

Abstract
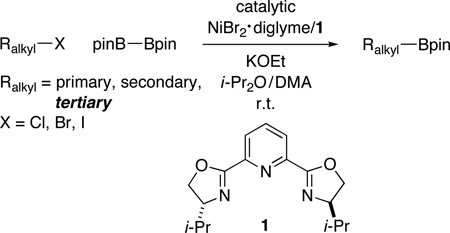
Through the use of a catalyst formed in situ from NiBr2•diglyme and a pybox ligand (both of which are commercially available), we have achieved our first examples of coupling reactions of unactivated tertiary alkyl electrophiles, as well as our first success with nickel-catalyzed couplings that generate bonds other than C–C bonds. Specifically, we have determined that this catalyst accomplishes Miyaura-type borylations of unactivated tertiary, secondary, and primary alkyl halides with diboron reagents to furnish alkylboronates, a family of compounds with substantial (and expanding) utility, under mild conditions; indeed, the umpolung borylation of a tertiary alkyl bromide can be achieved at a temperature as low as −10 °C. The method exhibits good functional-group compatibility and is regiospecific, both of which can be issues with traditional approaches to the synthesis of alkylboronates. In contrast to seemingly related nickel-catalyzed C–C bond-forming processes, tertiary halides are more reactive than secondary or primary halides in this nickel-catalyzed C–B bond-forming reaction; this divergence is particularly noteworthy in view of the likelihood that both transformations follow an inner-sphere electron-transfer pathway for oxidative addition.
INTRODUCTION
Alkylboron compounds have a broad spectrum of applications, ranging from cancer medicine (Velcade™; bortezomib)1 to organometallic partners in cross-coupling reactions.2 Alkylboranes are most often prepared via the hydroboration of olefins3 or the transmetalation of highly reactive organolithium/organomagnesium reagents with electrophilic boron species.4 These methods have significant limitations, such as a regioselectivity issue in the case of hydroborations of many internal olefins and functional-group incompatibility in the case of transmetalations.
During the past several years, we have endeavored to enlarge the scope of cross-coupling reactions of alkyl electrophiles.5,6 Until now, we have focused our attention exclusively on C–C bond-forming processes, establishing that an array of organometallic reagents can be coupled efficiently with primary and secondary (but not tertiary) electrophiles. We recently decided to attempt to expand these coupling reactions to generate bonds other than C–C bonds. In view of the need for additional, complementary methods for the synthesis of alkylboranes,7,8 we undertook the challenge of achieving Miyaura borylations of unactivated alkyl electrophiles to provide C–B bonds,9 a transformation that could enable regiospecific and late-stage incorporation of boron into organic molecules in an umpolung process.10
At the time that we initiated this investigation, there were no reports of such reactions; however, earlier this year, two groups independently described copper catalysts that effect borylations of unactivated primary and secondary alkyl electrophiles at room temperature or above.11 In this report, we provide a nickel-catalyzed method that accomplishes Miyaura reactions of an array of unactivated alkyl halides (eq 1); particularly noteworthy is our observation that, for the first time for borylations and for our studies of nickel catalysis, unactivated tertiary electrophiles can serve as effective coupling partners.
 |
(1) |
RESULTS AND DISCUSSION
Although we were unable to obtain acceptable yields for nickel-catalyzed C–B bond formation using a variety of methods that we had developed for C–C bond formation,12 we determined that, under the appropriate conditions, a nickel/pybox catalyst can achieve the desired Miyaura borylation by pinB–Bpin (pin = pinacolato = OCMe2Me2CO) of a wide array of electrophiles at room temperature.13 Interestingly, in our previous studies of Suzuki reactions of alkyl electrophiles, bidentate ligands (a bipyridine, an aminoalcohol, or a 1,2-diamine) have always been optimal;14 tridentate pybox ligands have only proved to be the ligand of choice for Negishi reactions.15
In the case of unactivated primary alkyl electrophiles, bromides and iodides are suitable coupling partners (Table 1, entries 1 and 2). On the other hand, the corresponding chlorides and tosylates are not effectively borylated under these conditions (<10% yield), although activated chlorides couple with the diboron reagent with useful efficiency (entries 3 and 4).16 Both of the catalyst components, NiBr2•diglyme and pybox ligand 1, are commercially available and can be handled in the air.
Table 1.
Nickel-Catalyzed Borylation of Unactivated and Activated Primary Alkyl Halides
Yield of purified product (average of two experiments).
Yield determined by GC analysis versus a calibrated internal standard.
Many primary alkylboranes can be synthesized conveniently via existing methods that have acceptable functional-group compatibility, such as the hydroboration of olefins. Unfortunately, hydroboration is a less-effective approach in the case of most secondary alkylboranes, due to the generation of regioisomers (e.g., consider the products of Table 2, entries 1–3 and 6–10). This issue is circumvented when a Miyaura borylation strategy is employed instead. Thus, NiBr2•diglyme/pybox 1 catalyzes the room-temperature coupling of pinB–Bpin with unactivated alkyl iodides (Table 2, entries 1 and 2) and bromides (entries 3–10) to furnish the desired secondary alkylboranes in good yield.17 A variety of functional groups, including an olefin, a carbamate, an aniline, an amide, and a sulfonamide, are compatible with this method. Under the standard conditions, borylations of activated, but not unactivated, secondary chlorides proceed smoothly (entries 11 and 12). Our nickel-catalyzed method is not limited to the use of pinB–Bpin as the borylating agent (eq 2).18
Table 2.
Nickel-Catalyzed Borylation of Unactivated and Activated Secondary Alkyl Halides (for the reaction conditions, see Table 1)
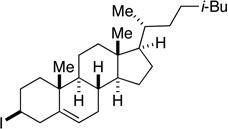
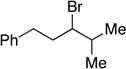
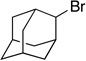
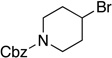
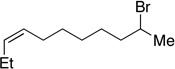
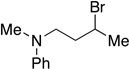
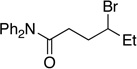
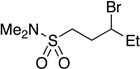
Yield of purified product (average of two experiments).
Diastereoselectivity:3:1 β:α.
Starting material: 97:3 Z:E; product: 95:5 Z:E.
Reaction conditions: see Table 3.
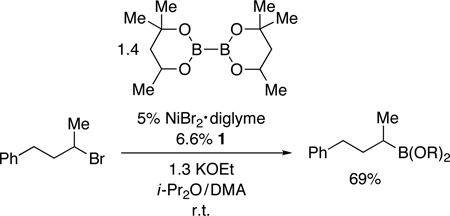 |
(2) |
Although we have reported progress in the development of nickel-based catalysts for cross-coupling primary and secondary alkyl electrophiles with a range of organometallic nucleophiles, we have not described any success with tertiary electrophiles. Correspondingly, the recent copper-catalyzed Miyaura borylation methods of Steel/Marder/Liu11a and Ito11b have not proved applicable to tertiary electrophiles (two examples: 0% yield and 17% yield). We were therefore pleased to determine that NiBr2•diglyme/pybox 1 catalyzes not only the borylation of primary (Table 1) and secondary (Table 2), but also tertiary (Table 3), halides.
Table 3.
Nickel-Catalyzed Borylation of Unactivated Tertiary Alkyl Halides
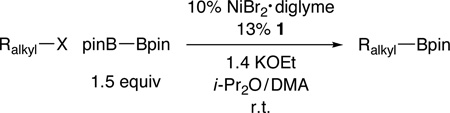
Yield of purified product (average of two experiments).
Diastereoselectivity: 11:1 cis:trans.
As illustrated in Table 3,19 nickel-catalyzed borylation of unactivated tertiary alkyl iodides furnishes the desired tertiary alkylboranes in good-to-modest yields (entries 1 and 2). Couplings of cyclic (entries 3–5) and acyclic (entries 6–9) bromides proceed smoothly in the presence of an array of functional groups, including an ether, an ester, an alkyl chloride, and an olefin. Good diastereoselectivity is observed in a Miyaura reaction of a substituted cyclohexane (11:1; entry 4). Remarkably, the borylation of a tertiary alkyl bromide can be achieved in excellent yield at −10 °C (eq 3), the lowest temperature that we have employed to date for a nickel-catalyzed coupling of any unactivated alkyl halide. None of these tertiary alkylboranes can be accessed via olefin hydroboration.
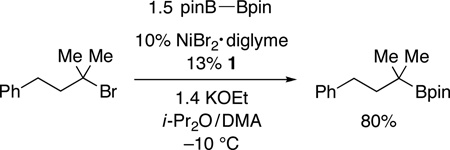 |
(3) |
Intrigued by our ability to couple tertiary alkyl halides for the first time, we examined the relative rates of product formation in a series of competition experiments between a tertiary, a secondary, and a primary alkyl bromide (Figure 1). Interestingly, the more substituted the alkyl bromide, the more borylation product was formed.
Figure 1.

Nickel-catalyzed C–B bond formation: More substituted electrophiles are more reactive.
These results are even more striking when contrasted with the data for a corresponding set of competition experiments for a nickel/pybox-catalyzed Negishi cross-coupling (Figure 2).20 In the case of this C–C bond-forming process, we observe essentially no product derived from reaction of the tertiary alkyl bromide, and there is a modest preference for coupling a primary, rather than a secondary, bromide.
Figure 2.

Nickel-catalyzed C–C bond formation: A tertiary halide is unreactive.
For our nickel-catalyzed cross-coupling reactions, we have hypothesized that the mechanism outlined in Figure 3 is operative when unactivated alkyl halides are employed as electrophiles.5a,21,22,23 If our Miyaura borylations (Tables 1–3) proceed through an analogous pathway, then the difference in reactivity as a function of nucleophile (boron- vs. carbon-based in Figures 1 vs. 2) may be attributable to the disparate reaction profiles of intermediates G, H, and/or I as a function of Nu.24
Figure 3.

Outline of a possible mechanism for nickel-catalyzed coupling reactions.
In the case of nickel-catalyzed C–C bond-forming reactions of unactivated alkyl halides, all of the data that we have accumulated to date are consistent with an inner-sphere electron-transfer pathway for oxidative addition (Figure 3). For our Miyaura borylations, our observations are also most consistent with a radical (versus an SN2 or a direct-insertion) mechanism. For example, the enhanced reactivity with greater substitution illustrated in Figure 1 can be rationalized by a radical pathway wherein the stability of R• (see H in Figure 3) plays a key role in determining the course of the reaction. Furthermore, borylation of either exo- or endo-2-bromonorbornane provides the exo product with >20:1 diastereoselectivity (eq 4).
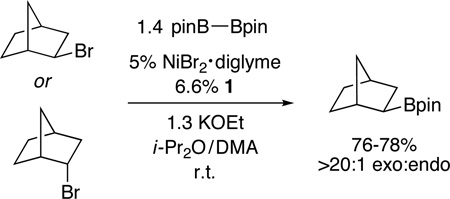 |
(4) |
For an inner-sphere electron-transfer pathway for oxidative addition, the anticipated order of reactivity as a function of the leaving group is I > Br > Cl > OTs. Consistent with this, unactivated alkyl chlorides and tosylates are unreactive under our standard borylation conditions (vide supra). In the case of corresponding alkyl iodides and bromides, product formation proceeds at qualitatively similar rates; we have made analogous observations for nickel-catalyzed Suzuki reactions, likely due to transmetalation (not oxidative addition) being the turnover-limiting step of the catalytic cycle.25 To gain insight into the relative reactivity of the halides in our Miyaura borylations, we conducted competition experiments between pairs of electrophiles. As illustrated in Figure 4, the nickel/pybox catalyst reacts with a high preference for an alkyl iodide in the presence of a bromide, as well as an alkyl bromide in the presence of a chloride, consistent with the expectations for an inner-sphere electron-transfer mechanism for oxidative addition.26,27,28
Figure 4.

Nickel-catalyzed C–B bond formation: Relative reactivity as a function of the halide.
CONCLUSIONS
In conclusion, we have developed a readily available nickel catalyst that achieves the room-temperature Miyaura borylation of a diverse set of alkyl electrophiles, including the first effective couplings of tertiary halides (at a temperature as low as −10 °C). This umpolung process displays good functional-group compatibility and enables the regiospecific generation of a wide array of alkylboranes, a family of compounds with substantial and growing utility, thereby addressing key shortcomings of traditional methods for their synthesis. Mechanistic investigations reveal that this nickel-catalyzed C–B bond-forming process exhibits a reactivity profile (tertiary>secondary>primary halide) distinct from seemingly related nickel-catalyzed C–C bond-forming reactions (tertiary<secondary, primary halide); yet, a range of observations are consistent with a common pathway for oxidative addition of the alkyl halide to nickel (inner-sphere electron-transfer). Additional studies of nickel-catalyzed couplings of alkyl electrophiles, including new reactions of tertiary electrophiles, couplings with other non-carbon-based nucleophiles, and enantioselective transformations, are underway.
Supplementary Material
ACKNOWLEDGMENT
Support has been provided by the National Institutes of Health (National Institute of General Medical Sciences, grant R01–GM62871). We thank Dr. Ashraf Wilsily for experimental assistance.
Footnotes
ASSOCIATED CONTENT
Supporting Information.
Experimental procedures and compound characterization data. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
REFERENCES
- 1.For leading references, see: Einsele H. Recent Results in Cancer Research. 2010;184:173–187. doi: 10.1007/978-3-642-01222-8_12.
- 2.For reviews, see: Chemler SR, Trauner D, Danishefsky SJ. Angew. Chem. Int. Ed. 2001;40:4544–4568. doi: 10.1002/1521-3773(20011217)40:24<4544::aid-anie4544>3.0.co;2-n. Doucet H. Eur. J. Org. Chem. 2008:2013–2030. Jana R, Pathak TP, Sigman MS. Chem. Rev. 2011;111:1417–1492. doi: 10.1021/cr100327p.
- 3.For overviews, see: Zaidlewicz M. In: Kirk–Othmer Encyclopedia of Chemical Technology. Seidel A, editor. Vol. 13. Hoboken: John Wiley & Sons; 2005. pp. 631–684. Matteson DS. Science of Synthesis. Vol. 6. Claustal: Georg Thieme Verlag; 2004. pp. 5–79. Fu GC. In: Transition Metals for Organic Synthesis. Bolm C, Beller M, editors. Chapter 1.5. New York: VCH; 2004.
- 4.For an overview and leading references, see: Hall DG. In: Boronic Acids. Hall DG, editor. Vol. 1. Wiley–VCH: Weinheim; 2011. pp. 1–133.
- 5.For recent reports and leading references, see: Wilsily A, Tramutola F, Owston NA, Fu GC. J. Am. Chem. Soc. 2012;134:5794–5797. doi: 10.1021/ja301612y. Oelke AJ, Sun J, Fu GC. J. Am. Chem. Soc. 2012;134:2966–2969. doi: 10.1021/ja300031w.
- 6.For reviews, see: Glasspoole BW, Crudden CM. Nature Chem. 2011;3:912–913. doi: 10.1038/nchem.1210. Glorius F. Angew. Chem. Int. Ed. 2008;47:8347–8349. doi: 10.1002/anie.200803509. Rudolph A, Lautens M. Angew. Chem. Int. Ed. 2009;48:2656–2670. doi: 10.1002/anie.200803611.
- 7.For an overview of the synthesis and applications of alkylboronates and related compounds, see: Hall DG, editor. Boronic Acids. Wiley–VCH: Weinheim; 2011.
- 8.For a few recent examples of the synthesis and applications of tertiary boronate esters, see: Bagutski V, Elford TG, Aggarwal VK. Angew. Chem. Int. Ed. 2011;50:1080–1083. doi: 10.1002/anie.201006037. Elford TG, Nave S, Sonawane RP, Aggarwal VK. J. Am. Chem. Soc. 2011;133:16798–16801. doi: 10.1021/ja207869f. Guzman-Martinez A, Hoveyda AH. J. Am. Chem. Soc. 2010;132:10634–10637. doi: 10.1021/ja104254d. O’Brien JM, Lee K-s, Hoveyda AH. J. Am. Chem. Soc. 2010;132:10630–10633. doi: 10.1021/ja104777u. Stymiest JL, Bagutski V, French RM, Aggarwal VK. Nature. 2008;456:778–782. doi: 10.1038/nature07592.
- 9.For leading references to metal-catalyzed borylations of aryl, alkenyl, and activated alkyl (allyl and benzyl) electrophiles, see: Ishiyama T, Miyaura N. In: Boronic Acids. Hall DG, editor. Vol. 1. Wiley–VCH: Weinheim; 2011. pp. 135–169. Miyaura N. Bull. Chem. Soc. Jpn. 2008;81:1535–1553.
- 10.For an early review, see: Seebach D. Angew. Chem. Int. Ed. 1979;18:239–258.
- 11.(a) Yang C-T, Zhang Z-Q, Tajuddin H, Wu C-C, Liang J, Liu J-H, Fu Y, Czyzewska M, Steel PG, Marder TB, Liu L. Angew. Chem. Int. Ed. 2012;51:528–532. doi: 10.1002/anie.201106299. [DOI] [PubMed] [Google Scholar]; (b) Ito H, Kubota K. Org. Lett. 2012;14:890–893. doi: 10.1021/ol203413w. [DOI] [PubMed] [Google Scholar]
- 12.For example, see: Zultanski SL, Fu GC. J. Am. Chem. Soc. 2011;133:15362–15364. doi: 10.1021/ja2079515.
- 13.Under the conditions employed in Tables 1–3, in the absence of NiBr2•diglyme, no borylation (<1% yield) of unactivated primary, secondary, or tertiary halides was observed.
- 14.For example, see: Zhou J, Fu GC. J. Am. Chem. Soc. 2004;126:1340–1341. doi: 10.1021/ja039889k. González-Bobes F, Fu GC. J. Am. Chem. Soc. 2006;128:5360–5361. doi: 10.1021/ja0613761. Saito B, Fu GC. J. Am. Chem. Soc. 2007;129:9602–9603. doi: 10.1021/ja074008l.
- 15.For example, see Zhou J, Fu GC. J. Am. Chem. Soc. 2003;125:14726–14727. doi: 10.1021/ja0389366. (b) ref 5b.
- 16.Under the same conditions, borylations of a primary allylic and a primary benzylic bromide were not efficient.
- 17.Notes: (a) Under the standard conditions: when a borylation was conducted in a capped vial under an atmosphere of air, the yield of the product was unaffected; the addition of 0.10 equiv of water led to a small (5%) decrease in yield; use of TBME, CPME, Et2O, or DME as the solvent led to a somewhat lower yield (Table 2, entry 3: 66–85%); functional groups such as a TIPS-substituted alkyne, an enolizable ketone, an aryl fluoride, and a trifluoromethyl group were compatible with the borylation process, whereas a nitro group and an aryl iodide were not; a highly hindered unactivated secondary bromide (t-BuCHBrCH2CH2Ph), an unactivated secondary chloride, an unactivated secondary tosylate, and an activated secondary bromide ((1-bromoethyl)benzene) were not suitable coupling partners; for the coupling illustrated in entry 3 of Table 2, use of 2.5% NiBr2•diglyme/3.3% ligand 1 provided the product in 85% yield, whereas use of 1.0% NiBr2•diglyme/1.3% ligand 1 furnished the alkylboronate in 69% yield; in the case of entry 2 of Table 2, use of the enantiomer of ligand 1 led to a 2:1 (β:α) mixture of diastereomers. (b) The Brønsted basicity of the reaction medium is substantially attenuated by complexation of the alkoxide to boron; thus, when enantioenriched 1,2-diphenylbutan-1-one was added to a borylation process, it could be recovered at the end of the reaction in essentially quantitative yield (>95%) and with little erosion in ee (<10%; complete racemization occurs in the absence of pinB–Bpin).
- 18.Under our standard borylation conditions, use of pinacolborane instead of pinB–Bpin did not generate a significant quantity of the desired product.
- 19.Under the standard conditions: the catalytic borylation illustrated in entry 6 of Table 3 proceeded in 70% yield on a gram scale, whereas use of 5% NiBr2•diglyme/6.6% ligand 1 furnished a 56% yield on a gram scale; if TBME, CPME, Et2O, or DME were employed as the solvent, a significantly lower yield was observed; an unactivated tertiary chloride, an activated tertiary chloride, and a highly hindered tertiary alkyl bromide were not useful coupling partners.
- 20.These are the conditions reported in: Zhou J, Fu GC. J. Am. Chem. Soc. 2003;125:14726–14727. doi: 10.1021/ja0389366.
- 21.For example, see: Lu Z, Wilsily A, Fu GC. J. Am. Chem. Soc. 2011;133:8154–8157. doi: 10.1021/ja203560q.
- 22.For noteworthy early mechanistic studies by others, see: Jones GD, Martin JL, McFarland C, Allen OR, Hall RE, Haley AD, Brandon RJ, Konovalova T, Desrochers PJ, Pulay P, Vicic DA . J. Am. Chem. Soc. 2006;128:13175–13183. doi: 10.1021/ja063334i. Lin X, Phillips DL. J. Org. Chem. 2008;73:3680–3688. doi: 10.1021/jo702497p. Phapale VB, Bunuel E, Garcia-Iglesias M, Cardenas DJ. Angew. Chem. Int. Ed. 2007;46:8790–8795. doi: 10.1002/anie.200702528.
- 23.For reviews and leading references on possible mechanisms for nickel-catalyzed cross-couplings of alkyl electrophiles, see: Hu X. Chem. Sci. 2011;2:1867–1886. Phapale VB, Cardenas DJ. Chem. Soc. Rev. 2009;38:1598–1607. doi: 10.1039/b805648j.
- 24.One would anticipate that a Ni–BX2 or a Ni–BX3 intermediate would exhibit different reactivity from a Ni–alkyl, due to the trivalency of boron (Ni–BX2) or the charge associated with a boron “ate” complex (Ni–BX3).
- 25.For example, see: Lu Z, Fu GC. Angew. Chem. Int. Ed. 2010;49:6676–6678. doi: 10.1002/anie.201003272.
- 26.For related observations for nickel-catalyzed Suzuki reactions of unactivated alkyl halides, see ref 25.
- 27.Use of a stereochemical probe (see ref 14b) provided results fully consistent with a radical intermediate in our nickel-catalyzed Miyaura borylations of unactivated alkyl electrophiles. Thus, both of the secondary bromides underwent cyclization/borylation to furnish the same ratio of diastereomers as observed for reductive cyclizations with Bu3SnH, consistent with a common intermediate for the two processes (a secondary alkyl radical that adds to the pendant olefin).
- 28.In a preliminary investigation, we have obtained moderate ee (61%), but modest yield, in a nickel/pybox-catalyzed enantioselective borylation of a racemic halide. For leading references to applications of enantioenriched alkylboronates in organic synthesis, see: Scott HK, Aggarwal VK. Chem. Eur. J. 2011;17:13124–13132. doi: 10.1002/chem.201102581. Crudden CM, Glasspoole BW, Lata CJ. Chem. Commun. 2009:6704–6716. doi: 10.1039/b911537d.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.