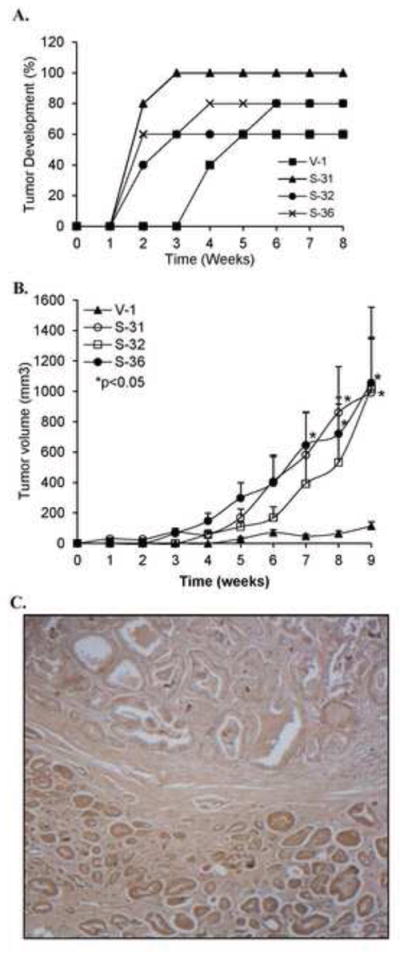
Effect of elevated p66Shc protein expression on PCa tumorigenicity. (A) p66Shc cDNA transfected stable subclone cells (S-31, S-32 and S-36) and control V-1 cells with 1×106 cells each in 0.1 ml medium plus 0.1 ml Matrigel were injected into 5 male nude mice per group. (B) 2×106 cells each in 0.1 ml medium plus 0.1 ml Matrigel were injected into 5 female nude mice per group. The tumor growth was monitored by a caliper weekly. Tumor volume was shown as Mean ± S.E. (S-31: 8 & 9 weeks; S-36: 7, 8 & 9 weeks vs. V-1, *p<0.05) (C) Immunohistochemical staining for p66Shc protein in human prostate tissue archival specimen using anti-p66Shc antibody. Note the prostatic adenocarcinoma (bottom) with higher levels of p66Shc protein as compared to benign prostatic acini (top). Original magnification x200.
p66Shc via ROS mediates DHT-stimulated cell proliferation
To establish the requirement of ROS production by p66Shc protein in DHT-induced PCa cell proliferation, AS LNCaP C-33 cells that were transiently transfected with cDNA encoding WT p66Shc, W134F redox mutant or S36A mutant proteins or empty vector alone were cultured with or without DHT. While W134F p66Shc mutant can translocate to mitochondria, it cannot induce ROS production [13, 26]. On the contrary, S36A p66Shc mutant is retained in cytosol and cannot translocate into mitochondria [13]. Fig. 5A (column #3, 5, 7) showed that, in the absence of DHT, elevated expression of WT p66Shc protein, but not W134F or S36A mutant, correlated with increased cell growth as seen in Fig. 2A. DHT significantly stimulated the proliferation of control cells transfected with vector alone (p<0.05; column #2 vs. #1, Fig. 5A), but not the WT p66Shc cDNA-transfected cell with rapid basal cell growth (column #4 vs. #3, Fig. 5A). Further, expression of p66Shc W134F mutant (column #6), but not S36A mutant (column #8), significantly abolished DHT-stimulated cell proliferation (Fig. 5A). Similar results were observed in MDA PCa2b cells that DHT-induced cell proliferation was significantly blocked by p66Shc W134F mutant (data not shown). Thus, ROS production by p66Shc in mitochondria is involved in DHT-stimulated PCa cell growth.
Fig. 5.
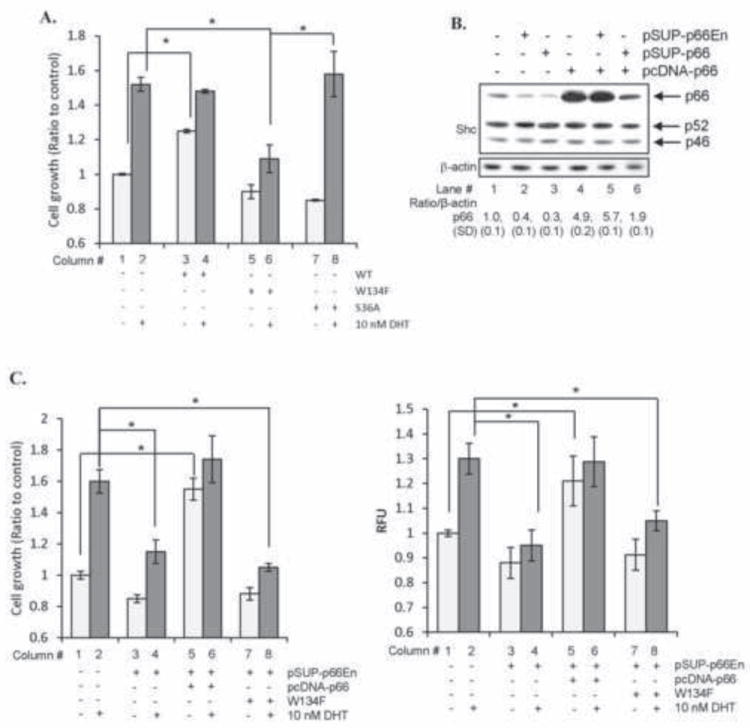
p66Shc via ROS mediates DHT-stimulated cell proliferation. (A) LNCaP C-33 cells were plated at a density of 1×104 cells/cm2 in duplicates. Cells were transfected with either WT, W134F mutant or S36A mutant cDNA of p66Shc. Control cells were transfected with pcDNA 3.1 alone. Cells were then steroid-starved for 48 h and treated with either ethanol or 10 nM DHT. Cell numbers were analyzed after 48 h of treatment. Data shown is the representative of two sets of independent experiments (*p<0.05). (B) Immunoblot analyses were performed for p66Shc protein levels in LNCaP C-33 cells transiently transfected with either shRNA against endogenous p66Shc (pSUP-p66En), p66Shc-specific shRNA (pSUP-p66) or p66Shc cDNA (pcDNA-p66). (C) As described as (A), LNCaP C-33 cells in duplicates were co-transfected with shRNA against endogenous p66Shc (pSUP-p66En) and WT or W134F mutant of p66Shc cDNA. Control cells were transfected with pSUP or pSUP-p66En alone. All cells were further co-transfected with pECFP cDNA to normalize the transfection efficiency. Cells were then steroid-starved for 48 h and subsequently treated with ethanol or 10 nM DHT. Cell proliferation was performed by counting the cells. Results were normalized to the control pSUP vector-transfected cells treated with ethanol. (left panel). ROS was measured after 48 h of DHT treatment using DCF-DA assay. Relative fluorescence units (RFU) were normalized to pECFP fluorescence to nullify the differences in the transfection efficiency (right panel). Data shown is the Mean ± S.E. of one set of representative results from two sets of independent experiments (*p<0.05).
We further performed p66Shc knock-in experiments in p66Shc-knocked-down LNCaP C-33 cells and analyzed its effect on cell growth by DHT. The shRNA to endogenous p66Shc (pSUP-p66En) knockdown endogenous p66Shc alone and allows the expression of exogenous p66Shc driven by the CMV promoter (pcDNA-p66) (lane #5 vs #2, Fig. 5B; [13]); while shRNA to N-terminal coding region (pSUP-p66Shc) can knockdown both endogenous and exogenous p66Shc (lane #6, Fig. 5B). DHT-stimulated cell proliferation was significantly reduced in endogenous p66Shc knocked-down cells (lane #2, Fig. 5B and column #2 vs. #4, Fig. 5C). Elevated expression of exogenous WT p66Shc in endogenous p66Shc-knocked-down cells (lane #5, Fig. 5B) restored their rapid basal cell proliferation, similar to DHT-treated control cells (column #2 vs. #5, Fig. 5C), and DHT could no longer significantly stimulate the growth of these cells (column #5 vs. #6, Fig. 5C). Ectopic expression of W134F mutant in these endogenous p66Shc-knocked-down cells failed to increase either the basal or the DHT-induced cell proliferation (column #7 & #8, Fig. 5C). These p66Shc effects on cell growth were closely associated with the corresponding ROS production (Fig. 5C, right panel). These data together indicate that the ROS production ability of WT p66Shc plays a critical role in mediating DHT-stimulated PCa cell proliferation.
Androgens, p66Shc and ROS signaling activate ErbB-2 tyrosine phosphorylation for cell proliferation
In pursuit of a common downstream signaling molecule that couples androgens with ROS towards cell proliferation, we analyzed Tyr-P profile in C-33 cells upon various stimulations. In DHT- and H2O2-treated C-33 cells, there was a consistent increase in Tyr-P of proteins with molecular weights between ~100 kDa and ~250 kDa (Upper panels, Figs. 6A & 6B). The 185 kDa phosphoprotein with a consistent increase in Tyr-P reacted with anti-ErbB-2 Ab, indicating it to be the ErbB-2 kinase (Lower panels, Figs. 6A & 6B).
Fig. 6.
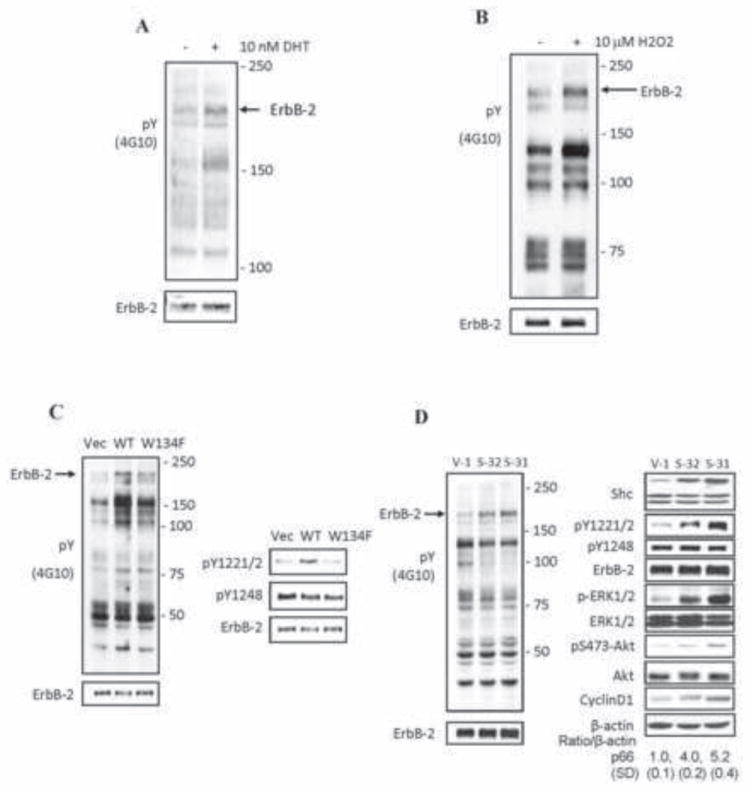
DHT, H2O2 and p66Shc activate ErbB-2 tyrosine phosphorylation in AS PCa cells. (A) LNCaP C-33 cells of 8×103 cells/cm2 were seeded in regular medium for 48 h. After steroid starved for 48 h, cells were treated with 10 nM DHT and harvested after 24 h. Immunoblotting analyses were performed on the total lysate with anti-Tyr(P) antibody (4G10). After stripping, the membrane was re-hybridized with anti-ErbB-2 protein Ab (lower panel). Arrow marks the position of ErbB-2 protein. (B) LNCaP cells were seeded in regular culture medium and then maintained in a SR condition for 48 h. Cells were fed with fresh SR medium and exposed to 10 μM H2O2 for 16 h. Total cell lysate proteins were analyzed for the total tyrosine phosphorylation profile. The membrane was stripped and then re-hybridized for anti-ErbB-2 Ab (lower panel). Arrow points out ErbB-2 position. (C) LNCaP C-33 cells were transiently transfected with WT p66Shc or its W134F redox-defective mutant cDNA. Control cells were transfected with vector alone (Vec). Cells were steroid starved for 48 h and then harvested. The total tyrosine phosphorylation profile (left panel) and the ErbB-2 site-specific phosphorylation (right panel) were analyzed by anti-p-Tyr Ab (4G10) or Ab to the site-specific ErbB-2 phosphorylation. Arrow (left panel) indicates the position of ErbB-2. The ErbB-2 protein was analyzed by immunoblotting after stripping the membrane (lower panel). (D) p66Shc cDNA-transfected LNCaP C-33 stable subclone cells (S-32 and S-31) and vector-alone transfected control cells (V-1) were plated at a density of 8×103 cells/cm2 in regular medium for 48 h. Cells were harvested and the total cell lysate proteins were analyzed for (left panel) the total tyrosine phosphorylation profile, and (right panel) Shc, cyclin D1 protein, the site-specific phosphorylation levels of ErbB-2, ERK and Akt proteins. Arrow (left panel) marks the position of ErbB-2 protein.
Similarly, increased p66Shc expression by transient transfection with WT cDNA in LNCaP C-33 cells (Fig. 6C) and its stable subclone S-31 and S-32 cells (Fig. 6D), ErbB-2 Tyr-P level was elevated (Left panels, Figs. 6C & 6D). On the other hand, expression of p66Shc W134F redox-defective mutant only had a marginal effect on the increase of overall ErbB-2 tyrosine phosphorylation level (Fig. 6C, left panel). Further, in p66Shc WT cDNA transiently transfected cells (Fig. 6C) and its S-31 & S-32 stable subclones (Fig. 6D), ErbB-2 exhibited higher phosphorylation levels at Y1221/2, but not Y1248, than vector-alone (V-1) or redox inactive W134F mutant-transfected control cells (Fig. 6C & 6D, right panels). In p66Shc subclone cells, ERK and AKT were also activated by phosphorylation, correlating with cyclin D1 elevation (Fig. 6D). Thus, ErbB-2 functions as a downstream of steroid-p66Shc-ROS signaling in growth stimulation.
Growth stimuli inactivate the phosphatase activity in cells
In growth factor-treated cells, ROS is produced that inhibit the PTP activity by reversible oxidation resulting in the activation of its corresponding tyrosine kinase(s) [7-10]. We analyzed androgen effect on PTP activity by quantifying intracellular phosphatase activity with a traditional AcP assay using pNPP as a substrate in the presence or absence of L(+)-tartrate, a classical phosphatase inhibitor [37, 38]. As shown in Fig. 7A, DHT treatment of LNCaP C-33 cells led to an average of about 35% decrease in intracellular TSP activity, but not TRP activity. H2O2 treatment also decreased TSP activity, but not TRP activity, in a dose-dependent manner with about 30% inhibition by 20 μM H2O2 (Fig. 7B). Increased expression of p66Shc in S-31 and S-32 stable subclone cells was also associated with down-regulation of approximately 40% TSP, but not TRP activity (Fig. 7C).
Fig. 7.
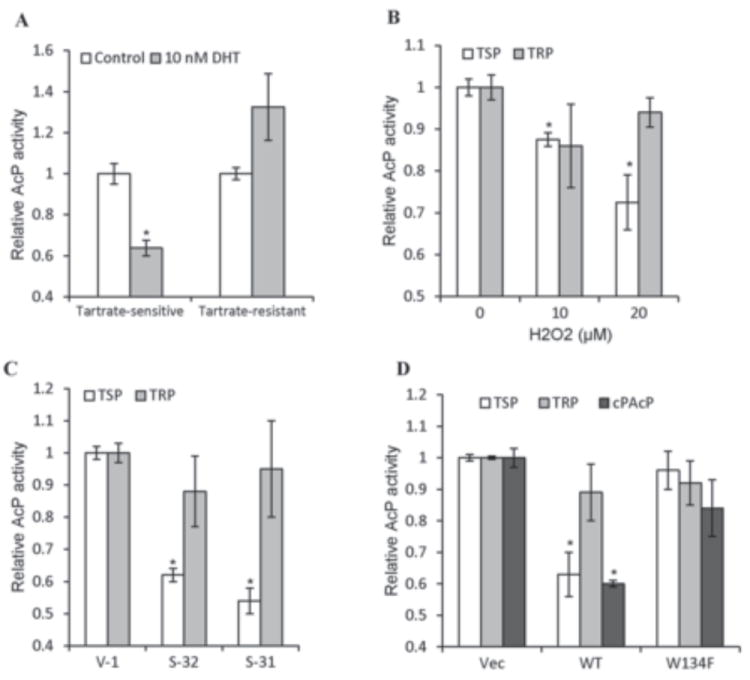
Acid phosphatase activities in steroid- and H2O2-treated and p66Shc cDNA-transfected C-33 cells. (A) LNCaP C-33 cells were seeded in regular medium for 3 days and then steroid-starved for 2 days. Cells were fed with fresh SR medium and treated with 10 nM DHT for 2 days. Total cell lysate proteins were used to analyze L-(+)-tartrate-sensitive (TSP) and tartrate-resistant AcP (TRP) activities in each cell lysate. Similar results were obtained from at least three sets of independent experiments. (n=2×3, *p<0.05). (B) LNCaP C-33 cells were seeded in regular medium for 3 days and then steroid-starved for 2 days. Cells were fed with fresh SR medium and treated with H2O2 for 24 h. Total cell lysate proteins were used to analyze H2O2 effect on TSP and TRP activities. Similar results were obtained from 3 sets of independent experiments. (*p<0.05). (C) Total cell lysate proteins of p66Shc stable subclone S-31 and S-32 cells plus control V-1 cells were analyzed for TSP and TRP phosphatase activity. Similar results were obtained from at least three sets of independent experiments. (*p<0.05). (D) LNCaP C-33 cells were transiently transfected with WT p66Shc (WT) or its W134F redox-defective mutant cDNA (W134F). Another control cells were transfected with vector alone (Vec). After 48 h, cells were harvested for analyzing TSP and TRP activities. An aliquot of each cell lysate proteins was immunoprecipitaed by anti-PAcP Ab and the immunocomplexes were analyzed for cPAcP specific phosphatase activity. Similar results were obtained from at least three sets of independent experiments. (*p<0.01).
While ROS can inactivate several tyrosine phosphatases in different cells; in LNCaP C-33 cells, cPAcP represents more than 90% of total TSP as well as protein tyrosine phosphatase activity [37, 38, 42]. In these cells, cPAcP functions as a neutral PTP and dephosphorylates ErbB-2 at its tyrosyl residues [15, 18, 39]. Further, our preliminary results revealed that cPAcP activity is the most sensitive phosphatase activity to DHT (data not shown). Hence, we focused our efforts on investigating the effect of p66Shc-ROS on cPAcP activity in LNCaP C-33 cells. The TSP and the cPAcP-specific activity in the anti-PAcP immunocomplex were similarly down-regulated by approximately 40% in WT p66Shc cDNA transiently transfected cells, but not in W134F mutant cDNA transfected cells (Fig. 7D). Thus, in androgen- as well as H2O2-treated and p66Shc-transfected cells, cPAcP, but not TRP, activity is decreased, and importantly, p66Shc-induced down-regulation of cPAcP activity requires its redox property.
p66Shc down regulates cPAcP activity by reversible oxidation
We explored the regulatory mechanism of p66Shc on cPAcP activity after immunoprecipitating from S-31 and S-32 cells. Fig. 8A showed that increased expression of p66Shc protein in S-31 and S-32 subclone cells led to the down-regulation of cPAcP activity, as seen for TSP activity in those same cells (Fig. 7C). Importantly, incubation of these cell lysate proteins with 10 mM DTT significantly restored PAcP activity (Fig. 8A). Further, co-transfection of antioxidant enzyme glutathione peroxidase (GPx1) cDNA with WT p66Shc cDNA blocked the inhibitory effect of p66Shc protein on TSP activity (Fig. 8B). Similarly, in H2O2-treated C-33 cells, cPAcP specific activity was decreased, following the H2O2 dose response fashion, with about 40% inhibition by 20 μM H2O2; while the 50 kDa cPAcP protein level was not decreased in H2O2-treated cells after normalizing to β-actin as a loading control (Fig. 8C).
Fig. 8.
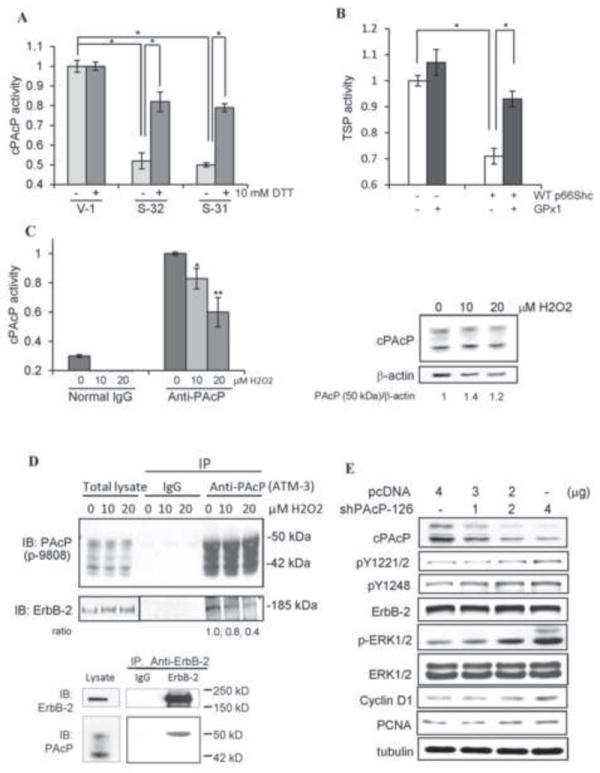
Oxidation-induced inactivation of cPAcP in p66Shc cDNA-transfected PCa cells. (A) The total cell lysate proteins from S-31, S-32 and V-1 cells were prepared for immunoprecipitation of cPAcP. The immunocomplexes was then incubated with or without 10 mM DTT for 10 min at room temperature. The cPAcP-specific activity in the immunocomplexes was quantified. Similar results were obtained from at least three sets of independent experiments. The data shown is one set of representative results. (*p<0.05). (B) LNCaP C-33 cells were transiently co-transfected with WT p66Shc (WT) cDNA plus glutathione peroxidase 1 (GPx1) cDNA. Control cells were transfected with p66Shc cDNA, GPx1 cDNA or the vector alone. After 48 h, cells were harvested for analyzing TSP activity. Similar results were obtained from two sets of independent experiments. (*p<0.05). (C) LNCaP C-33 cells were seeded in regular medium and then maintained in SR medium for 2 days. After being treated with 0, 10 and 20 μM H2O2 for 24 h, total cell lysate proteins were used to immunoprecipitate cPAcP by anti-PAcP Ab. Normal IgG was used as a control for immunoprecipitation. The AcP activity in the immunocomplex was quantified (left panel, n=2×3; *p<0.05; **p<0.01). The total cell lysate proteins were analyzed for cPAcP protein levels. β-actin protein level was used as a loading control. (D) Interaction of cPAcP and ErbB-2 in LNCaP C-33 cells. (Upper panel) After attachments for 3 days in regular medium, LNCaP C-33 cells were maintained in a SR medium for 48 h and exposed to 0, 10 and 20 μM H2O2. Cells were then harvested, lysed and immunoprecipitated by anti-PAcP Ab (#ATM-3) or normal IgG as a control. The immunoprecipitate was used for western blotting with anti-PAcP Ab (Sigma #P-9808) and anti-ErbB-2 Ab (C-18). Similar results were obtained from three sets of independent experiments. (Lower panel) ErbB-2 was immunoprecipitated from steroid-starved LNCaP C-33 cell lysate proteins using anti-ErbB-2 mouse IgG (9G6 Ab) coupled to protein G-Sepharose beads. The immunoprecipitate complexes were analyzed by western blotting with anti-ErbB-2 rabbit IgG (C-18 Ab) and anti-PAcP mouse Ab (Sigma, USA). Only the 50 kD form of PAcP was seen to be co-immunoprecipitated with ErbB-2 even after prolonged exposure. The data is the representative of five sets of independent experiments. (E) Effect of transient knockdown of cPAcP by shRNA in LNCaP C-33 cells on tyrosine phosphorylation of ErbB-2. LNCaP C-33 cells were transiently transfected with different amounts of PAcP shRNA-126 plasmids. Control cells were transfected with vector containing scrambled oligonucleotides (pcDNA). Total lysate proteins were analyzed for cPAcP protein levels, the site-specific ErbB-2 phosphorylation, ERK phosphorylation, cyclin D1 and PCNA protein levels. Tubulin was used as a loading control.
ErbB-2 is shown as an in vivo substrate of cPAcP [14, 18, 39], and its Tyr-P inversely correlates with cPAcP activity upon various treatments (Fig. 6 vs. Figs. 7D, 8A & 8C). We analyzed H2O2 effect on the interaction of cPAcP with ErbB-2. In a non-permissive SR growth condition, in the absence of H2O2, cPAcP and ErbB-2 were co-immunoprecipitated by anti-PAcP Ab (upper panel, Fig. 8D). Treatment with H2O2 reduced the amount of interaction between cPAcP and ErbB-2 for about 60% by 20 μM H2O2 (upper panel, Fig. 8D). It should be noted that the 50 kDa protein is the mature glycosylated PAcP protein; while the 42 kDa protein is an intermediate form during biosynthesis [15, 37]. The reciprocal co-immunoprecipitation with anti-ErbB-2 Ab showed that only the mature 50 kDa PAcP binds to ErbB-2 in PCa cells (lower panel, Fig. 8D). The other forms did not appear even after a prolonged period of exposure of the X-ray film to the western blot (data not shown). The ErbB-2 dephosphorylation by cPAcP was further shown that in cPAcP-knocked down C-33 cells by shRNA, ErbB-2 was activated by increased Tyr-P at Y1221/2 and also Y1248, and ERK activation by phosphorylation, correlating with increased cell proliferation shown by cyclin D1 and PCNA elevation (Fig. 8E) [15]. The data collectively indicated that ROS generated by p66Shc down regulates cPAcP specific activity by reversible oxidation and dissociates it from interacting with ErbB-2; subsequently, ErbB-2 is activated by increasing Tyr-P, which results in activating of downstream signaling and increased PCa cell proliferation.
DISCUSSION
In this communication, our results reveal a novel ROS-Tyr-P pathway in transducing non-genomic androgen action on cell growth regulation. While p66Shc can be involved in androgen action, the molecular mechanisms require further investigations [13]. Our data first showed p66Shc via ERK1/2 signaling in regulating cell proliferation (Fig. 3). Our data further revealed that androgens, via elevated p66Shc [41], induce oxidation-inactivation of PTPs to mediate PCa cell proliferation (Fig. 8). In PCa cells, p66Shc-ROS signaling down-regulates cPAcP specific activity (Figs. 7D, 8A, 8C), which reduces its interaction with ErbB-2 (Fig. 8D), and subsequently ErbB-2 is activated by Tyr-P (Fig. 6), leading to cell proliferation. Similarly, knockdown cPAcP expression by shRNA resulted in ErbB-2 activation, and increased cell growth (Fig. 8E). Inhibition of cPAcP activity by oxidation is confirmed by GPx1 co-expression with p66Shc or by incubating cell lysate proteins of WT p66Shc cDNA-transfected cells with 10 mM DTT in that both these reducing environments restore cPAcP activity (Fig. 8A & 8B). Furthermore, p66Shc (W134F) redox-defective mutant has no effect on inactivating cPAcP (Fig. 7D) or activating ErbB-2 (Fig. 6C). Collectively, our study, for the first time, reveals a novel molecular mechanism by which androgens via reversible oxidation of PTPs transduce their non-genomic signaling to up-regulate cell proliferation. Our study also sheds light on the importance of p66Shc and ErbB-2 as critical signaling molecules in this process, and provides a mechanistic explanation of observations on clinical specimens that p66Shc protein level is higher in prostate carcinomas than in respective non-cancerous cells (Fig. 4C).
We would like to point it out that the lack of significant difference in the xenograft tumor size between S-32 subclone vs. V-1 subclone is at least in part due to the variation in S-32-induced tumor sizes (Fig. 4B). While, it is known that elevated p66Shc protein can translocate into mitochondria and interact with Cyt c for ROS production [13, 22, 26]; p66Shc can interact with Rac1 at plasma membrane for ROS production [27]. Further experiments are required to determine the mechanism of p66Shc-induced tyrosine phosphorylation signaling for cell proliferation.
While inactivation of PTPs by growth factors through oxidation has been reported [7-10], steroid action via redox to Tyr-P signaling on cell proliferation remains unknown. In PCa cells, TSP but not TRP activity is sensitive to both steroid-induced and ROS-mediated inactivation (Fig. 7 and [37, 38]). Since cPAcP represents the major TSP as well as PTP activity in differentiated prostate epithelia and its activity is sensitive to DHT treatment [37, 38, 42], and also since cPAcP can interact with ErbB-2 in PCa cells [17-19], we explored the role of cPAcP in DHT-p66Shc-ROS-mediated proliferation of PCa cells. Treatment of PCa cells with androgens results in increased ROS production and cell proliferation (Figs. 1A, 1B & 1C and [13]) and down regulation of cPAcP activity [18, 37-39]. Recent studies have shed light on the role of ROS in the inactivation of cysteine-dependent PTPs in the process of growth stimulation by growth factors [7, 9]. While oxidation of histidine residue in superoxide dismutase has been reported, its biological significance in growth regulation is not known [12]. cPAcP is an excellent candidate to explore this mode of regulation because cPAcP is a histidine-dependent, but cysteine-independent, PTP and regulates growth signaling in PCa cells [11, 19]. Nevertheless, in cells, there are many PTPs that are sensitive to ROS. It is thus possible that ROS may inactivate other tyrosine phosphatases leading to the activation of kinase signaling. Since our studies involved the cross-talk between androgen, ROS and tyrosine phosphorylation, we initially analyzed DHT effect on other PTP activities. Our preliminary data showed that while cPAcP activity is very sensitive to DHT treatment; Shp1 and Shp2 activities are only marginally affected upon DHT treatment (data not shown). In parallel, PTP1B is up-regulated by DHT (data not shown) [43]. Further experiments are required to clarify if ROS and/or DHT inactivates other tyrosine phosphatases, leading to the activation of tyrosine kinase signaling for cell proliferation. In summary, our study clearly reveals a novel role of a histidine-dependent PTP, such as cPAcP, that responds to ROS signals, which resulted in the activation of tyrosine kinase, i.e., ErbB-2, by phosphorylation and cell proliferation.
PAcP has a key histidine residue (His12) at its active site that serves as a phosphate acceptor [19, 44], and thus cPAcP functions as a neutral histidine-dependent PTP inside PCa cells [14, 17-19]. While His12 is critical for both its AcP and PTP activities of PAcP [19, 44, 45]; one cysteine residue (Cys183) within the close proximity of the active domain is not critical for its activity [17, 19]. In fact, several prokaryotic and eukaryotic histidine-dependent PTPs have inactive cysteine near their active sites and share a sequence similarity with cPAcP [11]. Apparently, due to their close functional and active site sequence homology with PAcP, we faced the difficulty to express cPAcP in prokaryotes with high copy number plasmids in our preliminary studies (Zelivianski S. and Lin M.F., unpublished data). Thus, PAcP can serve as a prototype model for studying the regulation of histidine-dependent PTP activity.
Formation of oxo-histidine from histidine has been shown to occur under oxidative stress conditions in cells [12]. Thus, we hypothesize that ROS converts the active site histidine residue (His12) of PAcP into oxo-histidine that inhibits its phosphate acceptor function and down regulates PAcP activity. Nevertheless, due to the reversibility of cPAcP activity by DTT (Fig. 8A), despite the fact that the active site cysteine residue (e.g. Cys183) is not a critical determinant for PTP activity of PAcP; oxidation of cysteine residue leading to conformational changes in active site of PAcP resulting in its inability to bind and/or dephosphorylate ErbB-2 could not be overruled currently. Direct biochemical studies are required for addressing the reversible formation of oxo-histidine in cPAcP.
In human PCa cells, while different post-translationally modified PAcP forms are seen in the immunoblots of total cell lysate (Fig. 8D), only the 50 kDa mature form of PAcP protein interacts with ErbB-2 (Fig. 8D) [18, 39]. In parallel, ErbB-2 can be activated by phosphorylation at multi tyrosine residues, including Y1221/2 and Y1248 [18, 46], and human and rat PAcP preferentially dephosphorylate ErbB-2 at pY1221/2 and also pY1248 [18, 19, 39, 45]. In p66Shc-elevated cells, where cPAcP is inactivated by ROS (Figs. 7 & 8), ErbB-2 is activated by phosphorylation at Y1221/2, but not Y1248 (Fig. 6C & 6D). Kinetic analyses revealed that knockdown cPAcP correlates with ErbB-2 activation initially at Y1221/2 phosphorylation and followed by Y1248 phosphorylation [18]. Activation of ErbB-2 at these tyrosine residues can lead to ERK-MAPK (Figs. 6D & 8E, and [18, 46]) and AKT activation (Fig. 6D and [18]). In fact, p66Shc also mediates growth stimulation through activation of ERK1/2 in PCa cells (Fig. 3 and [36]), and ERK1/2 are activated in some CR PCa clinical specimens [47]. While the biological significance of signal activation by each individual residue of ErbB-2 in PCa growth stimulation is not known, it is clinically important to elucidate whether p66Shc-ROS-cPAcP signaling selectively favors pY1221/2-mediated signaling in PCa cells cultured under SR growth conditions, as p66Shc overexpression and tyrosine phosphorylation experiments in the current study were performed in a SR condition, mimicking androgen ablation practiced in clinics.
In conclusion, our study reveals a novel molecular mechanism by which androgens up-regulate PCa cell proliferation at least in part by stabilizing p66Shc protein [41] to increase intracellular ROS production, which can reversibly oxidize PTPs and subsequently can activate ErbB-2 signaling (Fig. 9). This non-genomic growth signaling by androgens depends on p66Shc and its redox property. Our data further point towards the possibility of ROS being a critical mediator of androgen-induced carcinogenesis in prostate cells. Evidently, elevated p66Shc protein in PCa cells enhances their tumorigenicity in xenograft animal models. Our data also provide a mechanistic explanation of the biological significance in the observation that p66Shc protein level is elevated in several steroid hormone-related clinical carcinomas and correlates with their tumor progression.
Fig. 9.
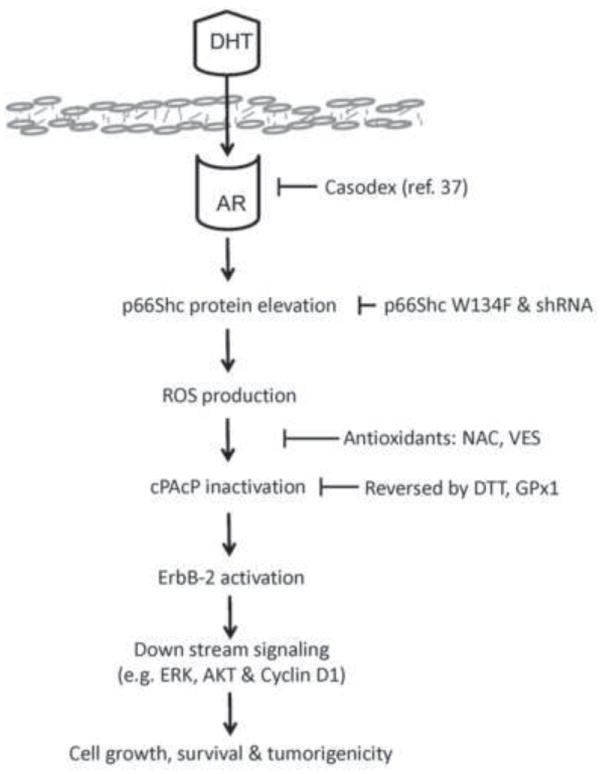
A schematic representation of androgens up-regulating PCa cell proliferation via p66Shc-ROS signal pathway. Androgen-stimulated PCa cell proliferation occurs at least in part through stabilization of p66Shc protein that results in increased ROS production, which leads to reversible cPAcP inactivation and ErbB-2 activation, followed by increased cell growth, survival and tumorigenicity.
Highlights.
A novel p66Shc-ROS-Tyr-P pathway mediates androgen action in PCa cells.
Elevated p66Shc protein in PCa cells increases cell growth and tumorigenicity.
p66Shc-ROS signaling down-regulates PTP activity and activates Tyr-P pathway.
Acknowledgments
This study was supported in part by grants CA88184 and CA125661 (NIH), PC050769 and PC074289 (DOD), #2010-18 (LB506, NE DHHS), and Nebraska Research Initiative (NE). We thank technical supports from the UNMC Flow Cytometry core facility which is supported by UNMC Eppley Cancer Center Core grant (P30CA036727, NIH). We thank Dr. Tsai-Der Chuang for performing shPAcP-126 transfection experiments.
Abbreviations
- Ab
antibody
- AcP
acid phosphatase
- AI
androgen-independent
- AR
androgen receptor
- AS
androgen sensitive
- cPAcP
cellular prostatic acid phosphatase
- Cyt C
Cytochrome C
- DHT
5α-dihydrotestosterone
- EGF
epidermal growth factor
- EGFR
EGF receptor
- ErbB-2
human epidermal growth factor receptor-2
- FBS
fetal bovine serum
- MEF
mouse embryo fibroblast
- NAC
N-acetyl cysteine
- PCa
prostate cancer
- pNPP
para-nitrophenyl phosphate
- PTP
protein tyrosine phosphatase
- pTyr
phosphotyrosine
- ROS
Reactive oxygen species
- Shc
Src homolog and collagen homolog protein
- SR
steroid-reduced
- Tyr-P
tyrosine phosphorylation
- TSP
L(+)-tartrate-sensitive acid phosphatase
- TRP
L(+)-tartrate-resistant acid phosphatase
- VES
vitamin E succinate
- WT
wild type
Footnotes
Disclosure summary:
The authors have nothing to declare.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Lopez-Lazaro M. Dual role of hydrogen peroxide in cancer: possible relevance to cancer chemoprevention and therapy. Cancer Lett. 2007;252:1–8. doi: 10.1016/j.canlet.2006.10.029. [DOI] [PubMed] [Google Scholar]
- 2.Clerkin JS, Naughton R, Quiney C, Cotter TG. Mechanisms of ROS modulated cell survival during carcinogenesis. Cancer Lett. 2008;266:30–36. doi: 10.1016/j.canlet.2008.02.029. [DOI] [PubMed] [Google Scholar]
- 3.Veeramani S, Lin MF. Role of reactive oxygen species in carcinogenesis. In: Banarjee R, editor. Redox Biochemistry. New Jersey: John Wiley & Sons Inc; 2007. pp. 212–218. [Google Scholar]
- 4.Lim SD, Sun C, Lambeth JD, Marshall F, Amin M, Chung L, Petros JA, Arnold RS. Increased Nox1 and hydrogen peroxide in prostate cancer. Prostate. 2005;62:200–207. doi: 10.1002/pros.20137. [DOI] [PubMed] [Google Scholar]
- 5.Oberley TD, Zhong W, Szweda LI, Oberley LW. Localization of antioxidant enzymes and oxidative damage products in normal and malignant prostate epithelium. Prostate. 2000;44:144–155. doi: 10.1002/1097-0045(20000701)44:2<144::aid-pros7>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- 6.Szatrowski TP, Nathan CF. Production of large amounts of hydrogen peroxide by human tumor cells. Cancer Res. 1991;51:794–798. [PubMed] [Google Scholar]
- 7.Lou YW, Chen YY, Hsu SF, Chen RK, Lee CL, Khoo KH, Tonks NK, Meng TC. Redox regulation of the protein tyrosine phosphatase PTP1B in cancer cells. FEBS J. 2008;275:69–88. doi: 10.1111/j.1742-4658.2007.06173.x. [DOI] [PubMed] [Google Scholar]
- 8.Meng TC, Buckley DA, Galic S, Tiganis T, Tonks NK. Regulation of insulin signaling through reversible oxidation of the protein-tyrosine phosphatases TC45 and PTP1B. J Biol Chem. 2004;279:37716–37725. doi: 10.1074/jbc.M404606200. [DOI] [PubMed] [Google Scholar]
- 9.Meng TC, Fukada T, Tonks NK. Reversible oxidation and inactivation of protein tyrosine phosphatases in vivo. Mol Cell. 2002;9:387–399. doi: 10.1016/s1097-2765(02)00445-8. [DOI] [PubMed] [Google Scholar]
- 10.Tonks NK. Protein tyrosine phosphatases: from genes, to function, to disease. Nat Rev Mol Cell Biol. 2006;7:833–846. doi: 10.1038/nrm2039. [DOI] [PubMed] [Google Scholar]
- 11.Veeramani S, Lee MS, Lin MF. Revisiting histidine-dependent acid phosphatases: a distinct group of tyrosine phosphatases. Trends Biochem Sci. 2009;34:273–278. doi: 10.1016/j.tibs.2009.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Uchida K, Kawakishi S. Identification of oxidized histidine generated at the active site of Cu,Zn-superoxide dismutase exposed to H2O2. Selective generation of 2-oxo-histidine at the histidine 118. J Biol Chem. 1994;269:2405–2410. [PubMed] [Google Scholar]
- 13.Veeramani S, Yuan TC, Lin FF, Lin MF. Mitochondrial redox signaling by p66Shc is involved in regulating androgenic growth stimulation of human prostate cancer cells. Oncogene. 2008;27:5057–5068. doi: 10.1038/onc.2008.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lin MF, Lee MS, Zhou XW, Andressen JC, Meng TC, Johansson SL, West WW, Taylor RJ, Anderson JR, Lin FF. Decreased expression of cellular prostatic acid phosphatase increases tumorigenicity of human prostate cancer cells. J Urol. 2001;166:1943–1950. [PubMed] [Google Scholar]
- 15.Veeramani S, Yuan TC, Chen SJ, Lin FF, Petersen JE, Shaheduzzaman S, Srivastava S, MacDonald RG, Lin MF. Cellular prostatic acid phosphatase: a protein tyrosine phosphatase involved in androgen-independent proliferation of prostate cancer. Endocr Relat Cancer. 2005;12:805–822. doi: 10.1677/erc.1.00950. [DOI] [PubMed] [Google Scholar]
- 16.Zylka MJ, Sowa NA, Taylor-Blake B, Twomey MA, Herrala A, Voikar V, Vihko P. Prostatic acid phosphatase is an ectonucleotidase and suppresses pain by generating adenosine. Neuron. 2008;60:111–122. doi: 10.1016/j.neuron.2008.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Igawa T, Lin FF, Rao P, Lin MF. Suppression of LNCaP prostate cancer xenograft tumors by a prostate-specific protein tyrosine phosphatase, prostatic acid phosphatase. Prostate. 2003;55:247–258. doi: 10.1002/pros.10240. [DOI] [PubMed] [Google Scholar]
- 18.Chuang TD, Chen SJ, Lin FF, Veeramani S, Kumar S, Batra SK, Tu Y, Lin MF. Human prostatic acid phosphatase, an authentic tyrosine phosphatase, dephosphorylates ErbB-2 and regulates prostate cancer cell growth. J Biol Chem. 2010;285:23598–23606. doi: 10.1074/jbc.M109.098301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang XQ, Lee MS, Zelivianski S, Lin MF. Characterization of a prostate-specific tyrosine phosphatase by mutagenesis and expression in human prostate cancer cells. J Biol Chem. 2001;276:2544–2550. doi: 10.1074/jbc.M006661200. [DOI] [PubMed] [Google Scholar]
- 20.Migliaccio E, Giorgio M, Mele S, Pelicci G, Reboldi P, Pandolfi PP, Lanfrancone L, Pelicci PG. The p66shc adaptor protein controls oxidative stress response and life span in mammals. Nature. 1999;402:309–313. doi: 10.1038/46311. [DOI] [PubMed] [Google Scholar]
- 21.Migliaccio E, Mele S, Salcini AE, Pelicci G, Lai KM, Superti-Furga G, Pawson T, Di Fiore PP, Lanfrancone L, Pelicci PG. Opposite effects of the p52shc/p46shc and p66shc splicing isoforms on the EGF receptor-MAP kinase-fos signalling pathway. EMBO J. 1997;16:706–716. doi: 10.1093/emboj/16.4.706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nemoto S, Combs CA, French S, Ahn BH, Fergusson MM, Balaban RS, Finkel T. The mammalian longevity-associated gene product p66shc regulates mitochondrial metabolism. J Biol Chem. 2006;281:10555–10560. doi: 10.1074/jbc.M511626200. [DOI] [PubMed] [Google Scholar]
- 23.Orsini F, Migliaccio E, Moroni M, Contursi C, Raker VA, Piccini D, Martin-Padura I, Pelliccia G, Trinei M, Bono M, Puri C, Tacchetti C, Ferrini M, Mannucci R, Nicoletti I, Lanfrancone L, Giorgio M, Pelicci PG. The life span determinant p66Shc localizes to mitochondria where it associates with mitochondrial heat shock protein 70 and regulates trans-membrane potential. J Biol Chem. 2004;279:25689–25695. doi: 10.1074/jbc.M401844200. [DOI] [PubMed] [Google Scholar]
- 24.Ravichandran KS. Signaling via Shc family adapter proteins. Oncogene. 2001;20:6322–6330. doi: 10.1038/sj.onc.1204776. [DOI] [PubMed] [Google Scholar]
- 25.Ventura A, Maccarana M, Raker VA, Pelicci PG. A cryptic targeting signal induces isoform-specific localization of p46Shc to mitochondria. J Biol Chem. 2004;279:2299–2306. doi: 10.1074/jbc.M307655200. [DOI] [PubMed] [Google Scholar]
- 26.Giorgio M, Migliaccio E, Orsini F, Paolucci D, Moroni M, Contursi C, Pelliccia G, Luzi L, Minucci S, Marcaccio M, Pinton P, Rizzuto R, Bernardi P, Paolucci F, Pelicci PG. Electron transfer between cytochrome c and p66Shc generates reactive oxygen species that trigger mitochondrial apoptosis. Cell. 2005;122:221–233. doi: 10.1016/j.cell.2005.05.011. [DOI] [PubMed] [Google Scholar]
- 27.Khanday FA, Santhanam L, Kasuno K, Yamamori T, Naqvi A, Dericco J, Bugayenko A, Mattagajasingh I, Disanza A, Scita G, Irani K. Sos-mediated activation of rac1 by p66shc. J Cell Biol. 2006;172:817–822. doi: 10.1083/jcb.200506001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pandolfi S, Bonafe M, Di Tella L, Tiberi L, Salvioli S, Monti D, Sorbi S, Franceschi C. p66(shc) is highly expressed in fibroblasts from centenarians. Mech Ageing Dev. 2005;126:839–844. doi: 10.1016/j.mad.2005.03.004. [DOI] [PubMed] [Google Scholar]
- 29.Grossman SR, Lyle S, Resnick MB, Sabo E, Lis RT, Rosinha E, Liu Q, Hsieh CC, Bhat G, Frackelton AR, Jr, Hafer LJ. p66 Shc tumor levels show a strong prognostic correlation with disease outcome in stage IIA colon cancer. Clin Cancer Res. 2007;13:5798–5804. doi: 10.1158/1078-0432.CCR-07-0073. [DOI] [PubMed] [Google Scholar]
- 30.Lee MS, Igawa T, Chen SJ, Van Bemmel D, Lin JS, Lin FF, Johansson SL, Christman JK, Lin MF. p66Shc protein is upregulated by steroid hormones in hormone-sensitive cancer cells and in primary prostate carcinomas. Int J Cancer. 2004;108:672–678. doi: 10.1002/ijc.11621. [DOI] [PubMed] [Google Scholar]
- 31.Park YJ, Kim TY, Lee SH, Kim H, Kim SW, Shong M, Yoon YK, Cho BY, Park DJ. p66Shc expression in proliferating thyroid cells is regulated by thyrotropin receptor signaling. Endocrinology. 2005;146:2473–2480. doi: 10.1210/en.2004-1588. [DOI] [PubMed] [Google Scholar]
- 32.Xie Y, Hung MC. p66Shc isoform down-regulated and not required for HER-2/neu signaling pathway in human breast cancer cell lines with HER-2/neu overexpression. Biochem Biophys Res Commun. 1996;221:140–145. doi: 10.1006/bbrc.1996.0559. [DOI] [PubMed] [Google Scholar]
- 33.Davol PA, Bagdasaryan R, Elfenbein GJ, Maizel AL, Frackelton AR., Jr Shc proteins are strong, independent prognostic markers for both node-negative and node-positive primary breast cancer. Cancer Res. 2003;63:6772–6783. [PubMed] [Google Scholar]
- 34.Stevenson LE, Frackelton AR., Jr Constitutively tyrosine phosphorylated p52 Shc in breast cancer cells: correlation with ErbB2 and p66 Shc expression. Breast Cancer Res Treat. 1998;49:119–128. doi: 10.1023/a:1006007227747. [DOI] [PubMed] [Google Scholar]
- 35.Alam SM, Rajendran M, Ouyang S, Veeramani S, Zhang L, Lin MF. A novel role of Shc adaptor proteins in steroid hormone-regulated cancers. Endocr Relat Cancer. 2009;16:1–16. doi: 10.1677/ERC-08-0179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Veeramani S, Igawa T, Yuan TC, Lin FF, Lee MS, Lin JS, Johansson SL, Lin MF. Expression of p66(Shc) protein correlates with proliferation of human prostate cancer cells. Oncogene. 2005;24:7203–7212. doi: 10.1038/sj.onc.1208852. [DOI] [PubMed] [Google Scholar]
- 37.Lin MF, DaVolio J, Garcia-Arenas R. Expression of human prostatic acid phosphatase activity and the growth of prostate carcinoma cells. Cancer Res. 1992;52:4600–4607. [PubMed] [Google Scholar]
- 38.Lin MF, Lee CL, Clinton GM. Tyrosyl kinase activity is inversely related to prostatic acid phosphatase activity in two human prostate carcinoma cell lines. Mol Cell Biol. 1986;6:4753–4757. doi: 10.1128/mcb.6.12.4753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Meng TC, Lin MF. Tyrosine phosphorylation of c-ErbB-2 is regulated by the cellular form of prostatic acid phosphatase in human prostate cancer cells. J Biol Chem. 1998;273:22096–22104. doi: 10.1074/jbc.273.34.22096. [DOI] [PubMed] [Google Scholar]
- 40.Chen SJ, Karan D, Johansson SL, Lin FF, Zeckser J, Singh AP, Batra SK, Lin MF. Prostate-derived factor as a paracrine and autocrine factor for the proliferation of androgen receptor-positive human prostate cancer cells. Prostate. 2007;67:557–571. doi: 10.1002/pros.20551. [DOI] [PubMed] [Google Scholar]
- 41.Kumar S, Kumar S, Rajendran M, Alam SM, Lin FF, Cheng PW, Lin MF. Steroids up-regulate p66Shc longevity protein in growth regulation by inhibiting its ubiquitination. PLoS One. 2011;6:e15942. doi: 10.1371/journal.pone.0015942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li HC, Chernoff J, Chen LB, Kirschonbaum A. A phosphotyrosyl-protein phosphatase activity associated with acid phosphatase from human prostate gland. Eur J Biochem. 1984;138:45–51. doi: 10.1111/j.1432-1033.1984.tb07879.x. [DOI] [PubMed] [Google Scholar]
- 43.Lessard L, Labbe DP, Deblois G, Begin LR, Hardy S, Mes-Masson AM, Saad F, Trotman LC, Giguere V, Tremblay ML. PTP1B is an androgen receptor-regulated phosphatase that promotes the progression of prostate cancer. Cancer Res. 2012 doi: 10.1158/0008-5472.CAN-11-2602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Van Etten RL, Davidson R, Stevis PE, MacArthur H, Moore DL. Covalent structure, disulfide bonding, and identification of reactive surface and active site residues of human prostatic acid phosphatase. J Biol Chem. 1991;266:2313–2319. [PubMed] [Google Scholar]
- 45.Sharma S, Pirila P, Kaija H, Porvari K, Vihko P, Juffer AH. Theoretical investigations of prostatic acid phosphatase. Proteins. 2005;58:295–308. doi: 10.1002/prot.20335. [DOI] [PubMed] [Google Scholar]
- 46.Lee MS, Igawa T, Lin MF. Tyrosine-317 of p52(Shc) mediates androgen-stimulated proliferation signals in human prostate cancer cells. Oncogene. 2004;23:3048–3058. doi: 10.1038/sj.onc.1207451. [DOI] [PubMed] [Google Scholar]
- 47.Gioeli D, Mandell JW, Petroni GR, Frierson HF, Jr, Weber MJ. Activation of mitogen-activated protein kinase associated with prostate cancer progression. Cancer Res. 1999;59:279–284. [PubMed] [Google Scholar]