

Abstract
Glycogen synthase kinase 3 (GSK3) has been implicated in neurological disorders; therefore, it is not surprising that there has been an increased focus towards developing therapies directed to this kinase. Unfortunately, these current therapies have not taken into consideration the physiological role of GSK3 in crucial events like synaptic plasticity. With this in mind we will discuss the relationship of synaptic plasticity with GSK3 and tau protein and their role as potential targets for the development of therapeutic strategies. Finally, we will provide perspectives in developing a cocktail therapy for Alzheimer's treatment.
1. Introduction
Glycogen synthase kinase 3 (GSK3) is an evolutionarily conserved protein that is active in resting cells and is inhibited in response to activation of several distinct pathways such as the Wnt, insulin, and the growth factor pathway [1–7]. GSK3 activity is regulated by different mechanisms, including (a) phosphorylation at an N-terminal serine [7, 8], (b) through phosphorylation of a tyrosine residue [9], (c) through phosphorylation of a C-terminal serine residue [10], and (d) through disruption of the axin-β-catenin multiprotein complex [4, 11, 12]. The other requirement of GSK3 is that most of its substrates require prior phosphorylation at residue 4 or 5 amino acids C-terminal to the target residue [13].
GSK3 has two isoforms GSK3α and GSK3β, which are encoded by different genes [14]. In mouse, rat, and human, an alternative isoform (GSK3β2) that contains a 13-amino-acid insert near the catalytic domain was reported [15]. In opposition to GSK3α and GSK3β, GSK3β2 is specifically found in the nervous system and has been strongly linked to neurodevelopment [15].
In order to participate in all these events, GSK3 has a broad range of substrates: cyclic AMP response element-binding protein (CREB), neurogenin 2, SMAD1, NFkappaB, Myc, heat shock factor-1, cyclin D1, nuclear factor of activated T-cells and CCAAT/enhancer-binding proteins, c-Jun, β-catenin, and microtubule-associated proteins like MAP2 and tau [16–18]. GSK3 regulates some of these factors by controlling their protein levels. However, changes in GSK3 activity have been associated with neurodegenerative diseases, such as bipolar disorder, schizophrenia, and Alzheimer's disease (AD) [19]. Indeed, in AD the active form of GSK3β was found to be directly related to the hyperphosphorylation of tau present in paired helical filament (PHF)-tau of neurofibrillary tangles (NFTs) [20]. Importantly and due to the fact that most drugs bind and compete with ATP, there appears to be only a single amino acid difference (Glu196 in GSK3α, Asp133 in GSK3β) making it difficult to identify an inhibitor that can distinguish the two isoforms.
Overall, it is clear that GSK3 is related to AD development, and, more importantly, current data suggest that both isoforms (GSK3α and GSK3β) contribute to AD pathogenesis.
2. Tau Pathology and GSK3
Tau is an axonal protein that regulates microtubule stability [21]; however, during AD tau is abnormally phosphorylated and aggregates into NFTs [22, 23]. Tau has at least 45 phosphorylation sites, mostly located in the proline-rich region (P-region) (residues 172–251) and the C-terminal tail region (C-region) (residues 368–441) [24]. Tau phosphorylation at both of these regions affects its capacity to interact with microtubules [25]. In terms of AD pathology, the phosphorylation sites located in the C-terminal region seem to cause (a) abnormal folding and (b) protein cleavage, which together could lead to tau deposition [26–28]. Phosphorylation at some sites (Ser262) selectively impairs binding of tau to microtubules [29], whereas phosphorylation at other sites (Ser202) enhances tau polymerization [30]. Crucially, GSK3β has been linked to many of these sites [15, 31]. Therefore, emphasis has been placed particularly on GSK3β, rather than GSK3α. However, due to the lack of inhibitor's specificity, GSK3α has not been ruled out. Indeed, some studies have shown that GSK3α through Wnt signalling pathway is also related to tau pathology [32]. Furthermore, by specifically knocking down GSK3β, GSK3α was found to be related to AD pathology [33].
In sum, the current data shows that both isoforms GSK3α and GSK3β could be involved in tau phosphorylation.
3. GSK3 as the Therapeutic Target for AD
GSK3 is strongly implicated in neurodegeneration [34], and, not surprisingly, it has been postulated as a therapeutic target in the treatment of AD. Indeed, lithium which is a direct inhibitor of both GSK3β and GSK3α has been used in humans [35, 36]. The direct regulation of GSK3 also modifies cell survival as it is known for facilitating a variety of apoptotic mechanisms [35]. Similarly, in an attempt to reduce tau pathology, the GSK3 inhibitor [Tideglusib/NP-12 (Nypta)] is currently in clinical trial [37]. NP-12 has been designated as an orphan drug by the EU and US authorities and has been granted Fast Track status by the FDA (see http://www.noscira.com).
The rationale is simple; blocking GSK3 will lead to nonphosphorylated tau and, consequently, less tau deposition according to the current hypothesis. However, the importance of GSK3 for normal physiological cell functioning must be taken into consideration. In this regard, we recently found that phosphorylation of tau protein is critical in order for the protein to function as a negative feedback mechanism to prevent NMDA-receptor overexcitation (unpublished data). This data becomes crucial in this debate since NMDA deregulation plays a vital role in synaptic plasticity. Therefore, by simple blockade of GSK3 we could alter the homeostasis of synaptic plasticity among other important physiological functions. Furthermore, blocking GSK3 also raises the possibility of affecting gene expression and cell survival [17]. So, is GKS3 the desired therapeutic target for AD? Although the answer is far from being simplistic, normal physiological functions for the cell, together with the complexity of the phenomena [38], need to be taken into consideration before selecting AD pharmacological targets.
4. GSK3 as Crucial Node for Synaptic Plasticity
Synaptic plasticity has been proposed to play a central role in brain capacity to incorporate transient experiences into persistent memory traces. Synaptic transmission can be enhanced (long-term potentiation, LTP) or depressed (long-term depression, LTD) by activity, and these changes can persist from seconds to hours and days [39, 40]. Importantly, the affected intracellular pathways leading to LTP or LTD activation involve primarily GSK3 [41, 42]. Indeed, it has been shown that enhanced GSK3 signalling impairs hippocampal memory formation [43]. Specifically, GSK3 activity blocks synaptic LTP and induces LTD [43]. Furthermore, it was found that GSK3 during LTP involves activation of NMDA receptors and the PI3K-Akt pathway consequently disrupting the ability of synapses to undergo LTD [43]. Clearly, the data claims that GSK3 is a crucial node mediating the LTP to LTD transition. Therefore, the simple idea of blocking GSK3 in order to prevent the progression of AD seems to be overly simplistic.
5. Conclusion and Perspectives
The hypothesis that GSK3 plays a role in the aetiology of brain disorders is further nurtured by the fact that several genetic susceptibility factors for psychiatric disorders have key roles in neurodevelopment. Importantly, many of the genes are involved in GSK3 signaling [44, 45]. Furthermore, GSK3 is directly related to the pathogenesis of AD as tau kinase [31]. Overall, it seems clear that GSK3 has an integral role in the pathogenesis of AD. Therefore, GSK3 remains as therapeutic target. However, the secondary effects caused by GSK3 blockade should also be taken into consideration, especially knowing that synaptic dysfunction in addition to neuronal death can lead to cognitive failure associated with AD. With this in mind, therapies that focus on rescuing events like LTP rather than single blocking strategy could bring needed results.
In conclusion, we suggest that downstream targets of GSK3 are an interesting option. In other words, we proposed the use of cocktail drugs that could enhance LTP and reduce induction of LTD. For instance, drugs like memantine (NMDA receptor antagonist) [46], in combination with other drugs like okadaic acid (PP1 activator) [47] and/or pseudosubstrate for Akt [43], could be used in order to balance the activity of GSK3 and therefore tau phosphorylation (Figure 1). Together, this combinatorial approach may result in LTP promotion and synaptic improvement. After all, if the current strategies for AD treatment have shown little benefits, it is tempting to consider new therapeutic approaches that are aimed to improve memory formation.
Figure 1.
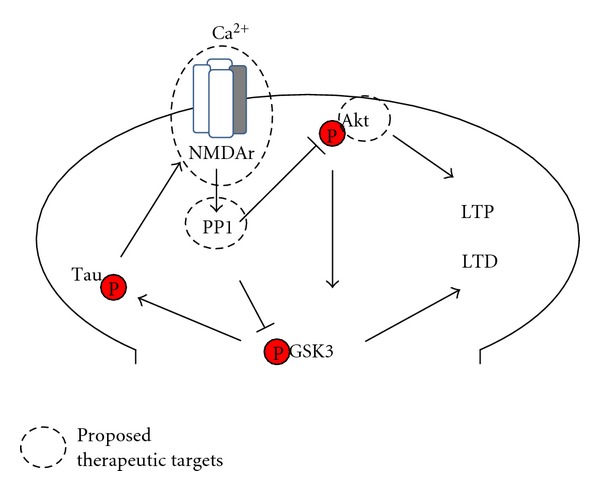
Cocktail drugs could balance the activity of GSK3 during AD. The role of PP1 and Akt in GSK3 activation, in combination with NMDA receptor, makes them important therapeutic targets. Calcium (Ca2+) enters via NMDA receptors, and this leads to activation of protein phosphatase 1 (PP1), a key enzyme in synaptically induced LTD. PP1 can dephosphorylate GSK3 that determines whether NMDA receptor activation induces LTD or inhibits LTD. PP1 can dephosphorylate Akt, resulting in GSK3 activation. GSK3, under the control of Akt and PP1, is a critical determinant of the direction of NMDA receptor-dependent plasticity. The active GSK3 isoforms contribute to phosphorylation of tau protein which is essential in order for the protein to function as a negative feedback mechanism to prevent NMDA-receptor overexcitation and synaptic failure.
Disclosure
George Perry is, or has in the past been, a paid consultant for and/or owns equity or stock options in Neurotez Pharmaceuticals, Panacea Pharmaceuticals, Takeda Pharmaceuticals, and Voyager Pharmaceuticals. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the paper apart from those disclosed.
Acknowledgments
The authors thank Jesse Jackson for critical comments. Work in the authors' laboratories is supported by Conacyt, Mexico; Alzheimer's Association, USA; Alzheimer Society, Canada; CIHR, Canada and FRSQ, Québec, Canada. This project was supported by grants from the National Center for Research Resources (5 G12RR013646-12), the National Institute on Minority Health and Health Disparities (G12MD007591) from the National Institutes of Health, and from the Research Centers in Minority Institutions (RCMI).
Abbreviations
- AD:
Alzheimer's disease
- GSK3:
Glycogen synthase kinase 3
- NFTs:
Neurofibrillary tangles
- LTP:
Long-term potentiation
- LTD:
Long-term depression
- NMDA:
N-methyl-D-aspartate receptor.
References
- 1.Wu D, Pan W. GSK3: a multifaceted kinase in Wnt signaling. Trends in Biochemical Sciences. 2010;35(3):161–168. doi: 10.1016/j.tibs.2009.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Metcalfe C, Bienz M. Inhibition of GSK3 by Wnt signalling—two contrasting models. Journal of Cell Science. 2011;124(21):3537–3544. doi: 10.1242/jcs.091991. [DOI] [PubMed] [Google Scholar]
- 3.Taelman VF, Dobrowolski R, Plouhinec JL, et al. Wnt signaling requires sequestration of glycogen synthase kinase 3 inside multivesicular endosomes. Cell. 2010;143(7):1136–1148. doi: 10.1016/j.cell.2010.11.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Papkoff J, Aikawa M. WNT-1 and HGF regulate GSK3β activity and β-catenin signaling in mammary epithelial cells. Biochemical and Biophysical Research Communications. 1998;247(3):851–858. doi: 10.1006/bbrc.1998.8888. [DOI] [PubMed] [Google Scholar]
- 5.Saito Y, Vandenheede JR, Cohen P. The mechanism by which epidermal growth factor inhibits glycogen synthase kinase 3 in A431 cells. Biochemical Journal. 1994;303(1):27–31. doi: 10.1042/bj3030027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cross DAE, Alessi DR, Vandenheede JR, McDowell HE, Hundal HS, Cohen P. The inhibition of glycogen synthase kinase-3 by insulin or insulin-like growth factor 1 in the rat skeletal muscle cell line L6 is blocked by wortmannin, but not by rapamycin: evidence that wortmannin blocks activation of the mitogen-activated protein kinase pathway in L6 cells between Ras and Raf. Biochemical Journal. 1994;303(1):21–26. doi: 10.1042/bj3030021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sutherland C, Leighton IA, Cohen P. Inactivation of glycogen synthase kinase-3β by phosphorylation: new kinase connections in insulin and growth-factor signalling. Biochemical Journal. 1993;296(1):15–19. doi: 10.1042/bj2960015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stambolic V, Woodgett JR. Mitogen inactivation of glycogen synthase kinase-3β in intact cells via serine 9 phosphorylation. Biochemical Journal. 1994;303(3):701–704. doi: 10.1042/bj3030701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lochhead PA, Kinstrie R, Sibbet G, Rawjee T, Morrice N, Cleghone V. A chaperone-dependent GSK3β transitional intermediate mediates activation-loop autophosphorylation. Molecular Cell. 2006;24(4):627–633. doi: 10.1016/j.molcel.2006.10.009. [DOI] [PubMed] [Google Scholar]
- 10.Thornton TM, Pedraza-Alva G, Deng B, et al. Phosphorylation by p38 MAPK as an alternative pathway for GSK3β inactivation. Science. 2008;320(5876):667–670. doi: 10.1126/science.1156037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wu G, Huang H, Abreu JG, He X. Inhibition of GSK3 phosphorylation of β-catenin via phosphorylated PPPSPXS motifs of Wnt coreceptor LRP6. PLoS ONE. 2009;4(3) doi: 10.1371/journal.pone.0004926.e4926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cadigan KM, Liu YI. Wnt signaling: complexity at the surface. Journal of Cell Science. 2006;119(3):395–402. doi: 10.1242/jcs.02826. [DOI] [PubMed] [Google Scholar]
- 13.Frame S, Cohen P, Biondi RM. A common phosphate binding site explains the unique substrate specificity of GSK3 and its inactivation by phosphorylation. Molecular Cell. 2001;7(6):1321–1327. doi: 10.1016/s1097-2765(01)00253-2. [DOI] [PubMed] [Google Scholar]
- 14.Woodgett JR. Molecular cloning and expression of glycogen synthase kinase-3/factor A. The EMBO Journal. 1990;9(8):2431–2438. doi: 10.1002/j.1460-2075.1990.tb07419.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mukai F, Ishiguro K, Sano Y, Fujita SC. Aternative splicing isoform of tau protein kinase I/glycogen synthase kinase 3β . Journal of Neurochemistry. 2002;81(5):1073–1083. doi: 10.1046/j.1471-4159.2002.00918.x. [DOI] [PubMed] [Google Scholar]
- 16.Al-Mulla F, Bitar MS, Al-Maghrebi M, et al. Raf kinase inhibitor protein RKIP enhances signaling by glycogen synthase kinase-3β . Cancer Research. 2011;71(4):1334–1343. doi: 10.1158/0008-5472.CAN-10-3102. [DOI] [PubMed] [Google Scholar]
- 17.Grimes CA, Jope RS. The multifaceted roles of glycogen synthase kinase 3β in cellular signaling. Progress in Neurobiology. 2001;65(4):391–426. doi: 10.1016/s0301-0082(01)00011-9. [DOI] [PubMed] [Google Scholar]
- 18.Soutar MPM, Kim WY, Williamson R, et al. Evidence that glycogen synthase kinase-3 isoforms have distinct substrate preference in the brain. Journal of Neurochemistry. 2010;115(4):974–983. doi: 10.1111/j.1471-4159.2010.06988.x. [DOI] [PubMed] [Google Scholar]
- 19.Jope RS, Roh MS. Glycogen synthase kinase-3 (GSK3) in psychiatric disease and therapeutic interventions. Current Drug Targets. 2006;7(11):1421–1434. doi: 10.2174/1389450110607011421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Leroy K, Boutajangout A, Authelet M, Woodgett JR, Anderton BH, Brion JP. The active form of glycogen synthase kinase-3β is associated with granulovacuolar degeneration in neurons in Alzheimers’s disease. Acta Neuropathologica. 2002;103(2):91–99. doi: 10.1007/s004010100435. [DOI] [PubMed] [Google Scholar]
- 21.Johnson GVW, Hartigan JA. Tau protein in normal and Alzheimer’s disease brain: an update. Journal of Alzheimer’s Disease. 1999;1(4-5):329–351. doi: 10.3233/jad-1999-14-512. [DOI] [PubMed] [Google Scholar]
- 22.Farias G, Cornejo A, Jiménez J, Guzmán L, Maccioni RB. Mechanisms of tau self-aggregation and neurotoxicity. Current Alzheimer Research. 2011;8(6):608–614. doi: 10.2174/156720511796717258. [DOI] [PubMed] [Google Scholar]
- 23.Johnson GVW, Stoothoff WH. Tau phosphorylation in neuronal cell function and dysfunction. Journal of Cell Science. 2004;117(24):5721–5729. doi: 10.1242/jcs.01558. [DOI] [PubMed] [Google Scholar]
- 24.Hanger DP, Anderton BH, Noble W. Tau phosphorylation: the therapeutic challenge for neurodegenerative disease. Trends in Molecular Medicine. 2009;15(3):112–119. doi: 10.1016/j.molmed.2009.01.003. [DOI] [PubMed] [Google Scholar]
- 25.Liu F, Li B, Tung EJ, Grundke-Iqbal I, Iqbal K, Gong CX. Site-specific effects of tau phosphorylation on its microtubule assembly activity and self-aggregation. European Journal of Neuroscience. 2007;26(12):3429–3436. doi: 10.1111/j.1460-9568.2007.05955.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mondragon-Rodriguez S, B.-I. G., García-Sierra F. The chronology and maturation of the neurofibrillary tangle pathology in Alzheimer’s disease is based on the state of phosphorylation, conformational changes, and cleavage of tau protein. Novascience Book. In press. [Google Scholar]
- 27.Mondragón-Rodríguez S, Basurto-Islas G, Santa-Maria I, et al. Cleavage and conformational changes of tau protein follow phosphorylation during Alzheimer’s disease. International Journal of Experimental Pathology. 2008;89(2):81–90. doi: 10.1111/j.1365-2613.2007.00568.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mondragón-Rodríguez S, Basurto-Islas G, Binder LI, García-Sierra F. Conformational changes and cleavage; are these responsible for the tau aggregation in Alzheimer’s disease? Future Neurology. 2009;4(1):39–53. [Google Scholar]
- 29.Fischer D, Mukrasch MD, Biernat J, et al. Conformational changes specific for pseudophosphorylation at serine 262 selectively impair binding of tau to microtubules. Biochemistry. 2009;48(42):10047–10055. doi: 10.1021/bi901090m. [DOI] [PubMed] [Google Scholar]
- 30.Rankin CA, Sun Q, Gamblin TC. Pseudo-phosphorylation of tau at Ser202 and Thr205 affects tau filament formation. Molecular Brain Research. 2005;138(1):84–93. doi: 10.1016/j.molbrainres.2005.04.012. [DOI] [PubMed] [Google Scholar]
- 31.Schaffer BAJ, Bertram L, Miller BL, et al. Association of GSK3B with Alzheimer disease and frontotemporal dementia. Archives of Neurology. 2008;65(10):1368–1374. doi: 10.1001/archneur.65.10.1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Asuni AA, Hooper C, Reynolds CH, Lovestone S, Anderton BH, Killick R. GSK3α exhibits β-catenin and tau directed kinase activities that are modulated by Wnt. European Journal of Neuroscience. 2006;24(12):3387–3392. doi: 10.1111/j.1460-9568.2006.05243.x. [DOI] [PubMed] [Google Scholar]
- 33.Phiel CJ, Wilson CA, Lee VMY, Klein PS. GSK-3α regulates production of Alzheimer’s disease amyloid-β peptides. Nature. 2003;423(6938):435–439. doi: 10.1038/nature01640. [DOI] [PubMed] [Google Scholar]
- 34.Kaytor MD, Orr HT. The GSK3β signaling cascade and neurodegenerative disease. Current Opinion in Neurobiology. 2002;12(3):275–278. doi: 10.1016/s0959-4388(02)00320-3. [DOI] [PubMed] [Google Scholar]
- 35.Jope RS, Bijur GN. Mood stabilizers, glycogen synthase kinase-3β and cell survival. Molecular Psychiatry. 2002;7(supplement 1):S35–S45. doi: 10.1038/sj.mp.4001017. [DOI] [PubMed] [Google Scholar]
- 36.Chiu CT, Chuang DM. Neuroprotective action of lithium in disorders of the central nervous system. Zhong Nan Da Xue Xue Bao Yi Xue Ban. 2011;36(6):461–476. doi: 10.3969/j.issn.1672-7347.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gravitz L. Drugs: a tangled web of targets. Nature. 2011;475(7355):S9–S11. doi: 10.1038/475S9a. [DOI] [PubMed] [Google Scholar]
- 38.Mondragón-Rodríguez S, Basurto-Islas G, Lee H-G, et al. Causes versus effects: the increasing complexities of Alzheimer’s disease pathogenesis. Expert Review of Neurotherapeutics. 2010;10(5):683–691. doi: 10.1586/ern.10.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Howland JG, Wang YT. Synaptic plasticity in learning and memory: stress effects in the hippocampus. Progress in Brain Research. 2008;169:145–158. doi: 10.1016/S0079-6123(07)00008-8. [DOI] [PubMed] [Google Scholar]
- 40.Ho VM, Lee J-A, Martin KC. The cell biology of synaptic plasticity. Science. 2011;334(6056):623–628. doi: 10.1126/science.1209236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Collingridge GL, Peineau S, Howland JG, Wang YT. Long-term depression in the CNS. Nature Reviews Neuroscience. 2010;11(7):459–473. doi: 10.1038/nrn2867. [DOI] [PubMed] [Google Scholar]
- 42.Appleby VJ, Corrêa SAL, Duckworth JK, et al. LTP in hippocampal neurons is associated with a CaMKII-mediated increase in GluA1 surface expression. Journal of Neurochemistry. 2011;116(4):530–543. doi: 10.1111/j.1471-4159.2010.07133.x. [DOI] [PubMed] [Google Scholar]
- 43.Peineau S, Taghibiglou C, Bradley C, et al. LTP inhibits LTD in the hippocampus via regulation of GSK3beta. Neuron. 2007;53(5):703–717. doi: 10.1016/j.neuron.2007.01.029. [DOI] [PubMed] [Google Scholar]
- 44.Kwok JB, Loy CT, Hamilton G, et al. Glycogen synthase kinase-3beta and tau genes interact in Alzheimer’s disease. Annals of Neurology. 2008;64(4):446–454. doi: 10.1002/ana.21476. [DOI] [PubMed] [Google Scholar]
- 45.Zhang N, Yu JT, Yang Y, Yang J, Zhang W, Tan L. Association analysis of GSK3B and MAPT polymorphisms with Alzheimer’s disease in Han Chinese. Brain Research. 2011;1391:147–153. doi: 10.1016/j.brainres.2011.03.052. [DOI] [PubMed] [Google Scholar]
- 46.Massoud F, Leger GC. Pharmacological treatment of Alzheimer disease. Canadian Journal of Psychiatry. 2011;56(10):579–588. doi: 10.1177/070674371105601003. [DOI] [PubMed] [Google Scholar]
- 47.Hernández F, Langa E, Cuadros R, Avila J, Villanueva N. Regulation of GSK3 isoforms by phosphatases PP1 and PP2A. Molecular and Cellular Biochemistry. 2010;344(1-2):211–215. doi: 10.1007/s11010-010-0544-0. [DOI] [PubMed] [Google Scholar]