

Abstract
Adenosine (ADO) released in the heart results in enhanced coronary blood flow and reduced catecholamine release and myocardial responsiveness to adrenergic stimulation (anti-adrenergic action). ADO release from the adrenergic-stimulated aged heart is less than that from the young adult heart. Because adrenergic signaling in the aged heart is impaired, this study was conducted to determine if reduced ADO release from the aged heart results from this reduced adrenergic responsiveness. Hearts of 3-4 mo (young adult) and 21-22 mo (aged) Fischer-344 rats were perfused with ADO deamination and re-phosphorylation inhibited. Coronary effluent ADO levels were determined. Cellular free ADO levels with and without sodium acetate (NaAc)-induced mitochondrial AMP synthesis were assessed using formed S-adenosylhomocysteine (SAH) in L-homocysteine thiolactone (L-HC) treated hearts. The activities of SAH-hydrolase were determined. Aged heart ADO release was 61% less than from young hearts. NaAc augmented young heart ADO release by 104%, while that of aged hearts remained unchanged. SAH synthesis was 51% and 56% lower in the aged heart in the absence and presence of NaAc, respectively, despite an 89% greater SAH hydrolase activity found in the aged hearts. Since synthesized AMP may be diverted to IMP and ultimately inosine by AMP deaminase, inosine release was determined. Aged heart inosine levels in the absence and presence of NaAc were 74% and 59% less than for the young hearts. It is concluded that a reduced mitochondrial AMP synthesis is in part responsible for the attenuation in ADO release from the adrenergic-stimulated aged heart.
Keywords: adenosine, S-adenosylhomocysteine, aged heart, SAH hydrolase, AMP deaminase, rat
INTRODUCTION
Adenosine as a metabolite of ATP is endogenously released into the interstitial space of the myocardium where it profoundly affects heart function by activating various adenosine receptor subtypes. This nucleoside has been reported to be involved in the stimulation of angiogenesis (Ethier et al., 1993), the modulation of glucose transport (Law and McLane, 1991), glycolysis (Finegan et al., 1992), electrical activity (El-Menyar and Gehani, 2010) and the control of neurotransmitter release (Lorbar et al., 2004). Adenosine also has been reported to enhance the activities of AMP-dependent protein kinase (Aymerich et al., 2006) and ATP-sensitive potassium channel (KATP) (Kirsch et al., 1990). Considerable study has been made of the role of adenosine in the attenuation of adrenergic drive in the myocardium (Dobson, 1983; Dobson et al., 2003).
Adenosine A1 receptors are G-protein coupled receptors that exhibit an anti-adrenergic action when activated, thereby limiting the responsiveness of the myocardium to the effects of β-adrenergic stimulation. This can be of particular importance in hearts that are exposed to elevated levels of adrenergic stimulation such as occurs during ischemic episodes (Schomig et al., 1991). Limiting the effectiveness of the β-adrenergic signal cascade to initiate target responses reduces cardiotoxic manifestations (Rona, 1985) that may result from high levels of circulating catecholamines (Fenton et al., 1995). Adrenergic signaling elements that are reduced include Gs-protein cycling (Fenton and Dobson, 2007), cAMP formation and activation of PKA (Dobson, 1983) that result in the β-adrenergic-elicited enhancement of protein phosphorylation (Fenton and Dobson, 1984), cardiomyocyte Ca2+ transient magnitude (Fenton et al., 2010) and sarcomere shortening (Fenton et al., 2009). Adenosine A1 receptors have also been found to exhibit a cardioprotective action by initiating the translocation of PKCε to RACK2 in the cardiomyocyte (Fenton et al., 2009; Fenton et al., 2010), a mechanism initiated by the activation of phospholipase C (Fenton et al., 2010) and culminating, in part, with an enhanced cellular PP2A activity (Liu and Hofmann, 2002; Tikh et al., 2008). Adenosine A2A receptors have been associated with an increase in ventricular Ca2+ transient magnitude and contractility (Dobson and Fenton, 1997; Dobson et al., 2008; Monahan et al., 2000).
The distribution of adenosine between the intracellular and interstitial compartments of the heart is attained via translocation of the nucleoside across the sarcolemma utilizing bi-directional Na+-independent equilibrative transporters (Yao et al., 2002) and Na+-dependent concentrative transporters (Gray et al., 2004). The interstitial concentration of adenosine in the aged heart has been determined to exceed that found in young adult hearts (Dobson and Fenton, 1993). Although the basis for this observation is presently unknown, it has been assumed to result from an aging-induced depression of the return transport of adenosine into the aged cardiomyocyte from the interstitial compartment (Lorbar et al., 1999).
The release of adenosine from adrenergically-stimulated aged hearts is significantly depressed as compared to that of the young adult myocardium (Lorbar et al., 1999). The present study was undertaken to obtain a better understanding of the mechanism underlying this depression. The absence of efficient outward-directed adenosine transport in the aged heart may reduce the ability of the intracellular nucleoside resulting from enhanced ATP metabolism to leave the myocardium. Thus, measuring the actual release of adenosine from the myocardium can be inconclusive. Therefore, the endogenous synthesis of S-adenosyl homocysteine was used to determine the estimate the levels of free intracellular adenosine attained (Deussen et al., 1988; Schrader et al., 1981). It is concluded that while adrenergic responsiveness of the myocardium may play a role in the reduced release of adenosine from the stressed aged heart, a reduced mitochondrial function that occurs with aging is also involved.
MATERIALS AND METHODS
Animals used in this study were maintained and used in accordance with recommendations in the Guide for the Care and Use of Laboratory Animals prepared by the Institute of Laboratory Animal Resources, National Research Council (DHEW Publication NIH #85-23, Rev. 1996) and the guidelines of the Institutional Animal Care and Use Committee of the University of Massachusetts Medical School. Sprague-Dawley rats were housed in rooms with 12 hr light/dark cycles and fed rodent chow and water ad lib.
Isolated Heart Preparations
Fischer-344 rats ages 3-4 months and 21-22 months were obtained from Harlan Sprague-Dawley (Indianapolis, IN). Hearts were removed from guillotined animals and placed in iced physiological saline (PS). Within 1 min of isolation, hearts were perfused via an aortic cannula at a constant rate as described previously (Fenton et al., 2000). Perfusion rates between the two groups were 14.76 ± 2.07 and 16.94 ± 2.47 ml/min for young and aged hearts, respectively. The perfusion saline (PS) contained 118 mM NaCl, 4.7 mM KCl, 2.5 mM CaCl2, 25 mM NaHCO3, 1.2 mM KH2PO4, 1.2 mM MgSO4, and 10 mM glucose with the pH maintained at 7.4 by gassing the PS with a 95% O2/5% CO2 mixture. Coronary perfusion pressures were measured by a pressure transducer attached to a side tube immediately above the aorta. Perfusion pressures ranged from 70 to 85 mm Hg. Hearts were paced at 420 contractions per minute. After isolation, hearts were allowed to stabilize for 15 min prior to initiating the experimental protocols. Agents were infused into the aortic cannula at 1% of the flow rate to achieve the desired PS concentrations. Upon termination of the experimental periods, hearts were frozen with liquid N2 cooled aluminum clamps and stored in liquid N2 until assayed.
Protocols
Upon stabilization of the heart, a one-min sample of coronary effluent was obtained, whereupon the infusion of EHNA (50 μM) and iodotubercidin (ITC; 2 μM) was initiated to inhibit cellular adenosine deaminase and adenosine kinase, respectively. At 4 min of infusion, a second one-min sample of coronary effluent was obtained whereupon the infusion of L-homocysteine thiolactone (L-HC; 1.0 mM) commenced. The EHNA/ITC infusion continued throughout the 10 min L-HC infusion. Consecutive one-min samples of coronary effluent were obtained for 6 min. At 10 min, hearts were rapidly frozen as described above. All fluid samples were immediately boiled upon collection to denature any proteins and subsequently air-dried and reconstituted with Milli-Q water prior to analysis by HPLC.
Experiments were repeated together with the infusion of sodium acetate (NaAc) to increase adenosine production by the heart (Achterberg et al., 1986). NaAc was infused at a concentration of 20 mM beginning at 5 min into the EHNA/iodotubercidin infusion and 1 min prior to initiation of L-HC infusion. Infusion continued throughout the experiment until freeze clamping of the heart.
HPLC analysis of perfusate and myocardium for S-adenosyl homocysteine (SAH), adenosine and inosine
Frozen heart samples (0.75 - 1.5 g) were homogenized on ice in 3.5 ml of 1N perchloric acid using a Polytron at setting 6. After removing 100 μl for protein determination using a bicinchoninic acid protein assay (Pierce, Rockford, IL) and bovine serum albumin as a standard. The remaining homogenate was centrifuged in an Eppendorff at top speed for 20 min. The supernatants were collected, neutralized to pH 7 with 1M KOH and filtered with 0.2 μm Acrodisc syringe filters (Pall, Ann Arbor, MI). Samples were analyzed for contained SAH, adenosine and inosine by HPLC using a mobile phase of 10 mM KH2PO4 (pH 4.0) and a 30 min linear gradient 1% MeOH to 10% MeOH. A Discovery C18 RP, 25 cm (5μm) column (Supelco, Sigma-Aldrich, St. Louis, MO) was used for the separation.
S-Adenosyl homocysteine hydrolase assay
Myocardial samples obtained from young and aged hearts were homogenized in a buffer composed of 25 mM HEPES, 1 mM EDTA, 2 mM DTT, 1 mM MgCl2, and 1x Complete protease cocktail (Roche, Indianapolis, IN). An aliquot was removed for protein determination as described above. After centrifugation of the homogenate in an Eppendorff centrifuge at full speed, 20 μl of the supernatant (100 μg protein) was added to 1.0 ml of reaction mixture containing 25 mM HEPES (pH 7.4), 2 mM MgCl2, 0.1 mM NaHPO4, 4 mM L-homocysteine thiolactone, 20 μM EHNA, and 50 μM adenosine. After a 20 min incubation in a shaker bath at 37° C, the reaction was stopped with 100 μl 1N perchloroacetic acid. Samples were neutralized with KOH, filtered and analyzed for contained SAH using HPLC as described above.
Immunoblotting
As described previously (Fenton et al., 2010), 130-150 mg of frozen rat heart were homogenized (PRO200 homogenizer, PRO Scientific, Oxford, CT) in 1.0 ml of homogenization buffer containing 25 mM HEPES, 1 mM EDTA, 2 mM DTT, 1 mM MgCl2, and 1x Complete protease cocktail. The homogenate was centrifuged at 15,000 × g for 20 min, whereupon the supernatant was collected and a sample removed for protein determination as described above. The remaining supernatant was supplemented with 300 mM Tris, 25% β-mercaptoethanol, and 0.5% bromophenol blue. Samples were boiled for 3-5 min, centrifuged for clarification and resolved using 12% SDS-PAGE. Each well contained 25-30 μg protein. Resolved proteins were transferred to nitrocellulose membranes and blotted against primary mouse monoclonal anti-SAH hydrolase (AHCYL-1) or rabbit polyclonal GAPDH. The secondary antibody was goat anti-rabbit conjugated to horseradish peroxidase. The chemiluminescence was determined with X-ray film and Western Lightning reagents (PerkinElmer, Waltham, MA). Exposure densities were quantified using U-SCAN-IT software (Silk Scientific, Orem, UT).
Statistical Methods
Data were analyzed using StatMost (Dataxiom, Los Angeles, CA). After applying one way ANOVA, additional analysis was conducted using Student-Newman-Keuls posthoc test. A P value of <0.05 was taken to indicate a statistically significant difference. All data are presented as means ± SE.
Materials
Buffer salts and NaAc were obtained from Fisher Scientific (Fair Lawn, NJ). EHNA hydrochloride and 5-iodotubercidin were purchased from Sigma-Aldrich. L-homocysteine thiolactone hydrochloride was obtained from Fluka (St. Louis, MO). S-adenosyl-L-homocysteine and adenosine used as HPLC standards were purchased from Sigma-Aldrich (St. Louis, MO). Antibodies were obtained from Abcam (San Fran cisco, CA). All gel electrophoretic reagents were obtained from Bio-Rad (Richmond, CA).
RESULTS
Adenosine release
Adenosine release rates from the myocardium were not significantly different between the young (839 ± 233 pmol/min) and aged (637 ± 167 pmol/min) hearts (Fig. 1). The enzyme inhibitors EHNA and iodotubercidin were administered to inhibit the loss of adenosine by deamination or phosphorylation, respectively. In the presence of these agents, the release of adenosine into the coronary effluent increased significantly by 756% and 335% for both young and aged hearts, respectively. Adenosine release in the presence of the inhibitors was 61% less in the aged heart when compared to that from the young hearts.
Figure 1.
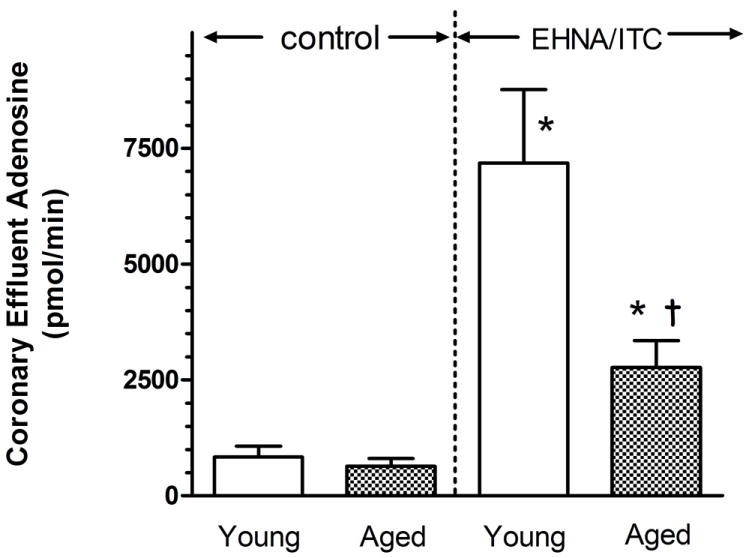
Effect of the inhibition of adenosine kinase and adenosine deaminase on min adenosine content of coronary effluent from young adult and aged rat hearts. After control 1 min coronary effluent samples were obtained, EHNA (50 μM) and iodotubercidin (ITC;2 μM) and were administered to the aortic perfusate to block adenosine deaminase and adenosine kinase, respectively. At 5 min, 1-min samples were again collected. Samples were analyzed with HPLC as described in the text. Values are means ± SE for 17 young and 14 aged hearts. * Mean is significantly different from the agent-free value (P<0.05). † Mean is significantly different from the inhibitor-treated young hearts (P<0.05).
Total adenosine contained in the coronary effluent during the initial six min of L-HC infusion was determined for young and aged hearts treated with EHNA and iodotubercidin (Fig. 2). In the absence of NaAc, 45.14 ± 13.60 and 37.04 ± 10.33 nmol adenosine were released from young and aged hearts, respectively. With the infusion of 20 mM NaAc into the aortic perfusate, adenosine release significantly increased by 104% in the young heart. The release of adenosine by the aged heart was unchanged when compared to the release in the absence of NaAc. The total effluent adenosine was a significant 76% less than that level found for the young heart.
Figure 2.
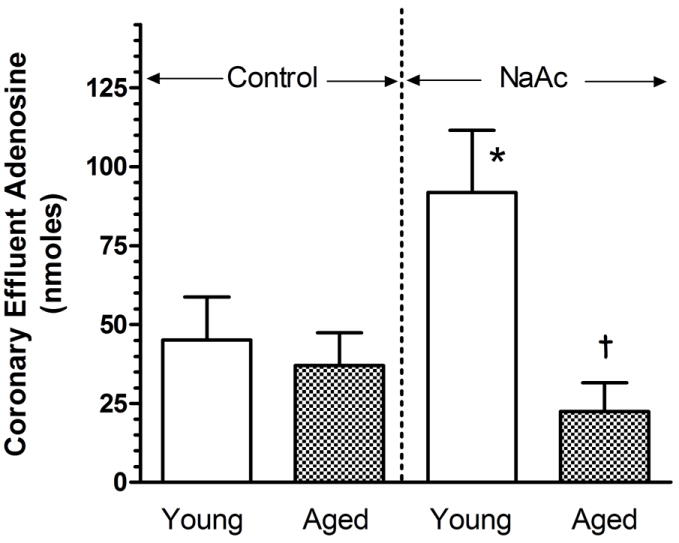
Effect of sodium acetate (NaAc) administration on adenosine release from young adult and aged rat hearts. Hearts were treated with EHNA and iodotubercidin for 5 min prior to the initiation of a 10 min L-homocysteine thiolactone (L-HC) administration. Hearts were subsequently perfused in the absence or presence of NaAc administered at 20 mM commencing 1 min prior to and during infusion of L-HC. Heart perfusates were collected during the first six minutes of L-HC infusion and the total adenosine contained during these 6 minutes reported. Values are means ± SE for 6-8 control hearts, and 6-9 NaAc-treated hearts. * Mean is significantly different from the control (P<0.05). † Mean is significantly different from the young NaAC value (P<0.05).
S-adenosyl homocysteine (SAH) synthesis by the intact perfused myocardium
The synthesis of SAH serves as an index of cellular free adenosine level in the myocardium (Deussen et al., 1988). In the presence or absence of NaAc, SAH was synthesized at a significantly lower rate in aged heart compared to young heart (Fig. 3). The rate of SAH synthesis in the young heart was determined to be 46.92 ± 5.37 nmol SAH/mg protein/min. This value was a significant 51% lower in the aged heart. With the administration of NaAc to enhance myocardial AMP synthesis and, therefore, ADO levels, SAH synthesis in the young heart was 52.29 ± 7.61 nmol SAH/mg protein/min, a value not different from that observed in the absence of NaAc. As without the administration of NaAc, SAH synthesis in the presence of NaAc was a significant 56% less in the aged heart.
Figure 3.
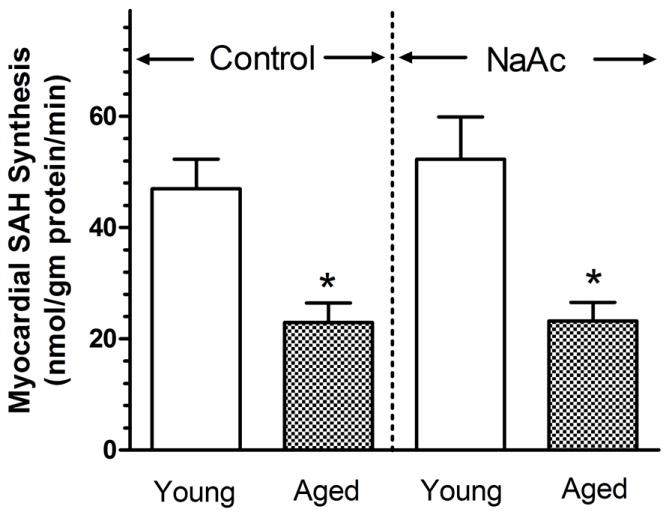
Effect of NaAc on the synthesis of S-adenosyl homocysteine (SAH) by the young adult and aged rat heart. Sodium acetate (NaAc) was administered at 20 mM for 1 min prior to and during infusion of L-homocysteine thiolactone(L-HC). EHNA and ITC were present to block adenosine deaminase and adenosine kinase, respectively, for 5 min prior to and throughout the NaAc infusion as described in the Fig. 1 legend. Values are means ± SE for 6-7 control hearts and 8-9 NaAc-treated hearts. * Means are significantly different from the appropriate young values (P<0.05).
SAH Hydrolase activity determined in vitro
The activity of this enzyme was significantly elevated in homogenates obtained from aged hearts (Fig. 4, top). The enzymatic activity in young hearts was 1.007 ± 0.112 nmol SAH produced/mg protein/min. This value was observed to be higher by 89% in the aged heart. However, the SAH hydrolase protein levels as determined by the ratio of optical densities for hydrolase and GAPDH indicate no significant difference between young and aged hearts (Fig. 4, bottom). This indicated that the increased activity in aged hearts did not result only from a greater presence of protein.
Figure 4.
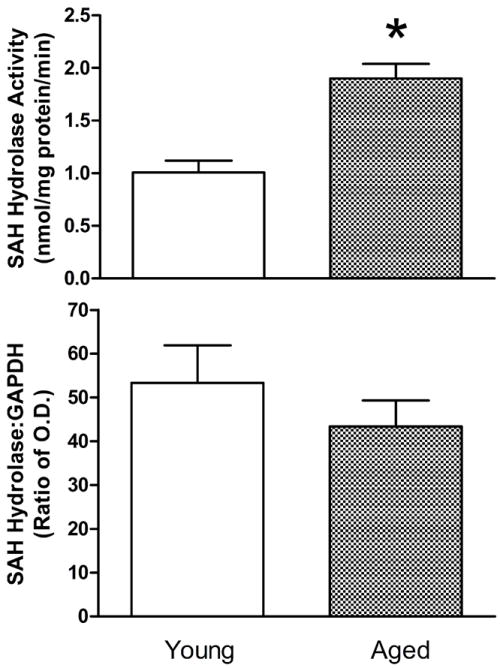
Effect of aging on myocardial SAH hydrolase activities (top) and protein levels (bottom) as determined from whole heart homogenates. Activity values are means ± SE of enzymatic activities determined in 6 hearts. SAH hydrolase protein ratio values are means ± SE of the ratios of optical densities for blot films of SAH hydrolase and GAPDH for 12 young and 7 aged rat hearts. *Mean is significantly different from the young adult value (P<0.05).
Coronary Inosine
Inosine release over a period of 6 min was found to be 29,742 ± 2,318 nmol from young hearts treated with EHNA, iodotubercidin and L-HC (Fig. 5). Inosine levels present in coronary effluents obtained from aged hearts was found to be 74% less than observed in young hearts. With the administration of NaAc to enhance cellular levels of AMP, inosine release from young hearts significantly increased by 42% above levels observed in the absence of NaAc. In aged hearts, while inosine release increased with NaAc treatment, the increase was not significant and only reached a level 59% less than that attained in the NaAc-treated young heart.
Figure 5.
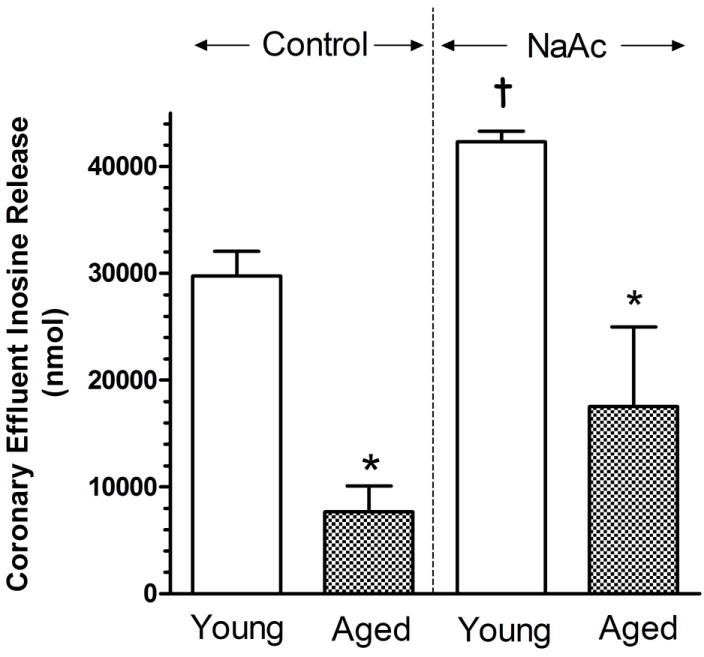
Effect of sodium acetate (NaAc) administration on inosine release into the coronary effluents of isolated and perfused young adult and aged rat hearts. NaAc was infused into the perfusion at 20 mM to hearts receiving EHNA, iodotubercidin and L-homocysteine thiolactone. Values are mean ± SE for 3 hearts. * Mean is significantly different from the appropriate young value (P<0.05). † Mean is significantly different from the comparative NaAc-free value (P<0.05).
DISCUSSION
Adenosine released from the adrenergic-stimulated heart has been found to be significantly reduced with aging (Lorbar et al., 1999). Because this nucleoside is involved extensively in the modulation of metabolic, electrical and functional aspects of the myocardium, a reduced release of adenosine can severely impact heart viability. For example, adenosine is a well-documented vasodilator (Morrison et al., 2002). An aging-induced reduction in adenosine release would result in an attenuation of the augmented coronary blood flow expected with adrenergic-elicited enhancement of interstitial adenosine levels (Fenton and Dobson, 1993). This would impact the oxygenation of the myocardium during a period of increased metabolic demand. Similarly, adenosine manifests an anti-adrenergic action by attenuating metabolic and functional responses of the myocardium to adrenergic stimulation as well as limiting catecholamine release from nerve terminals (Lorbar et al., 2004). Adrenergic neurotransmitters are released in the myocardium in response to ischemia (Schomig et al., 1991) creating a cellular environment potentially deleterious to the viability of the myocardium (Rona, 1985). A reduction in the adenosine levels attained during ischemia would encourage the development of arrhythmias and catecholamine-elicited necrosis in the aged myocardium (Schomig et al., 1991). An increase in released catecholamines would also be predicted with a reduction in adenosine (Lorbar et al., 2004). The difference in adenosine release between aged and young hearts can potentially impact the function and, indeed, survival of the aged heart.
Values for the minute release of adenosine from 3-4 mo young adult and 21-22 mo aged hearts in the absence of administered agents were found to be similar (Fig.1). It has been reported previously that the minute content of adenosine in coronary effluent collected from the aged myocardium significantly exceeds that from the young adult heart (Dobson et al., 1990; Lorbar et al., 1999). However, in these reports the effluents were obtained from constant-pressure perfused hearts, whereas in the present study hearts were constant-flow perfused. Although it is not known definitively how perfusion at constant flow vs. constant pressure may alter adenosine release, it is conceivable that constant pressure perfused hearts may be mildly hypoxic. In addition, the previously reported data were normalized to myocardial dry weight. In the present study dry weights weren’t obtained. However, using 1.33 as the ratio of dry aged to dry young heart weights (Dobson and Fenton, 1993), the present values for aged and young coronary effluent adenosine normalized for weight were still similar indicating that the method of perfusion most likely accounts for the present observations.
Adenosine formed in the heart is either deaminated to inosine by adenosine deaminase or salvaged by re-phosphorylation to AMP by adenosine kinase. The administration of EHNA and iodotubercidin to substantially inhibit these cellular enzymes (Achterberg et al., 1985) resulted in significant increases in the minute release of adenosine from both young adult and aged hearts after 5 min of inhibition (Figure 1). Preliminary experiments indicated that inhibition was maximal within the 5 min administration period. However, adenosine released by aged hearts was significantly less than that for young adult hearts, suggesting that the adenosine may have been trapped inside the cellular myocardium as a result of reduced transport (Lorbar et al., 1999). If this was the case, then intracellular levels of free adenosine might be expected to be elevated.
Intracellular free adenosine was estimated by measuring the synthesis of S-adenosylhomocysteine (SAH) in the presence of excess L-homocysteine (L-HC). AMP hydrolysis by cytosolic 5’-nucleotidase and the hydrolysis of SAH by the reversible cytosolic SAH-hydrolase are recognized as sources of intracellular adenosine in the heart (He et al., 1992; Lloyd and Schrader, 1993). Because the equilibrium constant of SAH-hydrolase favors the synthesis of SAH in the presence of an excess of L-HC, and SAH does not cross the cellular membrane, it has been proposed that the rate of SAH formation is directly proportional to the free cytosolic adenosine concentration when L-HC levels are saturating (Deussen et al., 1988; Loncar et al., 1997)}. After the enzymatic degradation and salvage of adenosine was inhibited, L-HC was administered to estimate differences in free adenosine levels between young and aged hearts. The total coronary effluent adenosine release over six minutes of L-HC infusion in the presence of EHNA and iodotubercidin was found to be similar between young adult and aged hearts. While the average min release for young adult hearts (7,523 pmol/min) was similar to the comparable min release reported in Fig. 1, the significant depression in min adenosine release from the aged heart as noted in the absence of L-HC was no longer observed. This effect of L-HC administration on adenosine release from the aged heart cannot be explained at this time. Surprisingly, the level of free adenosine present in the aged myocardium was significantly depressed (Fig 3), the opposite of what might be expected with only an impaired outward-directed nucleoside transport. These results would suggest a reduced cellular free adenosine level in the aged myocardium in the absence of adrenergic stimulation.
Adrenergic stimulation results in an enhanced release of adenosine from the young myocardium (Fenton and Dobson, 1993), a release that is reduced with aging (Lorbar et al., 1999). Although the synthesis of intracellular adenosine via stimulated metabolism of ATP could be explored using a β-adrenergic agonist, adrenergic signaling has been reported to be impaired in the aged myocardium (Farrell and Howlett, 2008). Reduced levels of adenosine present in the aged myocardium in response to adrenergic stimulation may result in part from an inadequacy of β-adrenergic receptor coupling and/or adenylyl cyclase activity. An alternative approach to elevating cellular adenosine levels involves the administration of sodium acetate (NaAc), which is known to elevate cellular AMP levels via the activity of mitochondrial acetyl-CoA synthetase activity without increasing ATP content (Randle et al., 1970; Taegtmeyer et al., 1980). Degradation of the resultant AMP by 5’-nucleotidase enhances cellular adenosine levels. In the presence of EHNA, iodoacetate, and excess L-HC, administration of NaAc resulted in a significant increase in the amount of adenosine released into the coronary effluent from the young heart as compared to a similar duration perfusion in absence of NaAc (Fig. 2). This increase in coronary effluent adenosine was entirely absent in the NaAc-stimulated aged heart. While an aging-induced reduction in efficient sarcolemmal adenosine transport would be an impediment to the movement of adenosine into the coronary effluent, a reduction in free cellular adenosine production would also result in a reduced adenosine available for transport into the vascular compartment of the aged heart. Data of Figure 3 suggests this might be the case. With the young heart, the levels of free adenosine were found to be roughly identical in the absence and presence of NaAc, suggesting that adenosine derived from AMP was efficiently removed from the cell. However, in the aged heart, myocardial SAH synthesis was significantly attenuated compared to the young heart suggesting that free adenosine levels in the presence of NaAc were depressed with aging. Alternately, it was possible that the activity of SAH hydrolase activity was depressed in the aged heart.
While SAH-hydrolase catalyzes the synthesis of S-adenosylhomocysteine from adenosine and L-homocysteine, it also influences S-adenosyl methionine-dependent transmethylation reactions.by catalyzing the metabolism of SAH to adenosine and homocysteine. If not metabolized by the hydrolase, excess SAH would serve as a negative feedback inhibitor to transmethylation thereby impacting cardiomyocyte function and viability (Leach et al., 2010). Thus, a reduced SAH-hydrolase activity in the aged heart would be detrimental to myocardial viability. Determination of the SAH-hydrolase activity in the aged heart revealed an enhanced activity of this enzyme when compared to that of the young enzyme (Fig 4) suggesting that the reduced formation of SAH in the aged heart was not a result of depressed SAH-hydrolase. This increase in activity did not result from an increased presence of SAH-hydrolase protein. It is to be noted that others have reported a decrease in SAH hydrolase activity with aging of erythrocytes (Bozzi et al., 1990). The basis for this difference is not known at present.
AMP in the cell is metabolized to adenosine by an AMP-specific cytosolic 5’-nucleotidase. AMP is also converted by AMP-deaminase (myoadenylate deaminase; AMP-d) to inosine monophosphate (IMP) which is subsequently hydrolyzed to inosine by an IMP-specific 5’-nucleotidase. AMP-d is an enzyme that is important to the availability of cellular adenosine for outward transport and may significantly impact the functioning of the cardiovascular system (Anderson et al., 2000; Finsterer et al., 2004; Rybakowska et al., 2010). A previous report from this laboratory indicated that the activity of AMP-d is labile and may increase in the aged cell in response to prolonged hypoxic stress (Reisert et al., 2002). The significantly lower SAH formation in the aged heart may reflect a reduced level of adenosine derived from AMP as a result of enhanced AMP-d activity. Assuming that in the presence of adenosine deaminase inhibition the amount of inosine released from the heart reflects the AMP-d activity, the proposed effect of aging on AMP-d is clearly not present. With adenosine deaminase inhibited, both in the absence and presence of NaAc to stimulate AMP production by the mitochondria via acetyl-CoA synthetase, inosine released from the aged heart was significantly depressed compared to that observed from the young heart. Inosine released with NaAc in the young heart increased as would be expected with an elevated AMP level. While the mean inosine release rose in the aged heart with NaAC, the increase was not significant. These data would suggest that AMP is not being shunted preferentially to IMP in the aged as compared to the young heart. Instead, they support the idea that AMP is not available in the aged heart to the same degree as in the young adult heart, whether stimulated or not by NaAc. This premise finds support in recent reports of depressed ATP levels in scenescent myocardium (Jullien et al., 1989) and mitochondrial dysfunction induced by aging (Chen and Knowlton, 2010; Preston et al., 2008; Trifunovic and Larsson, 2008).
In conclusion, adenosine released from aging hearts stimulated by adrenergic agonists is significantly reduced when compared to the release from young adult heart. Such a reduced presence of this nucleoside in the aged heart may adversely affect functionality and viability. Although prior evidence shows that sarcolemmal adenosine transport and adrenergic responsiveness are depressed in the aged heart, a reduced adenosine release may also reflect a depressed mitochondrial function resulting in less AMP available for hydrolysis to adenosine.
Acknowledgments
The authors wish to thank Lynne G. Shea for her excellent technical help in the conduct of these experiments.
Grant Sponsor: The National Institures of Health (NIH)
Grant Number: 5R03 AG30406
Footnotes
The authors have no conflict of interest to declare.
References
- Achterberg PW, Harmsen E, DeJong JW. Adenosine deaminase inhibition and myocardial purine release during normoxia and ischaemia. Cardiovasc Res. 1985;19:593–598. doi: 10.1093/cvr/19.10.593. [DOI] [PubMed] [Google Scholar]
- Achterberg W, Stroeve J, DeJong JW. Myocardial adenosine cycling rates during normoxia and under conditions of stimulated purine release. Biochem J. 1986;235:13–17. doi: 10.1042/bj2350013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson JL, Habashi J, Carlquist JF, Muhlestein JB, Horne BD, Bair TL, Pearson RR, Hart N. A common variant of the AMPD1 gene predicts improved cardiovascular survival in patients with coronary artery disease. J Amer Col Cardiol. 2000;36:1248–1252. doi: 10.1016/s0735-1097(00)00850-0. [DOI] [PubMed] [Google Scholar]
- Aymerich I, Foufelle F, Ferre P, Casado FJ, Pastor-Anglada M. Extracellular adenosine activates AMP-dependent protein kinase (AMPK) J Cell Sci. 2006;119:1612–1621. doi: 10.1242/jcs.02865. [DOI] [PubMed] [Google Scholar]
- Bozzi A, Furciniti-LaChiusa B, Strom R, Crifo C. S-Adenosylhomocysteine hydrolase and adenosine deaminase activities in human red cell ageing. Chinica Chimica Acta. 1990;189:81–86. doi: 10.1016/0009-8981(90)90237-m. [DOI] [PubMed] [Google Scholar]
- Chen L, Knowlton AA. Mitochondria and heart failure: new insights into an energetic problem. Minerva Cardioangiologica. 2010;58:213–229. [PMC free article] [PubMed] [Google Scholar]
- Deussen A, Borst M, Schrader J. Formation of S-adenosylhomocysteine in the heart. I: An index of free intracellular adenosine. Circ Res. 1988;63:240–249. doi: 10.1161/01.res.63.1.240. [DOI] [PubMed] [Google Scholar]
- Dobson JG., Jr Mechanism of adenosine inhibition of catecholamine-induced elicited responses in heart. Circ Res. 1983;52:151–160. doi: 10.1161/01.res.52.2.151. [DOI] [PubMed] [Google Scholar]
- Dobson JG, Jr, Fenton RA. Adenosine inhibition of β-adrenergic induced responses in aged hearts. Am J Physiol. 1993;265:H494–H503. doi: 10.1152/ajpheart.1993.265.2.H494. [DOI] [PubMed] [Google Scholar]
- Dobson JG, Jr, Fenton RA. Adenosine A2a receptor function in rat ventricular myocytes. Cardiovasc Res. 1997;34:337–347. doi: 10.1016/s0008-6363(97)00023-0. [DOI] [PubMed] [Google Scholar]
- Dobson JG, Jr, Fenton RA, Romano FD. Increased myocardial adenosine production and reduction of β-adrenergic contractile response in aged hearts. Circ Res. 1990;66:1381–1390. doi: 10.1161/01.res.66.5.1381. [DOI] [PubMed] [Google Scholar]
- Dobson JG, Jr, Shea LG, Fenton RA. β-Adrenergic and antiadrenergic modulation of cardiac adenylyl cyclase is influenced by phosphorylation. Am J Physiol. 2003;285:H1471–H1478. doi: 10.1152/ajpheart.00950.2002. [DOI] [PubMed] [Google Scholar]
- Dobson JG, Jr, Shea LG, Fenton RA. Adenosine A2A and β-adrenergic calcium transient and contractile responses in rat ventricular myocytes. Am J Physiol. 2008;295:H2364–H2372. doi: 10.1152/ajpheart.00927.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Menyar A, Gehani A. Adenosine-induced tachyarrhythmai and cardiac arrest. Future Cardiol. 2010;6:433–436. doi: 10.2217/fca.10.66. [DOI] [PubMed] [Google Scholar]
- Ethier MF, Chander V, Dobson JG., Jr Adenosine stimulates proliferation of human endothelial cells in culture. Am J Physiol. 1993;265:H131–H138. doi: 10.1152/ajpheart.1993.265.1.H131. [DOI] [PubMed] [Google Scholar]
- Farrell SR, Howlett SE. The age-related decrease in catecholamine sensitivity is mediated by β1-adrenergic receptors linked to a decrease in adenylate cyclase activity in ventricular myocytes from male Fischer 344 rats. Mech Ageing Devel. 2008;129:735–744. doi: 10.1016/j.mad.2008.09.017. [DOI] [PubMed] [Google Scholar]
- Fenton RA, Dickson EW, Meyer TE, Dobson JG., Jr Aging reduces the cardioprotective effect of ischemic preconditioning in rat heart. J Mol Cell Cardiol. 2000;32:1371–1375. doi: 10.1006/jmcc.2000.1189. [DOI] [PubMed] [Google Scholar]
- Fenton RA, Dobson JG., Jr Adenosine and calcium alter adrenergic-induced intact heart protein phosphorylation. Am J Physiol. 1984;246:H559–H565. doi: 10.1152/ajpheart.1984.246.4.H559. [DOI] [PubMed] [Google Scholar]
- Fenton RA, Dobson JG., Jr Hypoxia enhances isoproterenol-induced increase in heart interstitial adenosine depressing β-adrenergic contractile responses. Circ Res. 1993;72:571–578. doi: 10.1161/01.res.72.3.571. [DOI] [PubMed] [Google Scholar]
- Fenton RA, Dobson JG., Jr Adenosine A1 and A2A receptor effects on G-protein cycling in β-adrenergic stimulated ventricular membranes. J Cell Physiol. 2007;213:785–792. doi: 10.1002/jcp.21149. [DOI] [PubMed] [Google Scholar]
- Fenton RA, Galeckas KJ, Dobson JG., Jr Endogenous adenosine reduces depression of cardiac function induced by β-adrenergic stimulation during low flow perfusion. J Mol Cell Cardiol. 1995;27:2373–2383. doi: 10.1016/s0022-2828(95)92055-2. [DOI] [PubMed] [Google Scholar]
- Fenton RA, Komatsu S, Ikebe M, Shea LG, Dobson JG., Jr Adenoprotection of the heart involves phospholipase C-induced activation and translocation of PKCε to RACK2 in adult rat and mouse. Am J Physiol. 2009;297:H718–H725. doi: 10.1152/ajpheart.00247.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenton RA, Shea LG, Doddi C, Dobson JG., Jr Myocardial adenosine A1-receptor-mediated adenoprotection involves phospholipase C, PKC-ε, and p38 MAPK, but not HSP27. Am J Physiol. 2010;298:H1671–H1678. doi: 10.1152/ajpheart.01028.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finegan BA, Clanachan AS, Coulson CS, Lopaschuk GD. Adenosine modification of energy substrate use in isolated hearts perfused with fatty acids. Am J Physiol. 1992;262:H1501–H1507. doi: 10.1152/ajpheart.1992.262.5.H1501. [DOI] [PubMed] [Google Scholar]
- Finsterer J, Schoser B, Stollberger C. Myoadenylate-deaminase gene mutation associated with left ventricular hypertrabeculation/non-compaction. Acta Cardiol. 2004;59:453–456. doi: 10.2143/AC.59.4.2005215. [DOI] [PubMed] [Google Scholar]
- Gray MO, Zhou H-Z, Schafhalter-Zoppoth I, Zhu P, Mochly-Rosen D, Messing RO. Preservation of base-line hemodynamic function and loss of inducible cardioprotection in adult mice lacking protein kinase Cε. J Biol Chem. 2004;279:3596–3604. doi: 10.1074/jbc.M311459200. [DOI] [PubMed] [Google Scholar]
- He MX, Gorman MW, Romig GD, Sparks HVJ. Adenosine formation and myocardial energy status during graded hypoxia. J Mol Cell Cardiol. 1992;24:79–89. doi: 10.1016/0022-2828(92)91161-w. [DOI] [PubMed] [Google Scholar]
- Jullien T, Cand F, Fargier C, Verdetti J. Age-dependent differences in energetic status, electrical and mechanical performance of rat myocardium. Mech Ageing Devel. 1989;48:243–254. doi: 10.1016/0047-6374(89)90086-9. [DOI] [PubMed] [Google Scholar]
- Kirsch GE, Codma J, Birnbaumer L, Brown AM. Coupling of ATP-sensitive K+ channels to A1 receptors by G proteins in rat ventricular myocytes. Am J Physiol. 1990;259:H820–H826. doi: 10.1152/ajpheart.1990.259.3.H820. [DOI] [PubMed] [Google Scholar]
- Law WR, McLane MP. Adenosine enhances myocardial glucose uptake only in the presence of insulin. Metabolism. 1991;40:947–952. doi: 10.1016/0026-0495(91)90071-4. [DOI] [PubMed] [Google Scholar]
- Leach IM, van der Harst P, de Boer RA. Pharmacoepigenetics in heart failure. Curr Heart Fail Rep. 2010;7:83–90. doi: 10.1007/s11897-010-0011-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Hofmann PA. Antiadrenergic effects of adenosine A1 receptor-mediated protein phosphatase 2a activation in the heart. Am J Physiol. 2002;283:H1314–H1321. doi: 10.1152/ajpheart.00343.2002. [DOI] [PubMed] [Google Scholar]
- Lloyd HGE, Schrader J. Adenosine metabolism in the guinea pig heart: the role of cytosolic S-adenosyl-L-homocysteine hydrolase, 5’-nucleotidase and adenosine kinase. Eur Heart J. 1993;14(Suppl.I):27–33. [PubMed] [Google Scholar]
- Loncar R, Flesche CW, Deussen A. Determinants of the S-adenosylhomocysteine (SAH) technique for the local assessment of cardiac free cytosolic adenosine. J Mol Cell Cardiol. 1997;29:1289–1305. doi: 10.1006/jmcc.1996.0351. [DOI] [PubMed] [Google Scholar]
- Lorbar M, Chung ES, Nabi A, Skalova K, Fenton RA, Dobson JG., Jr Receptor subtypes involved in adenosine-mediated modulation of norepinephrine release from cardiac nerve terminals. Can J Physiol Pharmacol. 2004;82:1026–1031. doi: 10.1139/y04-108. [DOI] [PubMed] [Google Scholar]
- Lorbar M, Fenton RA, Duffy AJ, Graybill CA, Dobson JG., Jr Effect of aging on myocardial adenosine production, adenosine uptake and adenosine kinase activity in rats. J Mol Cell Cardiol. 1999;31:401–412. doi: 10.1006/jmcc.1998.0877. [DOI] [PubMed] [Google Scholar]
- Monahan TS, Sawmiller DR, Fenton RA, Dobson JG., Jr Adenosine A2a-receptor activation increases contractility in isolated perfused hearts. Am J Physiol. 2000;279:H1472–H1481. doi: 10.1152/ajpheart.2000.279.4.H1472. [DOI] [PubMed] [Google Scholar]
- Morrison RR, Talukder MA, Ledent C, Mustafa SJ. Cardiac effects of adenosine in A2A receptor knockout hearts: uncovering A2B receptors. Am J Physiol. 2002;282:H437–H444. doi: 10.1152/ajpheart.00723.2001. [DOI] [PubMed] [Google Scholar]
- Preston CC, Oberlin AS, Holmuhamedov EL, Gupta A, Sagar S, Syed RHK, Siddiqui SA, Raghavakaimal S, Terzic A, Jahangir A. Aging-induced alterations in gene transcripts and functional activity of mitochondrial oxidative phosphorylation complexes in the heart. Mech Ageing Devel. 2008;129:304–312. doi: 10.1016/j.mad.2008.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randle PJ, England PJ, Denton RM. Control of the tricarboxylate cycle and its interactions with glycolysis during acetate utilization in rat heart. Biochem J. 1970;117:677–695. doi: 10.1042/bj1170677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reisert PS, Dobson JG, Jr, Fenton RA. Anoxia-induced changes in purine nucleoside metabolism of in vitro aged human fibroblasts. Life Sci. 2002;70:1369–1382. doi: 10.1016/s0024-3205(01)01518-1. [DOI] [PubMed] [Google Scholar]
- Rona G. Catecholamine cardiotoxicity. J Mol Cell Cardiol. 1985;17:291–306. doi: 10.1016/s0022-2828(85)80130-9. [DOI] [PubMed] [Google Scholar]
- Rybakowska I, Bakula S, Klimek J, Milczarek R, Smolenski RT, Kaletha K. Cardiac muscle AMP-deaminase from a 10-year-old male heterozygous for the AMPD1 C34T mutation. Nucleosides, Nucleotides and Nucleic Acids. 2010;29:453–456. doi: 10.1080/15257771003741380. [DOI] [PubMed] [Google Scholar]
- Schomig A, Haass M, Richardt G. Catecholamine release and arrhythmias in acute myocardial ischemia. Eur Heart J. 1991;124:F38–F47. doi: 10.1093/eurheartj/12.suppl_f.38. [DOI] [PubMed] [Google Scholar]
- Schrader J, Schutz W, Bardenheuer H. Role of S-adenosylhomocysteine hydrolase in adenosine metabolism in mammalian heart. Biochem J. 1981;196:65–70. doi: 10.1042/bj1960065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taegtmeyer H, Hems R, Krebs HA. Utilization of energy-providing substrates in the isolated working rat heart. Biochem J. 1980;186:701–711. doi: 10.1042/bj1860701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tikh EI, Fenton RA, Dobson JG., Jr Adenosine A1 and A2A receptor regulation of protein phosphatase 2A in the murine heart. J Cell Physiol. 2008;216:83–90. doi: 10.1002/jcp.21375. [DOI] [PubMed] [Google Scholar]
- Trifunovic A, Larsson N-G. Mitochondrial dysfunction as a cause of ageing. Journal of Internal Medicine. 2008;263:167–178. doi: 10.1111/j.1365-2796.2007.01905.x. [DOI] [PubMed] [Google Scholar]
- Yao SYM, Ng AML, Vickers MF, Sundaram M, Cass CE, Baldwin SA, Young JD. Functional and molecular characterization of nucleobase transport by recombinant human and rat equilibrative nucleoside transporters 1 and 2. J Biol Chem. 2002;277:24938–24948. doi: 10.1074/jbc.M200966200. [DOI] [PubMed] [Google Scholar]