

Abstract

Glycation of biopolymers by glucose-derived α-oxo-aldehydes such as methylglyoxal (MG) is believed to play a major role in the complex pathologies associated with diabetes and metabolic disease. In contrast to the extensive literature detailing the formation and physiological consequences of protein glycation, there is little information about the corresponding phenomenon for DNA. To assess the potential contribution of DNA glycation to genetic instability, we prepared shuttle vectors containing defined levels of the DNA glycation adduct N2-(1-carboxyethyl)-2′-deoxyguanosine (CEdG) and transfected them into isogenic human fibroblasts that differed solely in the capacity to conduct nucleotide excision repair (NER). In the NER-compromised fibroblasts, the induced mutation frequencies increased up to 18-fold relative to background over a range of ~10-1400 CEdG adducts/105 dG, whereas the same substrates transfected into NER-competent cells induced a response that was 5-fold over background at the highest adduct density. The positive linear correlation (R2 = 0.998) of mutation frequency with increasing CEdG level in NER-defective cells suggested that NER was the primary if not exclusive mechanism for repair of this adduct in human fibroblasts. Consistent with predictions from biochemical studies using CEdG-substituted oligonucleotides, guanine transversions were the predominant mutation resulting from replication of MG-modified plasmids. At high CEdG levels, significant increases in the number of AT → GC transitions were observed exclusively in NER-competent cells (P < 0.0001). This suggested the involvement of an NER-dependent mutagenic process in response to critical levels of DNA damage, possibly mediated by error-prone Y-family polymerases.
Methylglyoxal (MG) is a highly reactive R-oxo-aldehyde produced endogenously via the nonenzymatic dephosphorylation of glyceraldehyde 3-phosphate and dihydroxyacetone phosphate.1 These reactions have been estimated to generate up to 0.1 mM MG/day in vivo, whereas the catabolism of threonine, the P450-mediated oxidation of ketone bodies, and the decomposition of carbohydrates and their corresponding Amadori products constitute important secondary sources of MG.1-4 Methylglyoxal is a mutagen in human cells5,6 and an effective inducer of apoptosis via the p38 MAPK pathway.7,8 Levels of MG are substantially elevated in both type 1 and 2 diabetes,9,10 and it has been proposed that this may contribute significantly to diabetes-related pathologies, including nephropathy, vascular impairment, and immune suppression.11-13 Methyglyoxal covalently modifies proteins, lipids, RNA, and DNA to form the corresponding advanced glycation end products (AGEs), resulting in modification of their normal function.14-20 Although there is a substantial body of literature about the formation of protein-AGE and its potential involvement in pathologies associated with diabetes, aging, cancer, and Alzheimer’s disease,21-23 information about the potential biological impact of DNA-AGEs is scant. In the absence of efficient removal by repair mechanisms, the accumulation of DNA-AGE may have genotoxic consequences and play an unrecognized role in human disease. Recent biochemical experiments using synthetic DNA containing the DNA-AGE N2-1-(carboxyethyl)-2′-deoxyguanosine (CEdG) have shown that this adduct can partially inhibit replicative DNA polymerases or induce miscoding during replication,24,25 suggesting that glycation of DNA may contribute to genetic instability and mutagenesis in vivo.
Although methylglyoxal and glyoxal adducts of DNA and RNA were initially described more than 40 years ago,17,26 it was only recently that these adducts were shown to occur endogenously in vivo. Immunological methods were initially used to demonstrate elevated levels of CEdG in kidney and aorta of diabetic and uremic patients relative to normal individuals.27 Recently, we described a mass spectrometric method for the determination of levels of CEdG in biological samples and used it to demonstrate significantly elevated levels of CEdG in diabetic animals relative to normoglycemic controls, as well as the first quantitative determination of a DNA-AGE in human breast tumors and normal tissue.28 Levels of CEdG were determined to be ~1–10 in 107 dG residues in tissue-extracted DNA, indicating that DNA glycation adducts may be widely distributed in vivo.
There is some confusion in the literature regarding the predominant adducts produced as a result of DNA glycation, shown in Figure 1. Cyclic imidazopurinone adduct 2 was initially described as the sole product resulting from reaction of MG with guanine;26 however, other laboratories have reported adduct 1 as the major isolated DNA-AGE.19,29 Still other workers have claimed bis-adduct 3 is the nearly exclusive product of DNA glycation.30 Large variations in the reaction conditions, particularly the stoichiometric ratio of oxo-aldehyde to DNA, pH, and reaction times, may account in part for this variability. In our hands, time course product studies of the reaction of guanine deoxynucleosides with MG provided evidence of the formation of all three products; however, LC-ESI-MS/MS analyses of reactions of MG with shuttle vector plasmid pSP189 indicated that CEdG 1 was the predominant thermodynamic adduct, although evidence of the kinetic formation of cyclic adduct 2 as its immediate precursor was also obtained. Adducts were quantified as the number of CEdG adducts per dG and were presumed to account for the induced biological response observed as a function of adduct density.
Figure 1.

Three primary adducts identified from the reaction of dG with methylglyoxal (MG). Asterisks denote chiral centers. Adducts 1 and 2 possess identical molecular weights (m/z 340), whereas adduct 3, corresponding to a bis-MG adduct, has an m/z of 412.
MATERIALS AND METHODS
Chemicals and Reagents
Methylglyoxal was prepared by acid-catalyzed hydrolysis of dimethyl pyruvaldehyde (Sigma-Aldrich, St. Louis, MO) and purified by vacuum distillation.31 Solutions were stored under argon at −80 °C prior to use. Concentrations of MG stock solutions were determined by 1H NMR integration of the methyl group at 2.09 ppm in d6-DMSO relative to a tert-butyl alcohol internal standard. Bovine phosphodiesterase II and alkaline phosphatase were purchased from Sigma-Aldrich. Nuclease P1 was purchased from U.S. Biologicals (Swampscott, MA).
Plasmid Vectors and Mammalian Fibroblasts
Shuttle vector pSP189 containing the supF amber suppressor tRNA mutation marker and Escherichia coli indicator strain MBM7070 [F-lacZ (am)CA7020, lacY1, hsdR−, hsdM+, araD139 Δ(araABC-leu)7679, galU, galK, rpsL, thi] were provided by M. Seidman.32 The pSP189 vector contains viral and bacterial genetic elements that allow replication in both SV40 permissive mammalian cells and E. coli. A unique 8 bp randomized “signature” sequence at the 3′ end of the supF gene allows for discrimination between clonal and independent mutational events.33 This eliminates the inadvertent scoring of “jackpot” mutations in the calculation of mutation frequencies.
Plasmids were isolated by the alkaline lysis method and purified by cesium chloride gradient centrifugation. Host cells consisting of DNA repair deficient human fibroblasts derived from a patient with the XP-G class 2 defect (XP3BR.SV) and repair-competent cells corrected by c-DNA complementation (XP2BI-pXPG1) were provided by A. Sarasin (Institut Gustave Roussy, Villejuif, France). Cells were cultured in Dulbecco’s modified Eagle’s high-glucose medium (Mediatech, Inc., Manassas, VA), supplemented with 4 mM l-glutamine, 100 units/mL penicillin G, 100 μg/mL streptomycin (Gibco, Life Technologies Corp., Carlsbad, CA), and 10% fetal bovine serum (Omega Scientific, Inc., Tarzana, CA). Cells were maintained under a 10% CO2 atmosphere in a humidified incubator. The complemented XP2BI-pXPG1 cells were also supplemented with 0.5 mg/mL Geneticin (G-418 sulfate; Gibco, Life Technologies Corp.) to ensure plasmid maintenance.
Preparation of CEdG-Substituted Plasmid DNA
Plasmid pSP189 (200 μg) was dissolved in ddH2O and reacted with 13, 20, 26, 40, or 53 mM MG at 37 °C for 60 min in a volume of 200 μL. Reactions were stopped by the addition of 300 μL of ddH2O, followed by the removal of excess MG using a Microcon Centrifugal Filter Device equipped with an Ultracel YM-30 membrane (Millipore, Billerica, MA) according to the manufacturer’s protocol. Removal of unreacted MG from plasmid reaction mixtures was monitored by quantitation of the O-(2,3,4,5,6-pentafluorobenzyl)hydroxylamine hydrochloride (PFBOA) derivative of MG by gas chromatography and mass spectrometry (GC-MS) using a Shimadzu GCMS-QP5000 instrument equipped with a Zebron ZB-WAX column (15m×0.25mm×0.5 μm) from Phenomenex (Torrance, CA) as previously described.34 Repeated centrifugal filtration was sufficient for the removal of all traces of unreacted MG. This was validated by the GC-MS method within the limits of detection of the PFBOA derivative (0.131 μM). Modified plasmids were then precipitated with EtOH and stored at −80 °C prior to adduct quantitation by LC-ESI-MS/MS.
Quantification of CEdG in DNA by LC-ESI-MS/MS
Quantitation of CEdG in pSP189 plasmid DNA using LC-ESI-MS/MS was accomplished using the stable isotope dilution method with (R)- and (S)-[15N5]CEdG standards prepared as previously described.28 Methylglyoxal-reacted pSP189 (60-100 μg) was suspended in 125 μL of purified ddH2O containing 16 mM sodium acetate buffer (pH 5.5), 1 mM zinc chloride, and 9.6 mM d-penicillamine. DNA was denatured at 95 °C for 5 min and cooled at 4 °C for 5 min and then hydrolyzed and dephosphorylated with 3 units of nuclease P1 (45 °C for 1 h), followed by 32 units of alkaline phosphatase and 1 unit of bovine phosphodiesterase II in 10 mM CaCl2 (45 °C for 7 h). The digest was diluted to a final volume of 420 μL with purified ddH2O.
A 5 μL aliquot of digest was further diluted to 100 μL prior to quantification of dG in plasmid DNA by HPLC integration using an Agilent 1100 HPLC system (Agilent Technologies, Palo Alto, CA) equipped with a Waters Spherisorb ODS-2 column (250 mm × 4.6 mm × 5 μm, 80 Å). Separation of mononucleosides was achieved using a gradient of acetonitrile in 8 mM sodium acetate buffer (pH 6.9) as described previously.28 Calibration curves were constructed with 2′-dG standards, and good linearity (R2 ≥ 0.99) over a range of 4-250 μg/mL was routinely achieved.
LC-ESI-MS/MS quantitation of CEdG was accomplished using an Agilent 1100 Capillary LC system in line with a 6410 triple quadrupole mass spectrometer in positive ion and multiple-reaction monitoring (MRM) modes. The detector settings were as follows: capillary voltage, 4 kV; source gas temperature, 350 °C; gas flow, 6 L/min; nebulizer pressure, 15 psi; dwell time, 200 ms; fragmentor voltage, 100 V; collision energy, 10. The mass transitions monitored for [15N5]CEdG and CEdG were m/z 345 → m/z 229 and m/z 340 → m/z 224, respectively. Digests were spiked with 4.76 ng/mL (R,S)-[15N5]CEdG and 10 μL of 86% phosphoric acid. Samples were loaded onto strata-X-C cation mixed mode columns (Phenomenex) preconditioned with a MeOH/CH3CN mixture (1:4) followed by 2% phosphoric acid, 0.1% phosphoric acid, and then MeOH using a flow rate of 1 mL/min. Nucleosides were eluted with 3% ammonium hydroxide in a MeOH/CH3CN mixture (1:4) and evaporated to dryness in a centrifugal concentrator. Samples were resuspended with 50 μL of 0.1% formic acid, and 20 μL was introduced via autosampler injection. Chromatography was accomplished on a Prodigy ODS C-18 (250 mm × 2.0 mm × 5 μm) column (Phenomenex) using isocratic elution with a mobile phase of 30% aqueous MeOH and 0.1% formic acid at a flow rate of 0.2 mL/ min. All samples were run in duplicate. The retention times for the R and S diastereomers of CEdG were 4.6 and 6.2 min, respectively. Quantitative data were extracted using Agilent’s Mass Hunter Quantitative Analysis software.
Mutagenesis Assay and Analysis of Mutations
Transfection of plasmids into human fibroblasts, recovery of pSP189, and mutant selection in bacteria were performed essentially as previously described with some recent modifications.35 XP-G or XP-G+ human fibroblasts were seeded at a density of 6 × 105 cells/100 mm plate 24 h prior to transfection. Plasmid DNA (6 μg) was transfected into human cells using the FuGene 6 Reagent (Roche diagnostics, Indianapolis, IN) and cultured for 72 h at 37 °C in a humidified incubator. Plasmid DNA was isolated using the alkaline lysis method. Unreplicated plasmids and RNA were removed by digestion with DpnI (New England BioLabs, Inc., Ipswich, MA) and RNase A (Roche diagnostics), respectively. DNA was recovered by ethanol precipitation with glycogen and introduced into the E. coli MBM7070 indicator strain by electroporation (GenePulser II, Bio-Rad, Hercules, CA).
Bacteria were plated on LB agar supplemented with carbenicillin (120 μg/mL), X-gal (5-bromo-4-chloro-3-indoyl β-d-galactopyranoside) (120 μg/mL), and IPTG (isopropyl β-d-thiogalactopyranoside) (40 μg/mL). The plates were incubated at 37 °C for 12-16 h prior to scoring for normal (blue) and mutant (white or light blue) colonies. Mutation frequency was calculated as (number of white and light blue colonies)/(total number of colonies). Mutant colonies were isolated and cultured to harvest plasmids for sequencing as previously described. Sequencing was performed by Laragen, Inc. (Los Angeles, CA). Statistical significance was calculated using the Fisher’s exact, two-sided test.
RESULTS
Synthesis and Characterization of CEdG-Containing Plasmids
DNA shuttle vectors containing varying amounts of CEdG were prepared for use as transfection substrates for examination of the relationship among adduct level, induced mutation frequency, and repair efficiency. The pSP189 shuttle vector was reacted with an excess of MG (from 20- to 90-fold excess relative to guanine) at 37 °C for 1 h to allow for the formation of DNA containing variable levels of DNA-AGEs. Plasmids were isolated, hydrolyzed, and dephosphorylated to mononucleosides using a standard protocol prior to LC-ESI-MS/MS analyses for dG adducts 1-3.
To provide chromatographic and mass spectrometric reference data for the analysis of potential AGEs in double-stranded DNA, products obtained from reaction of dG mononucleoside with MG were isolated and characterized by LC-ESI-MS/MS. In addition to the previously characterized CEdG diastereomers (1), adducts corresponding to cyclic adduct 2 and bis-MG adduct 3 were also observed in monomer reactions (Figure 1). Adduct 3 was detected as a multiplet of peaks corresponding to four potential diastereomers by scanning the MRM m/z 412 → m/z 296 transition. Cyclic adduct 2 had the same molecular weight and provided the same BH+ fragment ion as CEdG 1 and was tentatively identified as an earlier eluting multiplet corresponding to the common m/z 340 → m/z 224 transition. Attempts to isolate 2 from monomer reaction mixtures by HPLC were unsuccessful, as the diastereomeric mixture was observed to rapidly revert to dG and unidentified non-nucleic acid products. The presence of the cyclic diol containing 2 was confirmed by additional in situ reaction with sodium periodate,26 which resulted in nearly quantitative conversion of 2 to N2-acyl-dG 4 (Figure 2 and the Supporting Information). This was readily identifiable in the MS spectrum by a new m/z 310 → m/z 194 transition. An authentic standard of 4 was prepared using a modification of a previously published procedure to verify this assignment.36
Figure 2.

Oxidative diol cleavage of imidazopurinone 2 to N2-acyl-2′-deoxyguanosine 4 using periodate.
Quantitative determination by LC-ESI-MS/MS of nucleoside AGEs obtained from reactions of plasmid DNA with MG as described above revealed the nearly exclusive formation of (R)- and (S)-CEdG. The bis-MG adduct 3 was not detected in dsDNA following reactions with MG at concentrations up to 53 mM, whereas only trace amounts of cyclic adduct 2 were tentatively identified. Levels of this product were typically ~0.5-1.0% of the abundance of CEdG, and the integrated intensities for peaks potentially corresponding to the diastereomers of 2 were not observed to increase as a function of MG concentration. These results imply that variations in the mutation spectrum observed following transfection with the MG-treated plasmids were due primarily to DNA-AGE 1, (R,S)-CEdG, although some contribution from the cyclic precursor (2) could not be rigorously excluded.
The (R,S)-CEdG levels determined by LC-ESI-MS/MS following reaction with different concentrations of MG are shown in Figure 3. Relative to untreated plasmids, 1 h reactions of DNA using the indicated MG concentrations resulted in ~30-200-fold increases in CEdG adduct levels. Untreated plasmids isolated from E. coli displayed background levels of ~7-10 (R,S)-CEdG adducts/105 dGs, suggesting that CEdG may be a relatively abundant DNA adduct in prokaryotes. The yield of CEdG in plasmid DNA increased as a function of MG concentration, and the (R) and (S) diastereomers of CEdG were produced in approximately equivalent yields.
Figure 3.
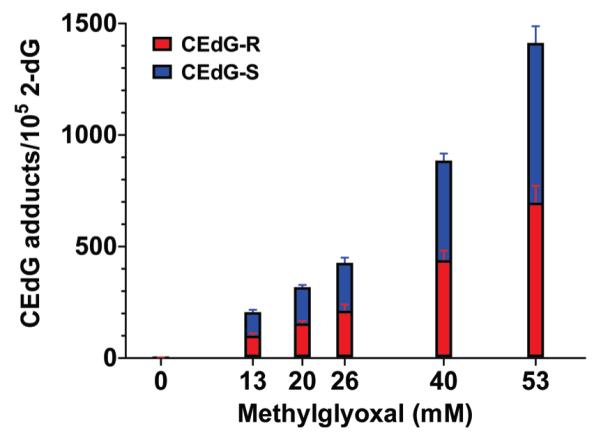
(R)- and (S)-CEdG adduct levels in pSP189 shuttle vectors determined by LC-ESI-MS/MS using the isotope dilution method following a 1 h reaction with the indicated concentrations of MG at 37 °C.
Dose-Dependent Induction of Mutations in Human Cells by CEdG as a Function of NER Status
Fibroblasts from a patient with xeroderma pigmentosum complementation group G (XP-G) and cells corrected for this defect by transfection of plasmids containing the XP-G gene (XP-G+) were used to examine the influence of DNA repair on CEdG-induced mutagenesis. The XP-G fibroblasts were completely deficient in NER while retaining alternative DNA repair pathways, whereas the complemented XP-G+ cells were normal in all aspects of repair. Plasmid pSP189 DNA containing the CEdG adduct levels shown in Figure 3 was transfected into XP-G and XP-G+ fibroblasts and allowed to undergo a single round of replication. This DNA was then isolated and transformed into an E. coli indicator strain to screen for mutations in the supF tRNA marker gene. The induced mutation frequencies as a function of adduct dose in both classes of human fibroblasts are shown in Figure 4. In the repair proficient XP-G+ cells, the mutation frequency was elevated nearly 3-fold over background at an adduct level of ~200 CEdG adducts/105 dGs. Increases in adduct levels up to 1400 CEdG adducts/105 dGs yielded a maximal 5-fold increase relative to background in repair-competent cells.
Figure 4.
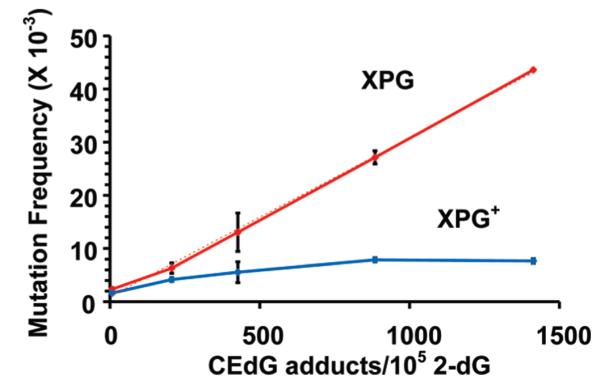
CEdG-induced mutation frequencies in the supF tRNA gene following replication of pSP189 containing the indicated CEdG adduct densities in NER deficient XP-G (red) or repair-competent XP-G+ (blue) human fibroblasts. The dotted line is the calculated linear regression (R2 = 0.998).
In contrast, the induced mutation frequency in the NER deficient fibroblasts increased linearly as a function of CEdG dose. At the highest CEdG levels examined, an 18.5-fold increase over background mutation frequency was observed. This linear relationship suggested that NER was the primary, if not exclusive, pathway for the repair of CEdG in these fibroblasts. If alternative pathways such as mismatch repair (MMR) were able to remove this adduct, a nonlinear correspondence with some attenuation in mutation frequencies at higher damage levels would have been expected, but this was not observed.
Mutation Spectra Induced by CEdG in XP-G and XP-G+ Human Cells
Mutants resulting from a single cycle of replication of pSP189 DNA containing elevated CEdG levels (~1400 CEdG adducts/105 dGs) in XP-G and XP-G+ cells were isolated from a bacterial screen and sequenced. Corresponding data from plasmids with background levels of CEdG (~7 CEdG adducts/105 dGs) were also obtained, and these results are presented in Table 1. Base substitutions in the supF tRNA marker gene comprised the major class of mutations in both cell lines. These occurred almost exclusively at guanines and were primarily transversions. A significant difference in the deletion frequencies between cell lines that was independent of the CEdG adduct level was observed. In the repair-competent XP-G+ line, deletions comprised ~10% of all mutations, consisting predominantly of large (>130 bp) deletions, whereas these were markedly absent in NER deficient fibroblasts.
Table 1.
CEdG-Induced Mutations in Human Fibroblasts Differing in NER Status
| NER Deficient (XP-G) |
NER Proficient (XP-G+) |
|||
|---|---|---|---|---|
| 7 CEdG adducts/ 105 2-dGsa |
1413 CEdG adducts/ 105 2-dGsb |
7 CEdG adducts/ 105 2-dGs |
1413 CEdG adducts/ 105 2-dGs |
|
| no. of independent plasmids analyzedc | 28 (100) | 68 (100) | 32 (100) | 40 (100) |
| no. of base substitutions | 28 (100) | 67 (99) | 29 (91) | 34 (85) |
| single | 26 (93) | 61 (90) | 27 (85) | 34 (85) |
| tandem | 0 | 1 (1) | 1 (3) | 0 |
| multiple | 2 (7) | 5 (8) | 1 (3) | 0 |
| no. of deletions | 0 | 1 (1)e | 3 (9)f | 6 (15) |
| single-base | 0 | 1 (1) | 0 | 2 (5) |
| larged | 0 | 0 | 3 (9) | 4 (10) |
| others | 0 | 0 | 0 | 0 |
| types and no. of base substitution mutations transitions |
7 (23)g | 13 (16)h | 6 (19)i | 15 (44) |
| G·C → A·T | 7 (23) | 10 (12) | 6 (19) | 7 (20.5) |
| A·T → G·C | 0 | 3 (4)j | 0 | 8 (23.5) |
| no. of transversions | 23 (77) | 67 (84) | 25 (81) | 19 (56) |
| G·C → T·A | 17 (57) | 48 (60) | 16 (52) | 12 (35) |
| G·C → C·G | 4 (13) | 15 (19) | 6 (19) | 4 (12) |
| A·T → T·A | 2 (7) | 0 | 1 (3) | 1 (3) |
| A·T → C·G | 0 | 4 (5) | 2 (6) | 2 (6) |
| total | 30 (100) | 80 (100) | 31 (100) | 34 (100) |
Background level of (R,S)-CEdG adducts.
Level of (R,S)-CEdG adducts obtained following reaction with 53 mM MG for 1 h.
Number of independent plasmids with alterations (percentage).
Large deletions encompass more than 130 bp, including and flanking the supF gene.
P = 0.0003 vs a high level of CEdG lesions in XP-G+ cells.
P > 0.05 vs a high level of CEdG in XP-G+ cells.
P > 0.05 vs a high level of CEdG in XP-G cells.
P < 0.0001 vs a high level of CEdG in XP-G+ cells.
P = 0.0002 vs a high level of CEdG in XP-G+ cells.
P < 0.0001 vs a high level of CEdG in XP-G+ cells.
A significant adduct-dependent change in the transition pattern was observed in NER proficient XP-G+ cells. Elevated levels of CEdG adducts induced a >2-fold increase in the level of transitions relative to background levels in this cell line (P = 0.0002), ascribed almost exclusively to A · T → G · C base substitutions. Transitions accounted for 44% of all mutations in XP-G+ fibroblasts at high adduct levels. A · T → G · C substitutions were not observed following replication of DNA containing background levels of CEdG, in either XP-G+ or XP-G cells. When considering only changes in the base substitution pattern, the NER deficient cells appeared to be remarkably insensitive to large variations in CEdG adduct burden, even though the mutation frequency increased by more than 18-fold at the larger dose. No change in the pattern of deletions, transversions, or transitions (P > 0.05) was observed at high adduct levels relative to background in the XP-G fibroblasts.
Sequence Map of supF tRNA Base Substitutions
A complete map of base substitutions within the supF tRNA gene observed in this study is shown in Figure 5. Mutations induced by high CEdG levels are presented above the supF tRNA sequence, whereas mutations resulting from transfection of plasmids with background adduct levels are shown below. Several prominent mutation hot spots, defined as three or more base substitutions at a single site, were observed. A few appear to be strongly induced at elevated CEdG adduct levels in NER deficient cells (colored red), such as those observed at G43, C46, and G87. Other potential CEdG-induced hot spots in XP-G fibroblasts were observed at G30-32 and G50. With the exception of the hot spot at G87, all base substitutions at these sites consisted of G · C → T · A/C · G transversions, consistent with the pattern of mispairing predicted from in vitro polymerase studies with CEdG containing oligonucleotides.24,25 Six of eight A · T → G · C transitions identified in NER-competent cells at high adduct doses were uniquely induced; i.e., there were no other mutations observed at those positions within the supF tRNA gene following replication in either XP-G or XP-G+ cells [A56 (×2), 63 (×2), 94, 108].
Figure 5.

Sequence map of mutations detected in the supF tRNA gene of pSP189 in XP-G and XP-G+ cells. Base substitutions above the sequence were observed following replication of plasmids containing 1.45 CEdG adducts/102 dGs, whereas those below were obtained using DNA containing background levels of 7 CEdG adducts/105 dGs. Tandem mutations are underlined.
DISCUSSION
Several complementary analytical methods, including 32P post-labeling,37 indirect immunostaining,27 and quantitative MS/MS,28 have been used to establish the presence of CEdG in human DNA. Initial measurements of adduct levels in human tissue using isotope dilution mass spectrometry range from 1 to 10 CEdG adducts/107 dGs, suggesting it is a relatively abundant adduct in vivo.28 These values are similar to those measured for the oxidative stress marker 8-hydroxy-2′-deoxyguanosine in urine and tissue.38
The miscoding properties of CEdG have been investigated using synthetic oligonucleotides and replicative bacterial polymerases to measure the steady-state kinetics of dNTP incorporation opposite this adduct.24,25 These experiments revealed the preferential pairing of purines opposite CEdG in a polymerase-independent manner, suggesting a preferred geometry of base pairing likely to favor transversions in vivo. These data support a role for CEdG in the mutations resulting from MG treatment of bacteria and mammalian cells, reported to be predominantly G transversions.5,39
Reaction of dG mononucleoside with MG resulted in the formation of the three previously reported adducts (Figure 1). At neutral pH, cyclic adduct 2 was formed rapidly, although it seemed to partially degrade over a period of 70 h (see the Supporting Information). The attempted isolation of 2 in our hands by HPLC resulted in the recovery of only dG. Frischmann et al. have shown that 2 is not noticeably formed during the reaction of MG with dG under conditions where unreacted MG is scavenged in situ using o-phenylenediamine.20 These observations suggest that cyclic adduct 2, formed from the direct condensation of MG with N1 and N2 of guanine, is likely formed as a kinetic product in equilibrium with MG and dG. We also found that addition of alkaline phosphate buffer to the MG/dG reaction mixture resulted in the rapid and quantitative conversion of 2 to the more thermodynamically stable CEdG (see the Supporting Information). A plausible mechanism for this transformation is shown in Figure 6. Consistent with these observations, the analogous cyclic adduct of glyoxal with dG has also been reported to be unstable.40,41 At pH >7, we additionally observed the rapid formation of bis-MG adduct 3 (see the Supporting Information).
Figure 6.

Proposed mechanism for formation of (R,S)-CEdG 1 via the cyclic adduct 2 arising from the equilibrium reaction of dG with MG. Modified from ref 20.
LC-ESI-MS/MS analyses of hydrolyzed and dephosphorylated nucleoside DNA-AGEs resulting from 1 h reactions of MG with plasmid DNA revealed that only (R,S)-CEdG was formed in a dose-dependent manner. No evidence of bis-MG adduct 3 in plasmid DNA was obtained. Because the bis-MG adduct of the dG monomer was stable for >40 h at pH 9.0 (see the Supporting Information), the failure to observe this product in plasmid DNA was not likely due to inherent chemical instability, but rather an insignificant rate of formation in double-stranded DNA. It is possible that trace amounts of 2 may have contributed to the observed mutation spectrum. However, the exhaustive removal of unreacted MG from plasmid reaction mixtures as well as the slightly alkaline conditions used for transfection would tend to drive the conversion of 2 to 1. We considered the possibility that the alkaline phosphatase protocol used to prepare DNA for MS/MS analyses may have resulted in underestimation of the amount of 2 in plasmid DNA;40 however, the use of an alternative acidic procedure42 did not reveal the presence of additional 2. Taken together, these observations indicate that the dose-dependent mutagenic effects observed were due to (R,S)-CEdG. Moreover, measurements of the intracellular pH of a variety of human cell lines have revealed that the nuclear compartment is slightly alkaline, ranging from pH 7.5 to 8.0.43 This would suggest that alkali labile 2 is not likely to persist in the nucleus in vivo, even under conditions that would favor its initial formation.
The availability of an ensemble of shuttle vector plasmids containing precisely defined levels of DNA damage made it possible to examine the effects of CEdG adduct density on mutation induction and the repair response. Moreover, this strategy obviated the potential issue of sequence context-dependent effects that can arise using single-adduct substituted plasmids, because DNA damage was randomly distributed throughout the marker gene. Transfection of pSP189 vectors bearing defined CEdG levels into fibroblasts differing solely in the ability to conduct NER revealed dramatic differences in induced mutation frequencies that indicated that NER is the primary, if not exclusive, pathway for repair of this adduct in DNA. It is striking that even though other DNA repair mechanisms were presumably active in the XP-G deficient cells, no attenuation of mutation frequency was observed with increasing adduct levels.
In the absence of alternative mechanisms for its removal, CEdG is predicted to accumulate to genotoxic levels in individuals with somatic or inherited defects in NER. This might be particularly important in patients with poor glucose control as a result of metabolic disease who display elevated levels of MG.10,11,44,45 Recent reports linking adiposity and diabetes with impairment of NER and DNA repair in general46,47 lend credence to a potential role for CEdG in contributing to somatic mutagenesis in these individuals. The accumulation of CEdG in patients compromised in their ability to repair it suggests a molecular mechanism that may explain in part the increased incidence of certain cancers associated with diabetes.48,49
Comparison of mutation spectra induced by high and low CEdG adduct levels in human fibroblasts differing in NER capacity indicated a secondary, indirect mutagenic pathway induced by this adduct. A >2-fold increase in the transition frequency was observed in XP-G+ cells at high adduct levels, a change ascribed solely to the sudden appearance of A · T → G · C base substitutions (Table 1). In contrast, in NER deficient XP-G cells, the induced pattern of base substitutions and deletions was unchanged relative to background following transfection of plasmids with elevated levels of CEdG, in spite of a >18-fold increase in the overall mutation frequency. Thus, the changes in the base substitution pattern observed in the XP-G+ cells at high adduct densities appeared to require a functional NER apparatus.
It initially appeared puzzling that the sole changes in the base substitution pattern induced by high CEdG adduct levels occurred at A · T rather than G · C base pairs. It was formally possible that this mutation arose as a result of replication past an adenine adduct(s) produced during reaction with high concentrations of MG. Frischmann et al. have described the formation of carboxyethyl-2′-deoxyadenosine (CEdA) in DNA in low yield following prolonged (1 week) reaction with a 20-fold excess of MG.20 However the fact that A · T → G · C transitions were not a significant feature of the mutation spectra when identically substituted plasmids were transfected into repair deficient XP-G cells suggests that this base substitution did not arise from replication past adenine adducts.
We considered the possibility that recruitment of an error-prone Y-family polymerase with poor fidelity opposite T or A could lead to the observed result. Evidence of the involvement of Y-family polymerases in the gap filling step of NER has recently been presented.50 However, most Y-family polymerases characterized to date induce mispairs at relatively low frequencies opposite undamaged DNA.51,52 An important exception to this general trend is pol ι, which exhibits a 10-fold preference for the incorporation of dGTP over dATP opposite T in template DNA.53-55 Recruitment of pol ι during “repair crisis” induced by high levels of CEdG could result in the observed increase in the level of A · T → G · C mutations as a result of this mispairing event during the gap-filling step of NER. The fact that significant increases in this level of base substitution only occurred in XP-G+ but not XP-G fibroblasts could imply a potential role for XP-G in the recruitment of pol ι.
Although CEdG is only modestly mutagenic in human cells possessing a fully functional NER pathway, it can induce mutations at high frequencies when this repair mechanism is ablated. It has become clear in recent years that substantial interindividual variability in the efficiency of NER exists, with the various xeroderma pigmentosum phenotypes representing one extreme of the range. It has been suggested that these NER polymorphisms may be biomarkers for cancer susceptibility.56,57 Because it appears that NER may be the primary, if not exclusive, mechanism for repair of CEdG in human cells, any loss of efficiency in NER cannot be offset by other repair pathways. Thus, the presence of NER polymorphisms that limit the efficiency of NER would tend to render these individuals susceptible to CEdG-induced genetic instability and possibly increase their risk for cancer.
Supplementary Material
ACKNOWLEDGMENT
The technical assistance of Dr. Bogdan Gugiu and Roger Moore of the Mass Spectrometry Core Facility of the City of Hope Comprehensive Cancer Center is gratefully acknowledged.
Funding Sources Supported by the City of Hope Comprehensive Cancer Center (P30 CA33572) and the California Breast Cancer Research Program for a predoctoral fellowship to D.T. (14GB-0162).
ABBREVIATIONS
- MG
methylglyoxal
- AGEs
advanced glycation end products
- CEdG
N2-(1-carboxyethyl)-2′-deoxyguanosine
- NER
nucleotide excision repair
- XP-G
xeroderma pigmentosum complementationgroup G
- LC-ESI-MS/MS
liquid chromatography-electrospray ionization-tandem mass spectrometry
- MRM
multiple-reaction monitoring
- MMR
mismatch repair
- CEdA
N2-(1-carboxyethyl)-2′-deoxyadenosine
Footnotes
Supporting Information. HPLC chromatograms of products resulting from reaction of dG with MG under different conditions. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- (1).Richard J. Acid-Base Catalysis of the Elimination and Isomerization Reactions of Triose Phosphates. J. Am. Chem. Soc. 1984;106:4926–4936. [Google Scholar]
- (2).Casazza JP, Felver ME, Veech RL. The metabolism of acetone in rat. J. Biol. Chem. 1984;259:231–236. [PubMed] [Google Scholar]
- (3).Homoki-Farkas P, Orsi F, Kroh LW. Methylglyoxal determination from different carbohydrates during heat processing. Food Chem. 1997;59:157–164. [Google Scholar]
- (4).Thornalley PJ, Langborg A, Minhas HS. Formation of glyoxal, methylglyoxal and 3-deoxyglucosone in the glycation of proteins by glucose. Biochem. J. 1999;344(Part 1):109–116. [PMC free article] [PubMed] [Google Scholar]
- (5).Murata-Kamiya N, Kamiya H, Kaji H, Kasai H. Methylglyoxal induces G:C to C:G and G:C to T:A transversions in the supF gene on a shuttle vector plasmid replicated in mammalian cells. Mutat. Res. 2000;468:173–182. doi: 10.1016/s1383-5718(00)00044-9. [DOI] [PubMed] [Google Scholar]
- (6).Hou SM, Nori P, Fang JL, Vaca CE. Methylglyoxal induces hprt mutation and DNA adducts in human T-lymphocytes in vitro. Environ. Mol. Mutagen. 1995;26:286–291. doi: 10.1002/em.2850260404. [DOI] [PubMed] [Google Scholar]
- (7).Liu BF, Miyata S, Hirota Y, Higo S, Miyazaki H, Fukunaga M, Hamada Y, Ueyama S, Muramoto O, Uriuhara A, Kasuga M. Methylglyoxal induces apoptosis through activation of p38 mitogen-activated protein kinase in rat mesangial cells. Kidney Int. 2003;63:947–957. doi: 10.1046/j.1523-1755.2003.00829.x. [DOI] [PubMed] [Google Scholar]
- (8).Fukunaga M, Miyata S, Higo S, Hamada Y, Ueyama S, Kasuga M. Methylglyoxal induces apoptosis through oxidative stress-mediated activation of p38 mitogen-activated protein kinase in rat Schwann cells. Ann. N.Y. Acad. Sci. 2005;1043:151–157. doi: 10.1196/annals.1333.019. [DOI] [PubMed] [Google Scholar]
- (9).Han Y, Randell E, Vasdev S, Gill V, Gadag V, Newhook LA, Grant M, Hagerty D. Plasma methylglyoxal and glyoxal are elevated and related to early membrane alteration in young, complication-free patients with Type 1 diabetes. Mol. Cell. Biochem. 2007;305:123–131. doi: 10.1007/s11010-007-9535-1. [DOI] [PubMed] [Google Scholar]
- (10).Nemet I, Turk Z, Duvnjak L, Car N, Varga-Defterdarovic L. Humoral methylglyoxal level reflects glycemic fluctuation. Clin. Biochem. 2005;38:379–383. doi: 10.1016/j.clinbiochem.2004.12.008. [DOI] [PubMed] [Google Scholar]
- (11).Bourrajjaj M, Stehouwer CD, van Hinsbergh VW, Schalkwijk CG. Role of methylglyoxal adducts in the development of vascular complications in diabetes mellitus. Biochem. Soc. Trans. 2003;31:1400–1402. doi: 10.1042/bst0311400. [DOI] [PubMed] [Google Scholar]
- (12).Beisswenger PJ, Howell SK, Nelson RG, Mauer M, Szwergold BS. R-Oxoaldehyde metabolism and diabetic complications. Biochem. Soc. Trans. 2003;31:1358–1363. doi: 10.1042/bst0311358. [DOI] [PubMed] [Google Scholar]
- (13).Price CL, Knight SC. Methylglyoxal: Possible link between hyperglycaemia and immune suppression? Trends Endocrinol. Metab. 2009;20:312–317. doi: 10.1016/j.tem.2009.03.010. [DOI] [PubMed] [Google Scholar]
- (14).Bulteau AL, Verbeke P, Petropoulos I, Chaffotte AF, Friguet B. Proteasome inhibition in glyoxal-treated fibroblasts and resistance of glycated glucose-6-phosphate dehydrogenase to 20 S proteasome degradation in vitro. J. Biol. Chem. 2001;276:45662–45668. doi: 10.1074/jbc.M105374200. [DOI] [PubMed] [Google Scholar]
- (15).Thornalley PJ, Battah S, Ahmed N, Karachalias N, Agalou S, Babaei-Jadidi R, Dawnay A. Quantitative screening of advanced glycation endproducts in cellular and extracellular proteins by tandem mass spectrometry. Biochem. J. 2003;375:581–592. doi: 10.1042/BJ20030763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Nakagawa K, Oak JH, Higuchi O, Tsuzuki T, Oikawa S, Otani H, Mune M, Cai H, Miyazawa T. Ion-trap tandem mass spectrometric analysis of Amadori-glycated phosphatidylethanolamine in human plasma with or without diabetes. J. Lipid Res. 2005;46:2514–2524. doi: 10.1194/jlr.D500025-JLR200. [DOI] [PubMed] [Google Scholar]
- (17).Staehelin M. Inactivation of virus nucleic acid with glyoxal derivatives. Biochim. Biophys. Acta. 1959;31:448–454. doi: 10.1016/0006-3002(59)90019-8. [DOI] [PubMed] [Google Scholar]
- (18).Bucala R, Model P, Cerami A. Modification of DNA by reducing sugars: A possible mechanism for nucleic acid aging and age-related dysfunction in gene expression. Proc. Natl. Acad. Sci. U.S.A. 1984;81:105–109. doi: 10.1073/pnas.81.1.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Papoulis A, al-Abed Y, Bucala R. Identification of N2-(1-carboxyethyl)guanine (CEG) as a guanine advanced glycosylation end product. Biochemistry. 1995;34:648–655. doi: 10.1021/bi00002a032. [DOI] [PubMed] [Google Scholar]
- (20).Frischmann M, Bidmon C, Angerer J, Pischetsrieder M. Identification of DNA adducts of methylglyoxal. Chem. Res. Toxicol. 2005;18:1586–1592. doi: 10.1021/tx0501278. [DOI] [PubMed] [Google Scholar]
- (21).Kalousova M, Zima T, Tesar V, Dusilova-Sulkova S, Skrha J. Advanced glycoxidation end products in chronic diseases: Clinical chemistry and genetic background. Mutat. Res. 2005;579:37–46. doi: 10.1016/j.mrfmmm.2005.03.024. [DOI] [PubMed] [Google Scholar]
- (22).van Heijst JW, Niessen HW, Hoekman K, Schalkwijk CG. Advanced glycation end products in human cancer tissues: detection of N-ε-(carboxymethyl)lysine and arg-pyrimidine. Ann. N.Y. Acad. Sci. 2005;1043:725–733. doi: 10.1196/annals.1333.084. [DOI] [PubMed] [Google Scholar]
- (23).Takeuchi M, Yamagishi S. Possible involvement of advanced glycation end-products (AGEs) in the pathogenesis of Alzheimer’s disease. Curr. Pharm. Des. 2008;14:973–978. doi: 10.2174/138161208784139693. [DOI] [PubMed] [Google Scholar]
- (24).Cao H, Jiang Y, Wang Y. Stereospecific synthesis and characterization of oligodeoxyribonucleotides containing an N2-(1-carboxyethyl)-2′-deoxyguanosine. J. Am. Chem. Soc. 2007;129:12123–12130. doi: 10.1021/ja072130e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Wuenschell GE, Tamae D, Cercillieux A, Yamanaka R, Yu C, Termini J. Mutagenic potential of DNA glycation: Miscoding by (R)- and (S)-N2-(1-carboxyethyl)-2′-deoxyguanosine. Biochemistry. 2010;49:1814–1821. doi: 10.1021/bi901924b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Shapiro R, Cohen BI, Shiuey SJ, Maurer H. On the reaction of guanine with glyoxal, pyruvaldehyde, and kethoxal, and the structure of the acylguanines. A new synthesis of N2-alkylguanines. Biochemistry. 1969;8:238–245. doi: 10.1021/bi00829a034. [DOI] [PubMed] [Google Scholar]
- (27).Li H, Nakamura S, Miyazaki S, Morita T, Suzuki M, Pischetsrieder M, Niwa T. N2-Carboxyethyl-2′-deoxyguanosine, a DNA glycation marker, in kidneys and aortas of diabetic and uremic patients. Kidney Int. 2006;69:388–392. doi: 10.1038/sj.ki.5000064. [DOI] [PubMed] [Google Scholar]
- (28).Synold T, Xi B, Wuenschell GE, Tamae D, Figarola JL, Rahbar S, Termini J. Advanced glycation end products of DNA: Quantification of N2-(1-carboxyethyl)-2′-deoxyguanosine in biological samples by liquid chromatography electrospray ionization tandem mass spectrometry. Chem. Res. Toxicol. 2008;21:2148–2155. doi: 10.1021/tx800224y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Ochs S, Severin T. Reaction of 2′-Deoxyguanosine with Glyceraldehyde. Liebigs Ann. Chem. 1994;1994:851–853. [Google Scholar]
- (30).Schneider M, Quistad GB, Casida JE. N2,7-Bis(1-hydroxy-2-oxopropyl)-2′-deoxyguanosine: Identical noncyclic adducts with 1,3-dichloropropene epoxides and methylglyoxal. Chem. Res. Toxicol. 1998;11:1536–1542. doi: 10.1021/tx9801256. [DOI] [PubMed] [Google Scholar]
- (31).Kellum MW, Oray B, Norton SJ. A convenient quantitative synthesis of methylglyoxal for glyoxalase I assays. Anal. Biochem. 1978;85:586–590. doi: 10.1016/0003-2697(78)90258-0. [DOI] [PubMed] [Google Scholar]
- (32).Kraemer KH, Seidman MM. Use of supF, an Escherichia coli tyrosine suppressor tRNA gene, as a mutagenic target in shuttle-vector plasmids. Mutat. Res. 1989;220:61–72. doi: 10.1016/0165-1110(89)90011-0. [DOI] [PubMed] [Google Scholar]
- (33).Parris CN, Seidman MM. A signature element distinguishes sibling and independent mutations in a shuttle vector plasmid. Gene. 1992;117:1–5. doi: 10.1016/0378-1119(92)90482-5. [DOI] [PubMed] [Google Scholar]
- (34).Lapolla A, Flamini R, Tonus T, Fedele D, Senesi A, Reitano R, Marotta E, Pace G, Seraglia R, Traldi P. An effective derivatization method for quantitative determination of glyoxal and methylglyoxal in plasma samples by gas chromatography/ mass spectrometry. Rapid Commun. Mass Spectrom. 2003;17:876–878. doi: 10.1002/rcm.992. [DOI] [PubMed] [Google Scholar]
- (35).Lim P, Sadre-Bazzaz K, Shurter J, Sarasin A, Termini J. DNA damage and mutations induced by arachidonic acid peroxidation. Biochemistry. 2003;42:15036–15044. doi: 10.1021/bi035555w. [DOI] [PubMed] [Google Scholar]
- (36).Narang SA, Jacob TM, Khorana HG. Studies on Polynucleotides. XLVI. The Synthesis of Hexanucleotides Containing the Repeating Trinucleotide Sequences Deoxycyctidylyl-(3′→′)-deoxyadenylyl-(3′→′)-deoxyadenosine and Deoxyguanylyl-(3′→′)-deoxyadenylyl-(3′→′)-deoxyadenosine. J. Am. Chem. Soc. 1965;87:2988–2995. doi: 10.1021/ja01091a031. [DOI] [PubMed] [Google Scholar]
- (37).Vaca CE, Fang JL, Conradi M, Hou SM. Development of a 32P-postlabelling method for the analysis of 2′-deoxyguanosine-3′-monophosphate and DNA adducts of methylglyoxal. Carcinogenesis. 1994;15:1887–1894. doi: 10.1093/carcin/15.9.1887. [DOI] [PubMed] [Google Scholar]
- (38).Valavanidis A, Vlachogianni T, Fiotakis C. 8-Hydroxy-2′ -deoxyguanosine (8-OHdG): A critical biomarker of oxidative stress and carcinogenesis. J. Environ. Sci. Health, Part C: Environ. Carcinog. Ecotoxicol. Rev. 2009;27:120–139. doi: 10.1080/10590500902885684. [DOI] [PubMed] [Google Scholar]
- (39).Murata-Kamiya N, Kaji H, Kasai H. Deficient nucleotide excision repair increases base-pair substitutions but decreases TGGC frameshifts induced by methylglyoxal in Escherichia coli. Mutat. Res. 1999;442:19–28. doi: 10.1016/s1383-5718(99)00054-6. [DOI] [PubMed] [Google Scholar]
- (40).Dennehy MK, Loeppky RN. Mass spectrometric methodology for the determination of glyoxaldeoxyguanosine and O6-hydroxyethyldeoxyguanosine DNA adducts produced by nitro-samine bident carcinogens. Chem. Res. Toxicol. 2005;18:556–565. doi: 10.1021/tx049802o. [DOI] [PubMed] [Google Scholar]
- (41).Wang H, Cao H, Wang Y. Quantification of N2-carboxymethyl-2′-deoxyguanosine in calf thymus DNA and cultured human kidney epithelial cells by capillary high-performance liquid chromatography: Tandem mass spectrometry coupled with stable isotope dilution method. Chem. Res. Toxicol. 2010;23:74–81. doi: 10.1021/tx900286c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Thornalley PJ, Waris S, Fleming T, Santarius T, Larkin SJ, Winklhofer-Roob BM, Stratton MR, Rabbani N. Imidazopurinones are markers of physiological genomic damage linked to DNA instability and glyoxalase 1-associated tumour multidrug resistance. Nucleic Acids Res. 2010;48:5432–5442. doi: 10.1093/nar/gkq306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Seksek O, Bolard J. Nuclear pH gradient in mammalian cells revealed by laser microspectrofluorimetry. J. Cell Sci. 1996;109(Part 1):257–262. doi: 10.1242/jcs.109.1.257. [DOI] [PubMed] [Google Scholar]
- (44).Turk Z. Glycotoxines, carbonyl stress and relevance to diabetes and its complications. Physiol. Res. 2010;59:147–156. doi: 10.33549/physiolres.931585. [DOI] [PubMed] [Google Scholar]
- (45).Queisser MA, Yao D, Geisler S, Hammes HP, Lochnit G, Schleicher ED, Brownlee M, Preissner KT. Hyperglycemia impairs proteasome function by methylglyoxal. Diabetes. 2010;59:670–678. doi: 10.2337/db08-1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Tyson J, Caple F, Spiers A, Burtle B, Daly AK, Williams EA, Hesketh JE, Mathers JC. Inter-individual variation in nucleotide excision repair in young adults: Effects of age, adiposity, micronutrient supplementation and genotype. Br. J. Nutr. 2009;101:1316–1323. doi: 10.1017/S0007114508076265. [DOI] [PubMed] [Google Scholar]
- (47).Blasiak J, Arabski M, Krupa R, Wozniak K, Zadrozny M, Kasznicki J, Zurawska M, Drzewoski J. DNA damage and repair in type 2 diabetes mellitus. Mutat. Res. 2004;554:297–304. doi: 10.1016/j.mrfmmm.2004.05.011. [DOI] [PubMed] [Google Scholar]
- (48).Hjartaker A, Langseth H, Weiderpass E. Obesity and diabetes epidemics: Cancer repercussions. Adv. Exp. Med. Biol. 2008;630:72–93. doi: 10.1007/978-0-387-78818-0_6. [DOI] [PubMed] [Google Scholar]
- (49).Nicolucci A. Epidemiological aspects of neoplasms in diabetes. Acta Diabetol. 2010;47:87–95. doi: 10.1007/s00592-010-0187-3. [DOI] [PubMed] [Google Scholar]
- (50).Ogi T, Limsirichaikul S, Overmeer RM, Volker M, Takenaka K, Cloney R, Nakazawa Y, Niimi A, Miki Y, Jaspers NG, Mullenders LHF, Yamashita S, Fousteri MI, Lehmann AR. Three DNA Polymerases, Recruited by Different Mechanisms, Carry Out NER Repair Synthesis in Human Cells. Mol. Cell. 2010;37:714–727. doi: 10.1016/j.molcel.2010.02.009. [DOI] [PubMed] [Google Scholar]
- (51).Zhang Y, Yuan F, Xin H, Wu X, Rajpal DK, Yang D, Wang Z. Human DNA polymerase κ synthesizes DNA with extraordinarily low fidelity. Nucleic Acids Res. 2000;28:4147–4156. doi: 10.1093/nar/28.21.4147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Johnson RE, Washington MT, Prakash S, Prakash L. Fidelity of human DNA polymerase η. J. Biol. Chem. 2000;275:7447–7450. doi: 10.1074/jbc.275.11.7447. [DOI] [PubMed] [Google Scholar]
- (53).Johnson RE, Washington MT, Haracska L, Prakash S, Prakash L. Eukaryotic polymerases ι and ζ act sequentially to bypass DNA lesions. Nature. 2000;406:1015–1019. doi: 10.1038/35023030. [DOI] [PubMed] [Google Scholar]
- (54).Tissier A, McDonald JP, Frank EG, Woodgate R. polι, a remarkably error-prone human DNA polymerase. Genes Dev. 2000;14:1642–1650. [PMC free article] [PubMed] [Google Scholar]
- (55).Zhang Y, Yuan F, Wu X, Wang Z. Preferential incorporation of G opposite template T by the low-fidelity human DNA polymerase ι. Mol. Cell. Biol. 2000;20:7099–7108. doi: 10.1128/mcb.20.19.7099-7108.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Benhamou S, Sarasin A. ERCC2/XPD gene polymorphisms and cancer risk. Mutagenesis. 2002;17:463–469. doi: 10.1093/mutage/17.6.463. [DOI] [PubMed] [Google Scholar]
- (57).Qiao Y, Spitz MR, Shen H, Guo Z, Shete S, Hedayati M, Grossman L, Mohrenweiser H, Wei Q. Modulation of repair of ultraviolet damage in the host-cell reactivation assay by polymorphic XPC and XPD/ERCC2 genotypes. Carcinogenesis. 2002;23:295–299. doi: 10.1093/carcin/23.2.295. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.