

Abstract
The incidence of thyroid cancer is increasing, especially papillary thyroid carcinoma (PTC), making it currently the fastest-growing cancer among women. Reasons for this increase remain unclear, but several risk factors including radiation exposure and improved detection techniques have been suggested. Recently, the induction of chromosomal fragile site breakage was found to result in the formation of RET/PTC1 rearrangements, a common cause of PTC. Chromosomal fragile sites are regions of the genome with a high susceptibility to forming DNA breaks and are often associated with cancer. Exposure to a variety of external agents can induce fragile site breakage, which may account for some of the observed increase in PTC. This paper discusses the role of fragile site breakage in PTC development, external fragile site-inducing agents that may be potential risk factors for PTC, and how these factors are especially targeting women.
1. Introduction
The incidence of thyroid cancer is dramatically rising in the Unites States and other countries. Thyroid cancer has increased steadily in the Unites States over the past several decades, and according to data from the National Cancer Institute's Surveillance, Epidemiology and End Results (SEER) database, incidences are now nearly three times that of the early 1970s [1–3]. Furthermore, for unknown reasons, thyroid cancer is three times more prevalent in women than men. Thyroid cancer is the sixth most common type of cancer among women and increasing more rapidly than any other cancer. The American Cancer Society estimates that 56,460 new cases of thyroid cancer (43,210 in women and 13,250 in men) will be diagnosed in the United States in 2012, with approximately 80% of patients below 65 years old [4].
The recent upsurge in thyroid cancer is not well understood. Rates of thyroid cancer diagnoses have increased the most among small (≤2 cm) thyroid nodules, which may be explained by the use of thyroid ultrasound for diagnosis beginning in the 1980s [2, 5]. However, it is believed that the increase in thyroid cancer is not solely based on diagnostic methods [6], but due to changes in other risk factors as well [7]. One possible contributory risk factor is exposure to ionizing radiation [8], either from external radiation, such as X-rays and γ-radiation, or internal radiation, from ingestion or inhalation of radioiodine. Increases in thyroid cancer have been well documented following exposure to high doses of radiation during medical procedures or following nuclear bomb explosions or nuclear reactor fallouts [9]. However, exposure to low doses of radiation from routine diagnostic X-ray procedures and in the workplace does not increase the risk of thyroid cancer development [9], suggesting radiation exposure is not the only risk factor. An increased body mass index (BMI) is also positively associated with thyroid cancer in both women and men [10], suggesting obesity is another risk factor for thyroid cancer.
Recently, it was observed that the chemical induction of DNA breakage at chromosomal fragile sites could result in the formation of RET/PTC1 rearrangements, one common mutation observed in papillary thyroid carcinoma (PTC) [11]. Chromosomal fragile sites are regions of the genome prone to DNA breakage and often coincide with mutations observed in cancer [12]. Exposure to a variety of external agents, including dietary, environmental, and chemotherapeutic agents, can induce breakage at fragile sites. Therefore, exposure to fragile-site-inducing conditions may be an additional risk factor for thyroid cancer development, specifically PTC. Interestingly, the increasing rates of thyroid cancer are almost entirely due to an increase in PTC [2, 3, 13]. In this paper, we will discuss the potential role of fragile sites in PTC and the external fragile-site-inducing agents that may be a risk factor for thyroid cancer.
2. Fragile Sites and Cancer
2.1. Chromosomal Fragile Sites
Chromosomal fragile sites are nonrandom loci that can be observed as gaps or breaks on metaphase chromosomes under conditions that partially inhibit DNA replication [14]. Fragile sites can further be defined as common or rare, based on the frequency of their occurrence in the population. Rare fragile sites consist of repeated sequence motifs, such as trinucleotide repeats, which are present in less than 5% of the population and are inherited in a Mendelian manner [15]. In contrast, common fragile sites are present in all individuals and therefore are a normal component of chromosomal architecture [16].
Fragile sites can be observed in culture through treatment with various chemicals. The majority of common fragile sites can be induced by aphidicolin (APH), an inhibitor of DNA polymerases α, β, and δ [17, 18]. Induction of other common fragile sites has been observed following treatment with bromodeoxyuridine (BrdU) or 5-azacytidine [19]. Most rare fragile sites are expressed through the removal of folate, while others show induction following treatment with distamycin-A or BrdU [15]. Additionally, common fragile site breakage can be induced or enhanced through exposure to various dietary or environmental chemicals, including chemotherapeutic agents [12] (details in Section 4).
Unlike rare fragile sites, no known consensus sequence exists for common fragile sites. However, several characteristics have been observed at many common fragile sites studied to date. These include being located within large genes [19] and within regions of the genome that replicate late in the S-phase [20–23]. Also, several fragile sites contain highly flexible AT-rich sequences [24, 25] and are predicted to form stable DNA secondary structures [24, 26, 27]. Also, the ATR (ataxia-telangiectasia and Rad3-Related-) dependent DNA repair pathway, which responds to stalled or collapsed replication forks, is known to be vital for maintaining stability at fragile sites [28–30]. One model for common fragile site breakage is that, under conditions of replication stress, stable DNA secondary structures form at fragile sites, blocking replication fork progression. If the ATR pathway fails to properly repair these stalled replication forks, this could result in DNA breakage within these regions. In addition to replication fork stalling, paucity of replication initiation in fragile site regions [31] and the presence of transcription-derived R-loops during DNA replication of fragile sites [32] may also be involved in the mechanism of fragility.
2.2. Role of Fragile Sites in Cancer
Studies over the past several decades have shown a correlation between fragile sites and cancer-specific chromosomal aberrations [33]. Many of the genes identified within fragile sites are known tumor suppressor genes or oncogenes [34]. Fragile sites have been identified as hot spots for sister chromatid exchange [35], viral integrations [36–41], and gene amplifications [42–45] in tumor cells. Additionally, mutational signatures of some unexplained homozygous deletions observed in cancer cell lines match those at fragile sites [46].
A comprehensive examination of all known simple chromosomal translocations in cancer revealed that 52% of these recurrent translocations had at least one breakpoint located within a fragile site [47]. Specifically, 40% of translocations had breakpoints within one gene located in a fragile site, while an additional 12% of translocations had breakpoints within both genes located in fragile sites (Figure 1). Furthermore, 65% of the breakpoints identified within fragile sites were within common—not rare—fragile sites, conferring a genetic risk among all individuals. Since this study only focused on simple translocations between two genes and not the participation of fragile sites in other more complex genomic rearrangements, the association of fragile sites with breakpoints in cancer may prove to be greater than estimated in this study.
Figure 1.
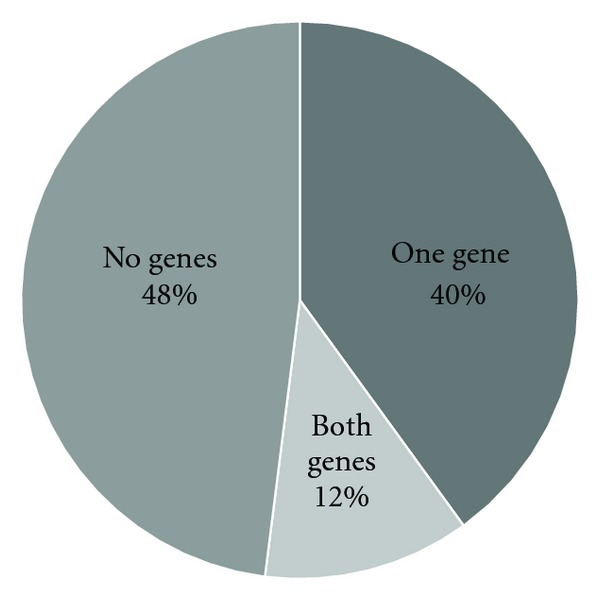
Percentage of breakpoints in genes involved in cancer-causing simple recurrent chromosomal translocations located within fragile sites.
In addition to the correlation between fragile sites and regions of the genome mutated in cancer, two studies have investigated the direct contribution of fragile site breakage to the formation of cancer-specific chromosomal aberrations. Durkin et al. observed deletions within the tumor suppressor gene FHIT, located within the most active common fragile site FRA3B, following treatment of human-mouse chromosome 3 somatic hybrid cells with APH. These deletions were consistent with those observed in esophageal, breast, and lung cancers [48]. Recently, we observed the formation of RET/PTC1 rearrangements, a translocation commonly observed in PTC, in a human thyroid epithelial cell line following treatment with the fragile-site-inducing chemicals APH, BrdU, and 2-aminopurine (2-AP) [11].
Individuals genetically predisposed to forming cancer also have higher levels of fragile site breakage. For example, Seckel syndrome—a rare genetic disorder in which patients exhibit high levels of chromosomal instability and cancer—is caused by low expression of the DNA repair protein ATR, due to a hypomorphic mutation in the ATR gene [49]. Cells from patients with Seckel syndrome have significantly higher levels of APH-induced fragile site breakage compared to normal individuals [50]. Another rare genetic disorder, Fanconi anemia (FA), is the result of mutations in various proteins involved in the FA double-strand DNA repair pathway. Patients with FA have elevated levels of chromosomal breakage and cancer [51]. Chromosomal breakpoints in blood lymphocytes from FA patients are preferentially located in fragile sites [52], and APH-induced fragile site breakage is significantly increased among these patients [53]. Proteins in both the ATR and FA DNA repair pathways are important in maintaining stability at fragile sites [12]. The WRN protein, which is phosphorylated by and colocalizes with ATR in response to replication fork arrest [54], is also vital for maintaining common fragile site stability, and this function requires WRN helicase activity [55]. Mutations in WRN can result in Werner syndrome, which is an autosomal recessive premature aging disease where individuals have a high predisposition to cancer development [56]. Interestingly, Japanese Werner syndrome patients have much higher levels of thyroid cancer, including PTC, than normal Japanese individuals [57]. Together these provide extreme examples for a genetic predisposition for fragile site breakage and cancer development.
Together these previous studies provide a strong link between fragile sites and cancer, whereby exposure to fragile-site-inducing conditions and/or a genetic predisposition could attribute to the development of various types of cancer.
3. Fragile Site Instability in PTC
3.1. RET/PTC Translocations in PTC
PTC is primarily responsible for the upsurge in thyroid cancer rates [3]. One mutation commonly observed in PTC is RET/PTC rearrangements, in which the RET oncogene translocates with a variety of genes that are constitutively expressed in the thyroid. RET (rearranged during transfection) encodes for a cell membrane receptor tyrosine kinase that responds to ligands of the glial cell line-neurotropic factor (GDNF) family, activating cell growth and survival pathways [58]. Expression of RET in the thyroid is high in neural-crest-derived C cells but not in follicular cells, where RET/PTC rearrangements result in its activation through expression of fusion proteins leading to tumorigenesis.
While prevalence of RET/PTC rearrangements is variable among different studies, overall these translocations are found in 30–40% of adult and 50–60% of pediatric PTC tumors [59]. To date, 12 RET/PTC rearrangements have been reported, all involving RET [60]. The two most common subtypes are RET/PTC1 and RET/PTC3, where RET is translocated with CCDC6 and NCOA4, respectively [61]. One known risk factor for RET/PTC rearrangement formation is exposure to radiation, where the incidence of RET/PTC rearrangements in PTC patients increases to 60–70% regardless of age [59]. RET/PTC3 rearrangements have shown a strong correlation with radiation exposure, where multiple studies indicated these translocations in 63–75% of radiation-induced RET/PTC-positive pediatric PTC tumors [62–65]. In contrast, RET/PTC1 rearrangements have been observed in 50–71% of RET/PTC-positive sporadic PTC tumors, while RET/PTC3 rearrangements were only observed in 13–42% of tumors [62, 66–68].
Spatial proximity of RET/PTC-participating genes is one of the factors contributing to cell specificity of the disease [69]. The interphase distance between RET and CCDC6 is shorter in normal human thyroid cells than in peripheral blood lymphocytes or in normal mammary epithelial cells [69]. Further, RET, CCDC6, and NCOA4 are all located on chromosome 10 and despite the predicted distance of the genes based on their location along chromosome 10, RET is located closer to NCOA4 and CCDC6 than would be expected, during interphase in normal human thyroid cells [70].
RET, CCDC6, and NCOA4, the genes participating in RET/PTC1 and RET/PTC3 rearrangements, are all located within common fragile sites. RET and NCOA4 are both located within the same APH-inducible common fragile site, FRA10G, while CCCD6 is located within the BrdU-induced common fragile site FRA10C. The location of these genes within fragile sites, the unexplained nature of sporadic PTC tumors containing translocations of these genes, and the increasing incidence of PTC tumors suggest that fragile site breakage may contribute to sporadic RET/PTC rearrangements.
3.2. Fragile-Site-Inducing Conditions Produce DNA Breaks in RET/PTC Rearrangement-Participating Genes and Generate RET/PTC1 Translocations
The idea that fragile site breakage contributes to RET/PTC rearrangement formation was directly demonstrated in our recent publication [11]. In this study, we first examined whether RET, CCDC6, and NCOA4 are true fragile sites, for example, sensitive to fragile-site-inducing conditions using fluorescence in situ hybridization (FISH). HTori-3 cells, a human thyroid epithelial cell line devoid of RET/PTC rearrangements, were treated with combinations of fragile-site-inducing chemicals known to induce the fragile sites containing the RET/PTC genes, and chromosomal breakage was measured at each gene based on the percent break of the corresponding FISH probe (Figure 2(a)). The presence of RET and NCOA4 in the APH-induced common fragile site FRA10G was investigated by treating HTori-3 cells with APH and 2-AP. The chemical 2-AP is a general inhibitor of ATR kinase, shown to increase fragile site breakage at FRA3B [29]. This treatment produced significant levels of chromosomal breakage at RET and only low levels at NCOA4 and CCDC6, indicating that RET and not NCOA4 is contained within FRA10G. The presence of CCDC6 within the BrdU-induced common fragile site FRA10C was tested by treating HTori-3 cells with BrdU and 2-AP. High levels of breakage were observed at CCDC6, but only low levels at RET and NCOA4 (Figure 2(a)), confirming the location of CCDC6 within FRA10C. These observations demonstrated that fragile-site-inducing chemicals consistent with the mode of induction for each fragile site can induce DNA breakage at RET and CCDC6 in thyroid cells. Furthermore, when HTori-3 cells were treated with all three chemicals at once, high levels of breakage were observed within RET and CCDC6 simultaneously (Figure 2(a)), indicating the possibility of RET/PTC1 chromosomal rearrangements.
Figure 2.
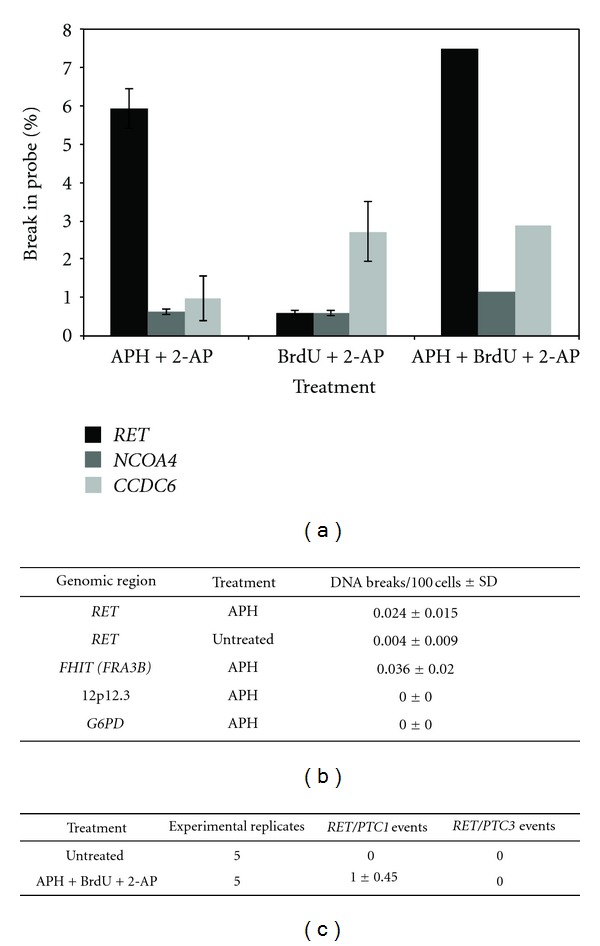
Fragile-site-inducing chemicals generate DNA breakage within RET/PTC genes and RET/PTC1 rearrangements. (a) Percentage of chromosomes showing disruption of RET, NCOA4, and CCDC6 following treatment of HTori-3 cells with fragile-site-inducing chemicals as detected by FISH. Error bars represent standard deviation. (b) The level of DNA breakage in HTori-3 cells at RET intron 11, FHIT intron 4, 12p12.3, and G6PD with or without APH treatment was detected using LM-PCR. (c) The formation of RET/PTC1 or RET/PTC3 rearrangement events was detected in HTori-3 cells using RT-PCR following treatment with fragile-site-inducing chemicals. Five experimental replicates were performed for each treatment and the average number of rearrangements detected per 106 cells per experiment is shown.
The major breakpoint cluster region of RET observed in RET/PTC tumor cells is intron 11 [71]. Using ligation-mediated-PCR (LM-PCR), we showed that APH treatment induced DNA breakage within intron 11 of RET in HTori-3 cells. The rate of DNA breakage at RET intron 11 with APH treatment was significantly greater than without treatment (P = 0.010, Figure 2(b)). Furthermore, APH-induced breakage in HTori-3 cells was specific to fragile sites, where APH treatment also induced DNA breakage within FHIT, located within the APH-inducible common fragile site FRA3B, but not in the nonfragile 12p12.3 region or in the G6PD gene, located within the non-APH-inducible rare folate-sensitive fragile site FRAXF (Figure 2(b)). Furthermore, the breakpoints located within intron 11 of RET were located near previously identified breakpoints in RET/PTC tumors [72, 73]. These results suggest that fragile site breakage within RET could lead to the generation of oncogenic fusion transcripts.
The generation of RET/PTC rearrangements following exposure to fragile-site-inducing chemicals was directly tested by treating HTori-3 cells with APH, BrdU, and 2-AP, and the presence of fusion RET/PTC1 or RET/PTC3 mRNA transcripts was detected by reverse transcription-PCR (RT-PCR). No RET/PTC rearrangements were detected without treatment, indicating an extremely low level of spontaneous rearrangement (Figure 2(c)). However, treatment with fragile-site-inducing chemicals resulted in RET/PTC1 rearrangement events with a frequency of one in 106 cells, but no RET/PTC3 rearrangement events (Figure 2(c)). These results were consistent with the chromosomal breakage observed in FISH analyses, where only the RET/PTC1 genes RET and CCDC6 exhibited high levels of breakage following treatment.
The data from this study provide direct evidence that fragile sites are involved in the generation of RET/PTC1 rearrangements in sporadic PTC tumors. Exposure to external factors that can induce fragile site breakage may play a role in the increasing incidence of PTC.
4. Effect of External Factors on Fragile Site Breakage
Aside from classic fragile-site-inducing chemicals like APH, DNA breakage at common fragile sites has been observed following exposure to many external agents, including dietary, environmental, and chemotherapeutic compounds (Table 1). Variability in fragile site breakage has been observed among individuals [87], with high levels being associated with cancer patients [88]. These variances may reflect differing exposures to external fragile-site-inducing agents, and such exposure may predispose an individual to a variety of cancers, including PTC.
Table 1.
External Fragile Site-Inducing/Enhancing Agents.
| Agents | Applications | References |
|---|---|---|
| Dietary and environmental | ||
| Atenolol | Hypertension drug | [74] |
| Benzene | Cigarette smoke, gasoline, pesticides, food, water | [75] |
| Caffeine | Dietary agent | [76, 77] |
| Carbon tetrachloride | Refrigerants, pesticides | [75] |
| Cigarette smoke | Dietary and environmental agent | [78, 79] |
| Diethylnitrosamine | Cigarette smoke, pesticides, food, beverage | [75] |
| Dimethyl sulfate | Dyes, drugs, perfumes, pesticides | [75] |
| Ethanol | Dietary agent | [80, 81] |
| Hypoxia | Low oxygen, tumor microenvironment | [82] |
| Pesticides | Dietary and environmental agent | [83–85] |
|
| ||
| Chemotherapeutics | ||
| 5-Azacytidine | Myelodysplastic syndrome, leukemia | [75] |
| Actinomycin D | Sarcoma, Wilms' tumor, germ cell, testicular, Melanoma, neuroblastoma, retinoblastoma | [75] |
| Bleomycin | Squamous cell, melanoma, sarcoma, testicular, Hodgkin's and non-Hodgkin's lymphoma | [75] |
| Busulfan | Chronic myelogenous leukemia | [75] |
| Camptothecin | Colon, rectal | [86] |
| Chlorambucil | Chronic lymphocytic leukemia, Hodgkin's and non-Hodgkin's lymphoma, breast, ovarian, testicular | [75] |
| Cytosine arabinoside | Leukemia, lymphoma | [75] |
| Floxuridine | Colon, kidney, stomach | [75] |
| Methotrexate | Breast, head and neck, lung, stomach, esophageal, sarcoma, non-Hodgkin's lymphoma, acute lymphoblastic leukemia | [75] |
4.1. Environmental and Dietary Fragile-Site-Inducing/Enhancing Chemicals
Numerous dietary and environmental chemicals can induce or enhance fragile site breakage (Table 1). Caffeine and ethanol are two dietary agents that can significantly increase the rate of fragile site breakage. Caffeine, an inhibitor of phosphoinositide 3-kinase-related kinases, including ATR and ATM (ataxia telangiectasia mutated), significantly enhances fragile site breakage in combination with APH or fluorodeoxyuridine (FUdR) [76, 77]. Similarly, ethanol enhances APH-induced fragile site breakage [80]. Cells from chronic alcohol users have an increased frequency of fragile site breakage compared to nondrinkers, which suggests that long-term alcohol use can induce fragile site expression [81].
Exposure to cigarette smoke, pesticides, or hypoxic conditions can also increase susceptibility to fragile site breakage. Peripheral blood lymphocytes from cigarette smokers have significantly greater levels of APH-induced fragile site breakage compared to nonsmokers [78]. Interestingly, peripheral blood lymphocytes from non-smokers and patients with small cell lung cancer who have stopped smoking both display lower levels of fragile site breakage following APH treatment than active smokers, suggesting this risk factor is reversible [79]. Individuals exposed to pesticides through occupational work, such as pesticide sprayers or flower collectors working in greenhouses, have increased levels of APH-induced fragile site breakage in their blood lymphocytes compared to control individuals, and these results persisted even a year later [83–85]. Furthermore, the pesticide-induced breakage was located within fragile sites containing breakpoints observed in non-Hodgkin's lymphoma and leukemia; consistent with this finding, increasing rates of hematopoietic cancers have been linked to pesticide exposure [89, 90]. Hypoxic conditions also enhance fragile site breakage with or without APH treatment in CMA32 Chinese hamster cells [82].
Dietary and environmental agents, carbon tetrachloride, dimethyl sulfate, benzene, and diethylnitrosamine all can induce fragile site breakage [75]. Carbon tetrachloride is used in refrigerants, pesticides, and industrial and manufacturing processes [91]. Formerly, this compound was used in cleaning fluids and fire extinguishers, but has since been banned from home use due to its carcinogenic properties. An increased risk of non-Hodgkin's lymphoma has been reported among individuals working in manufacturing, industrial, and laboratory jobs in which they are exposed to carbon tetrachloride. Dimethyl sulfate is used to manufacture organic chemicals including pesticides, dyes, drugs, perfumes, fabric softeners, and adhesives [91]. Dimethyl sulfate was also formerly used as a chemical weapon. Occupational exposure to this compound has been linked to cancers of the eye, bronchus, and lung.
Benzene is a known human carcinogen found in gasoline, pesticides, cigarette smoke, and industrial emissions and is a common contaminant detected in food and water [91]. According to the US Department of Health and Human Services, half of the national exposure to benzene comes from cigarette smoke, a known fragile-site-enhancing agent. The optimal concentrations of benzene (500 μg/mL) to induce fragile sites are relevant. Smoke from a smoldering cigarette yields 345–653 μg of benzene, and the average exposure for one hour of driving or riding in a car is about 40 μg benzene (even greater in highly congested areas) [91]. Therefore, the general population is exposed to a level of benzene comparable to the amount able to induce fragile sites, especially under long-term exposure.
Exposure to benzene due to occupation or geographic location has been linked to leukemia. Recently, Pellegriti et al. observed a significantly higher prevalence of PTC in the Sicilian province of Catania, located near the Mount Etna volcano, compared to other provinces, which could not be explained by industrial pollution or mild iodine deficiencies [92]. The authors suggest that exposure to environmental factors associated with the Mount Etna volcano may be responsible. Benzene can form following the incomplete combustion of organic materials in volcanoes and forest fires [93] and has been detected in lava gases emitted from Mount Etna [94], suggesting fragile site breakage due to benzene exposure may contribute to the increasing incidence of PTC in Catania. Furthermore, we have observed that treatment of HTori-3 cells with levels of benzene previously shown to induce fragile sites [75] results in a statistically significant increase in DNA breakage within RET intron 11, and this breakage was specific to fragile sites (unpublished observations).
Diethylnitrosamine (DEN) is found in pesticides, cigarette smoke, industrial pollution, drinking water, and foods and beverages [91]. DEN was previously used as a gasoline and lubricant additive, antioxidant, stabilizer in plastics, and in other manufacturing processes. Although there are no studies relating exposure to DEN with cancer susceptibility in humans, many studies in laboratory animals have shown that DEN exposure can result in the development of various tumors [91]. As with benzene, we found that DNA breakage within RET intron 11 was significantly increased in HTori-3 cells following exposure to dosages of DEN previously shown to induce fragile sites [75], and this breakage was specific to fragile sites (unpublished observations).
Atenolol is a common β-blocker used to treat hypertension, and peripheral blood lymphocytes from hypertensive patients taking this drug exhibit higher levels of chromatid and chromosomal breaks than normal individuals not taking atenolol; these breaks were preferentially located at fragile sites [74]. Furthermore, blood lymphocytes from patients taking atenolol have significantly more micronuclei than normal patients [95]. Although antihypertensive drugs have been evaluated for carcinogenic effects, studies performed to date may not be complete enough to rule out these drugs as potential cancer-causing agents [96]. Due to the prevalent usage of hypertensive drugs and the ability of atenolol to induce fragile site breakage, usage of such drugs may be an additional risk factor for cancer development, and extensive investigations are needed.
4.2. Chemotherapeutic Agents
Several chemotherapeutic agents are known to induce fragile site breakage including actinomycin D, bleomycin, busulfan, camptothecin, chlorambucil, cytosine arabinoside (cytarabine), 5-azacytidine, floxuridine, and methotrexate (Table 1) [75, 86]. The fragile site breakage observed following treatment with these chemotherapeutic agents was multiplied 3- to 8-fold by the addition of caffeine. These chemotherapeutics are commonly used to treat cancer, including leukemias and lymphomas (Table 1). Aside from killing cancer cells, residual doses of chemotherapy drugs can also lead to mutations in healthy cells that could result in a therapy-related second primary tumor. The plasma concentrations of cytarabine derived from the treatment dosage are comparable (~10 μM) or higher (depending on the regimens) [97], to the amount that causes fragile site induction (10 μM).
The rate of second primary cancers is on the rise, and they now account for one in six of all newly diagnosed cancers in the United States [98]. Thyroid cancer has been observed as a secondary cancer following treatment for various cancers, including Hodgkin's lymphoma [99, 100]. Patients with testicular cancer treated with chemotherapy and/or radiation also have a significantly elevated risk for developing thyroid cancer [101]. PTC has been observed in patients treated for osteosarcoma [57, 102–106], including children treated with chemotherapeutic agents (some of which are known to induce fragile sites, including bleomycin, actinomycin D, and methotrexate), but not radiation [106]. PTC was documented as a secondary malignancy following treatment of a pediatric rhabdomyosarcoma patient with only chemotherapeutic drugs, including actinomycin D [107]. PTC has also been observed as a second primary cancer in children treated with chemotherapy alone for acute lymphoblastic leukemia, neuroblastoma, and Ewing's sarcoma [108–111].
Many chemotherapeutic agents target DNA topoisomerases, acting as enzymatic poisons and resulting in an accumulation of double-strand DNA breaks in cells. DNA topoisomerase I activity is vital for common fragile site breakage [112, 113]. Camptothecin, one of the fragile-site-inducing chemotherapeutic agents, is a DNA topoisomerase I poison. The remaining fragile-site-inducing chemotherapeutic agents perturb DNA replication and/or RNA transcription in cells, the mode by which many fragile-site-inducing chemicals lead to chromosomal breakage. Besides the chemotherapeutic drugs already shown to induce fragile sites, many others work in a similar manner by inhibiting DNA topoisomerases and perturbing DNA replication or RNA transcription, suggesting additional drugs may have the ability to induce fragile site breakage.
Together, chemotherapeutic, dietary, and environmental agents represent a diverse spectrum by which individuals can be exposed to and increase their risk of fragile site breakage. Long-term exposure or exposure to significant doses of any of these agents, or a combination of different agents, can increase a person's susceptibility to cancer development, including PTC.
5. 5. PTC in Women
The American Cancer Society 2012 report indicated that the incidence of thyroid cancer among women is three times higher than men [4]. Thyroid cancer is the fastest-growing cancer and the sixth most common among women. Furthermore, the risk of thyroid cancer peaks earlier for women, where most women are diagnosed in the fourth and fifth decades of life compared to the sixth and seventh for men. The rate of PTC among women in the United States tripled between the early 1980s to the mid-2000s, accounting for over 75% of all thyroid cancers diagnosed during this time [3]. Despite this dramatic increase and strong disparity between women and men, there has been no explanation for these observations. However, various fragile-site-inducing factors may contribute to the difference in PTC incidence in women versus men (Figure 3).
Figure 3.
External fragile-site-inducing/enhancing agents as potential risk factors for increased PTC susceptibility in women.
One possible explanation for this gender disparity is hormonal differences between men and women. The numbers of chromosomal breaks and sister chromatid exchanges are elevated in women who are pregnant or taking oral contraceptives [114–117]. Furthermore, women currently taking oral contraceptives have an increased incidence of thyroid cancer, including a stronger association with PTC than other subtypes [118]. Variation in the frequency of APH-induced common fragile site breakage was also observed in women during different times of the menstrual cycle, with a significant increase during the luteal phase when progesterone and estradiol levels are at their highest [119]. Estrogen and progesterone levels fluctuate throughout the menstrual cycle, but hormonal birth control prevents ovulation by maintaining consistent hormone levels using synthetic estrogens and progestins. Since the development of oral hormonal contraceptives in 1960, the rate of usage has increased, such that over 10 million women in the United States ages 15 to 44 years currently take oral contraceptives [120]. Further, 82% of reproductive-age women have taken oral hormonal contraceptive pills at some point in their life. The increasing rate of oral contraceptive use among teenage girls ages 13 to 18 is particularly striking [121]. The percentage of women who have ever used high-dose oral contraceptives (emergency contraception) also rose from 4% to 10% from 2002 to 2008 [120]. The emergency contraceptive pill Plan B One-step, which contains 1.5 mg of levonorgestrel, a synthetic form of progesterone, has approximately 10-fold higher levels of the hormone than traditional contraceptives. These data, combined with the previous studies that hormone levels affect breakage within fragile sites, support a possible link between hormonal birth control use and increased fragile site breakage, but more work is needed to prove a causative role.
An increasing presence of women in the workforce may also contribute to elevated incidence of PTC in women. In 1964, only 19 million women in the United States held jobs outside of the home, compared to 65 million women in 2010 [122]. During the 1970s and 1980s, employment in the utility, trade, and transportation industries was most popular among women, and currently these industries are ranked second. Working outside of the home, including commuting to and from work, increases a woman's exposure to environmental mutagens such as benzene and diethylnitrosamine, fragile-site-inducing chemicals found in gasoline fumes and industrial emissions. Furthermore, exposure to the same amount of benzene results in approximately 20% higher levels of benzene metabolites in women versus men [123], indicating an increased susceptibility to fragile site breakage in women.
In a study examining cancer incidence in US Air Force active duty personnel between 1989 and 2002, thyroid cancer was the third most frequent invasive cancer in women and four times more prevalent than in the general population [124]. Of even greater importance, overall cancer rates among U.S. Air Force personnel were significantly reduced relative to the general population, suggesting occupational exposure may contribute to the difference in thyroid cancer rates. Active duty Air Force personnel encounter unique occupational exposures that can induce fragile site breakage, including jet fuel, high altitudes, and chemical weapons.
Trends in cigarette smoking and alcohol consumption may also contribute to increased PTC among women. Cigarette smoking has been declining in the United States over the past several decades, but the rate of decrease varies between men and women. The number of adult men that smoke decreased by 11.1% between 1965 to 2009, but the number of women only decreased by 0.5% [125]. A high level of alcohol consumption among young women is also very striking. Nearly 66% of women aged 18–44 years consume alcohol, and 14% binge drink [126]. In 2009, approximately 75% of high school girls reported having drank alcohol and nearly half currently consumed alcohol, both higher proportions than in their male counterparts [127]. Also, between 1979 and 2003 binge drinking among women ages 21–23 dramatically increased—40% among college students and 20% among nonstudents—compared to a 10% decrease among men [128]. Alcohol metabolism also differs between men and women, such that when drinking the same quantity of alcohol as men, women have a higher blood alcohol content, which enhances the potential of fragile site breakage.
Due to the significant increase in PTC diagnoses among women in the past several decades, especially among younger women, it is essential to investigate the mechanisms behind this trend. Several factors—such as hormone levels, increased exposure to environmental agents at the workplace, and behaviors like cigarette usage and alcohol consumption—suggest fragile site breakage may contribute to some of the increased incidence of PTC among women. However, more work is needed to clarify these risk factors.
6. Conclusion
The dramatic increase of thyroid cancer in the past several decades is alarming, and it is the fastest-growing cancer among women. A variety of potential risk factors for thyroid cancer, including radiation exposure, improved detection techniques, and obesity, have been proposed. Herein we provide evidence for an additional risk factor, chromosomal fragile site breakage. Chromosomal fragile sites are stable under normal conditions, but DNA breakage at these sites can be induced through exposure to many external agents. The genes involved in RET/PTC1 rearrangements, RET and CCDC6, are located within common fragile sites, and direct evidence shows that induction of fragile sites results in chromosomal breakage within these genes and ultimately leads to the formation of RET/PTC1 rearrangements. RET/PTC1 rearrangements are commonly seen in PTC tumors and are especially represented in case of sporadic PTC. Therefore, exposure to external fragile-site-inducing conditions could account for some sporadic PTC tumors and may contribute to the increase in PTC incidence.
Various external agents can induce fragile site breakage, including dietary, environmental, and chemotherapeutic compounds. Furthermore, women are especially susceptible to many of these agents, and trends over the past several decades support the involvement of fragile sites in the increased incidence of PTC in women. Therefore exposure to external fragile-site-inducing agents, along with genetic factors, may predispose an individual to PTC development, making fragile sites an additional potential risk factor for thyroid cancer. Further work is needed to elucidate the impact of fragile-site-inducing agents in the formation of PTC, such that the treatment of patients can be tailored and further growth in thyroid cancer incidence can be reduced.
Acknowledgment
This work was supported by the National Institutes of Health (RO1CA85826 and RO1CA113863 to Y.-H. Wang. and T32GM095440 to L. W. Dillon.).
References
- 1.Chen AY, Jemal A, Ward EM. Increasing incidence of differentiated thyroid cancer in the United States, 1988–2005. Cancer. 2009;115(16):3801–3807. doi: 10.1002/cncr.24416. [DOI] [PubMed] [Google Scholar]
- 2.Davies L, Welch HG. Increasing incidence of thyroid cancer in the United States, 1973–2002. Journal of the American Medical Association. 2006;295(18):2164–2167. doi: 10.1001/jama.295.18.2164. [DOI] [PubMed] [Google Scholar]
- 3.Enewold L, Zhu K, Ron E, et al. Rising thyroid cancer incidence in the United States by demographic and tumor characteristics, 1980–2005. Cancer Epidemiology Biomarkers and Prevention. 2009;18(3):784–791. doi: 10.1158/1055-9965.EPI-08-0960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.ACS. Cancer Facts and Figures 2012. Atlanta, Ga, USA: American Cancer Society; 2012. [Google Scholar]
- 5.Kent WDT, Hall SF, Isotalo PA, Houlden RL, George RL, Groome PA. Increased incidence of differentiated thyroid carcinoma and detection of subclinical disease. Canadian Medical Association Journal. 2007;177(11):1357–1361. doi: 10.1503/cmaj.061730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang Y, Zhu Y, Risch HA. Changing incidence of thyroid cancer. Journal of the American Medical Association. 2006;296(11):p. 1350. doi: 10.1001/jama.296.11.1350-a. [DOI] [PubMed] [Google Scholar]
- 7.How J, Tabah R. Explaining the increasing incidence of differentiated thyroid cancer. Canadian Medical Association Journal. 2007;177(11):1383–1384. doi: 10.1503/cmaj.071464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wartofsky L. Increasing world incidence of thyroid cancer: increased detection or higher radiation exposure? Hormones. 2010;9(2):103–108. doi: 10.14310/horm.2002.1260. [DOI] [PubMed] [Google Scholar]
- 9.Ron E, Lubin JH, Shore RE, et al. Thyroid cancer after exposure to external radiation: a pooled analysis of seven studies. Radiation Research. 1995;141(3):259–277. [PubMed] [Google Scholar]
- 10.Kitahara CM, Platz EA, Beane Freeman LE, et al. Obesity and thyroid cancer risk among U.S. men and women: a pooled analysis of five prospective studies. Cancer Epidemiology Biomarkers and Prevention. 2011;20(3):464–472. doi: 10.1158/1055-9965.EPI-10-1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gandhi M, Dillon LW, Pramanik S, Nikiforov YE, Wang YH. DNA breaks at fragile sites generate oncogenic RET/PTC rearrangements in human thyroid cells. Oncogene. 2010;29(15):2272–2280. doi: 10.1038/onc.2009.502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dillon LW, Burrow AA, Wang YH. DNA instability at chromosomal fragile sites in cancer. Current Genomics. 2010;11(5):326–337. doi: 10.2174/138920210791616699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Albores-Saavedra J, Henson DE, Glazer E, Schwartz AM. Changing patterns in the incidence and survival of thyroid cancer with follicular phenotype—papillary, follicular, and anaplastic: a morphological and epidemiological study. Endocrine Pathology. 2007;18(1):1–7. doi: 10.1007/s12022-007-0002-z. [DOI] [PubMed] [Google Scholar]
- 14.Richards RI. Fragile and unstable chromosomes in cancer: causes and consequences. Trends in Genetics. 2001;17(6):339–345. doi: 10.1016/s0168-9525(01)02303-4. [DOI] [PubMed] [Google Scholar]
- 15.Sutherland GR. Rare fragile sites. Cytogenetic and Genome Research. 2003;100(1-4):77–84. doi: 10.1159/000072840. [DOI] [PubMed] [Google Scholar]
- 16.Glover TW. Common fragile sites. Cancer Letters. 2006;232(1):4–12. doi: 10.1016/j.canlet.2005.08.032. [DOI] [PubMed] [Google Scholar]
- 17.Cheng CH, Kuchta RD. DNA polymerase ε: aphidicolin inhibition and the relationship between polymerase and exonuclease activity. Biochemistry. 1993;32(33):8568–8574. doi: 10.1021/bi00084a025. [DOI] [PubMed] [Google Scholar]
- 18.Glover TW, Berger C, Coyle J, Echo B. DNA polymerase α inhibition by aphidicolin induces gaps and breaks at common fragile sites in human chromosomes. Human Genetics. 1984;67(2):136–142. doi: 10.1007/BF00272988. [DOI] [PubMed] [Google Scholar]
- 19.Durkin SG, Glover TW. Chromosome fragile sites. Annual Review of Genetics. 2007;41:169–192. doi: 10.1146/annurev.genet.41.042007.165900. [DOI] [PubMed] [Google Scholar]
- 20.Handt O, Baker E, Dayan S, et al. Analysis of replication timing at the FRA10B and FRA16B fragile site loci. Chromosome Research. 2000;8(8):677–688. doi: 10.1023/a:1026737203447. [DOI] [PubMed] [Google Scholar]
- 21.Hellman A, Rahat A, Scherer SW, Darvasi A, Tsui LC, Kerem B. Replication delay along FRA7H, a common fragile site on human chromosome 7, leads to chromosomal instability. Molecular and Cellular Biology. 2000;20(12):4420–4427. doi: 10.1128/mcb.20.12.4420-4427.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Le Beau MM, Rassool FV, Neilly ME, et al. Replication of a common fragile site, FRA3B, occurs late in S phase and is delayed further upon induction: implications for the mechanism of fragile site induction. Human Molecular Genetics. 1998;7(4):755–761. doi: 10.1093/hmg/7.4.755. [DOI] [PubMed] [Google Scholar]
- 23.Pelliccia F, Bosco N, Curatolo A, Rocchi A. Replication timing of two human common fragile sites: FRA1H and FRA2G. Cytogenetic and Genome Research. 2008;121(3-4):196–200. doi: 10.1159/000138885. [DOI] [PubMed] [Google Scholar]
- 24.Mishmar D, Rahat A, Scherer SW, et al. Molecular characterization of a common fragile site (FRA7H) on human chromosome 7 by the cloning of a simian virus 40 integration site. Proceedings of the National Academy of Sciences of the United States of America. 1998;95(14):8141–8146. doi: 10.1073/pnas.95.14.8141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zlotorynski E, Rahat A, Skaug J, et al. Molecular basis for expression of common and rare fragile sites. Molecular and Cellular Biology. 2003;23(20):7143–7151. doi: 10.1128/MCB.23.20.7143-7151.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang H, Freudenreich CH. An AT-rich sequence in human common fragile site FRA16D causes fork stalling and chromosome breakage in S. cerevisiae . Molecular Cell. 2007;27(3):367–379. doi: 10.1016/j.molcel.2007.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shah SN, Opresko PL, Meng X, Lee MYWT, Eckert KA. DNA structure and the Werner protein modulate human DNA polymerase delta-dependent replication dynamics within the common fragile site FRA16D. Nucleic Acids Research. 2009;38(4):1149–1162. doi: 10.1093/nar/gkp1131.gkp1131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Arlt MF, Xu B, Durkin SG, Casper AM, Kastan MB, Glover TW. BRCA1 is required for common-fragile-site stability via its G2/M checkpoint function. Molecular and Cellular Biology. 2004;24(15):6701–6709. doi: 10.1128/MCB.24.15.6701-6709.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Casper AM, Nghiem P, Arlt MF, Glover TW. ATR regulates fragile site stability. Cell. 2002;111(6):779–789. doi: 10.1016/s0092-8674(02)01113-3. [DOI] [PubMed] [Google Scholar]
- 30.Durkin SG, Arlt MF, Howlett NG, Glover TW. Depletion of CHK1, but not CHK2, induces chromosomal instability and breaks at common fragile sites. Oncogene. 2006;25(32):4381–4388. doi: 10.1038/sj.onc.1209466. [DOI] [PubMed] [Google Scholar]
- 31.Letessier A, Millot GA, Koundrioukoff S, et al. Cell-type-specific replication initiation programs set fragility of the FRA3B fragile site. Nature. 2011;470(7332):120–124. doi: 10.1038/nature09745. [DOI] [PubMed] [Google Scholar]
- 32.Helmrich A, Ballarino M, Tora L. Collisions between replication and transcription complexes cause common fragile site instability at the longest human genes. Molecular Cell. 2011;44(6):966–977. doi: 10.1016/j.molcel.2011.10.013. [DOI] [PubMed] [Google Scholar]
- 33.Arlt MF, Durkin SG, Ragland RL, Glover TW. Common fragile sites as targets for chromosome rearrangements. DNA Repair. 2006;5(9-10):1126–1135. doi: 10.1016/j.dnarep.2006.05.010. [DOI] [PubMed] [Google Scholar]
- 34.Popescu NC. Genetic alterations in cancer as a result of breakage at fragile sites. Cancer Letters. 2003;192(1):1–17. doi: 10.1016/s0304-3835(02)00596-7. [DOI] [PubMed] [Google Scholar]
- 35.Glover TW, Stein CK. Induction of sister chromatid exchanges at common fragile sites. American Journal of Human Genetics. 1987;41(5):882–890. [PMC free article] [PubMed] [Google Scholar]
- 36.Wilke CM, Hall BK, Hoge A, Paradee W, Smith DI, Glover TW. FRA3B extends over a broad region and contains a spontaneous HPV16 integration site: direct evidence for the coincidence of viral integration sites and fragile sites. Human Molecular Genetics. 1996;5(2):187–195. doi: 10.1093/hmg/5.2.187. [DOI] [PubMed] [Google Scholar]
- 37.De Braekeleer M, Sreekantaiah C, Haas O. Herpes simplex virus and human papillomavirus sites correlate with chromosomal breakpoints in human cervical carcinoma. Cancer Genetics and Cytogenetics. 1992;59(2):135–137. doi: 10.1016/0165-4608(92)90207-o. [DOI] [PubMed] [Google Scholar]
- 38.Popescu NC, DiPaolo JA. Preferential sites for viral integration on mammalian genome. Cancer Genetics and Cytogenetics. 1989;42(2):157–171. doi: 10.1016/0165-4608(89)90084-8. [DOI] [PubMed] [Google Scholar]
- 39.Smith PP, Friedman CL, Bryant EM, McDougall JK. Viral integration and fragile sites in human papillomavirus-immortalized human keratinocyte cell lines. Genes Chromosomes and Cancer. 1992;5(2):150–157. doi: 10.1002/gcc.2870050209. [DOI] [PubMed] [Google Scholar]
- 40.Thorland EC, Myers SL, Gostout BS, Smith DI. Common fragile sites are preferential targets for HPV16 integrations in cervical tumors. Oncogene. 2003;22(8):1225–1237. doi: 10.1038/sj.onc.1206170. [DOI] [PubMed] [Google Scholar]
- 41.Thorland EC, Myers SL, Persing DH, et al. Human papillomavirus type 16 integrations in cervical tumors frequently occur in common fragile sites. Cancer Research. 2000;60(21):5916–5921. [PubMed] [Google Scholar]
- 42.Coquelle A, Pipiras E, Toledo F, Buttin G, Debatisse M. Expression of fragile sites triggers intrachromosomal mammalian gene amplification and sets boundaries to early amplicons. Cell. 1997;89(2):215–225. doi: 10.1016/s0092-8674(00)80201-9. [DOI] [PubMed] [Google Scholar]
- 43.Hellman A, Zlotorynski E, Scherer SW, et al. A role for common fragile site induction in amplification of human oncogenes. Cancer Cell. 2002;1(1):89–97. doi: 10.1016/s1535-6108(02)00017-x. [DOI] [PubMed] [Google Scholar]
- 44.Kuo MT, Vyas RC, Jiang LX, Hittelman WN. Chromosome breakage at a major fragile site associated with P- glycoprotein gene amplification in multidrug-resistant CHO cells. Molecular and Cellular Biology. 1994;14(8):5202–5211. doi: 10.1128/mcb.14.8.5202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Miller CT, Lin L, Casper AM, et al. Genomic amplification of MET with boundaries within fragile site FRA7G and upregulation of MET pathways in esophageal adenocarcinoma. Oncogene. 2006;25(3):409–418. doi: 10.1038/sj.onc.1209057. [DOI] [PubMed] [Google Scholar]
- 46.Bignell GR, Greenman CD, Davies H, et al. Signatures of mutation and selection in the cancer genome. Nature. 2010;463(7283):893–898. doi: 10.1038/nature08768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Burrow AA, Williams LE, Pierce LCT, Wang YH. Over half of breakpoints in gene pairs involved in cancer-specific recurrent translocations are mapped to human chromosomal fragile sites. BMC Genomics. 2009;10, article 59 doi: 10.1186/1471-2164-10-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Durkin SG, Ragland RL, Arlt MF, Mulle JG, Warren ST, Glover TW. Replication stress induces tumor-like microdeletions in FHIT/FRA3B. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(1):246–251. doi: 10.1073/pnas.0708097105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.O’Driscoll M, Ruiz-Perez VL, Woods CG, Jeggo PA, Goodship JA. A splicing mutation affecting expression of ataxia-telangiectasia and Rad3-related protein (ATR) results in Seckel syndrome. Nature Genetics. 2003;33(4):497–501. doi: 10.1038/ng1129. [DOI] [PubMed] [Google Scholar]
- 50.Casper AM, Durkin SG, Arlt MF, Glover TW. Chromosomal instability at common fragile sites in seckel syndrome. American Journal of Human Genetics. 2004;75(4):654–660. doi: 10.1086/422701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kitao H, Takata M. Fanconi anemia: a disorder defective in the DNA damage response. International Journal of Hematology. 2011;93(4):417–424. doi: 10.1007/s12185-011-0777-z. [DOI] [PubMed] [Google Scholar]
- 52.Schoder C, Liehr T, Velleuer E, et al. New aspects on chromosomal instability: chromosomal break-points in Fanconi anemia patients co-localize on the molecular level with fragile sites. International Journal of Oncology. 2010;36(2):307–312. [PubMed] [Google Scholar]
- 53.Howlett NG, Taniguchi T, Durkin SG, D’Andrea AD, Glover TW. The Fanconi anemia pathway is required for the DNA replication stress respone and for the regulation of common fragile site stability. Human Molecular Genetics. 2005;14(5):693–701. doi: 10.1093/hmg/ddi065. [DOI] [PubMed] [Google Scholar]
- 54.Franchitto A, Pichierri P. Protecting genomic integrity during DNA replication: correlation between Werner’s and Bloom’s syndrome gene products and the MRE11 complex. Human Molecular Genetics. 2002;11(20):2447–2453. doi: 10.1093/hmg/11.20.2447. [DOI] [PubMed] [Google Scholar]
- 55.Pirzio LM, Pichierri P, Bignami M, Franchitto A. Werner syndrome helicase activity is essential in maintaining fragile site stability. Journal of Cell Biology. 2008;180(2):305–314. doi: 10.1083/jcb.200705126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pichierri P, Ammazzalorso F, Bignami M, Franchitto A. The Werner syndrome protein: linking the replication checkpoint response to genome stability. Aging. 2011;3(3):311–318. doi: 10.18632/aging.100293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Goto M, Miller RW, Ishikawa Y, et al. Excess of rare cancers in Werner syndrome (adult progeria) Cancer Epidemiology, Biomarkers & Prevention. 1996;5:239–246. [PubMed] [Google Scholar]
- 58.Arighi E, Borrello MG, Sariola H. RET tyrosine kinase signaling in development and cancer. Cytokine and Growth Factor Reviews. 2005;16(4-5):441–467. doi: 10.1016/j.cytogfr.2005.05.010. [DOI] [PubMed] [Google Scholar]
- 59.Nikiforov YE. RET/PTC rearrangement in thyroid tumors. Endocrine Pathology. 2002;13(1):3–16. doi: 10.1385/ep:13:1:03. [DOI] [PubMed] [Google Scholar]
- 60.Nikiforova MN, E Nikiforov Y. Molecular genetics of thyroid cancer: implications for diagnosis, treatment and prognosis. Expert Review of Molecular Diagnostics. 2008;8(1):83–95. doi: 10.1586/14737159.8.1.83. [DOI] [PubMed] [Google Scholar]
- 61.Santoro M, Melillo RM, Fusco A. RET/PTC activation in papillary thyroid carcinoma: European Journal of Endocrinology prize lecture. European Journal of Endocrinology. 2006;155(5):645–653. doi: 10.1530/eje.1.02289. [DOI] [PubMed] [Google Scholar]
- 62.Nikiforov YE, Rowland JM, Bove KE, Monforte-Munoz H, Fagin JA. Distinct pattern of ret oncogene rearrangements in morphological variants of radiation-induced and sporadic thyroid papillary carcinomas in children. Cancer Research. 1997;57(9):1690–1694. [PubMed] [Google Scholar]
- 63.Fugazzola L, Pilotti S, Pinchera A, et al. Oncogenic rearrangements of the RET proto-oncogene in papillary thyroid carcinomas from children exposed to the Chernobyl nuclear accident. Cancer Research. 1995;55(23):5617–5620. [PubMed] [Google Scholar]
- 64.Klugbauer S, Lengfelder E, Demidchik EP, Rabes HM. High prevalence of RET rearrangement in thyroid tumors of children from Belarus after the Chernobyl reactor accident. Oncogene. 1995;11(12):2459–2467. [PubMed] [Google Scholar]
- 65.Klugbauer S, Jauch A, Lengfelder E, Demidchik E, Rabes HM. A novel type of RET rearrangement (PTC8) in childhood papillary thyroid carcinomas and characterization of the involved gene (RFG8) Cancer Research. 2000;60(24):7028–7032. [PubMed] [Google Scholar]
- 66.Bongarzone I, Vigneri P, Mariani L, Collini P, Pilotti S, Pierotti MA. RET/NTRK1 rearrangements in thyroid gland tumors of the papillary carcinoma family: correlation with clinicopathological features. Clinical Cancer Research. 1998;4(1):223–228. [PubMed] [Google Scholar]
- 67.Fenton CL, Lukes Y, Nicholson D, Dinauer CA, Francis GL, Tuttle RM. The ret/PTC mutations are common in sporadic papillary thyroid carcinoma of children and young adults. Journal of Clinical Endocrinology and Metabolism. 2000;85(3):1170–1175. doi: 10.1210/jcem.85.3.6472. [DOI] [PubMed] [Google Scholar]
- 68.Finn SP, Smyth P, O’Leary J, Sweeney EC, Sheils O. Comment: Ret/PTC chimeric transcripts in an Irish cohort of sporadic papillary thyroid carcinoma. Journal of Clinical Endocrinology and Metabolism. 2003;88(2):938–941. doi: 10.1210/jc.2002-021239. [DOI] [PubMed] [Google Scholar]
- 69.Nikiforova MN, Stringer JR, Blough R, Medvedovic M, Fagin JA, Nikiforov YE. Proximity of chromosomal loci that participate in radiation-induced rearrangements in human cells. Science. 2000;290(5489):138–141. doi: 10.1126/science.290.5489.138. [DOI] [PubMed] [Google Scholar]
- 70.Gandhi M, Medvedovic M, Stringer JR, Nikiforov YE. Interphase chromosome folding determines spatial proximity of genes participating in carcinogenic RET/PTC rearrangements. Oncogene. 2006;25(16):2360–2366. doi: 10.1038/sj.onc.1209268. [DOI] [PubMed] [Google Scholar]
- 71.Smanik PA, Furminger TL, Mazzaferri EL, Jhiang SM. Breakpoint characterization of the ret/PTC oncogene in human papillary thyroid carcinoma. Human Molecular Genetics. 1995;4(12):2313–2318. doi: 10.1093/hmg/4.12.2313. [DOI] [PubMed] [Google Scholar]
- 72.Bongarzone I, Butti MG, Fugazzola L, et al. Comparison of the breakpoint regions of ELE1 and RET genes involved in the generation of RET/PTC3 oncogene in sporadic and in radiation-associated papillary thyroid carcinomas. Genomics. 1997;42(2):252–259. doi: 10.1006/geno.1997.4685. [DOI] [PubMed] [Google Scholar]
- 73.Klugbauer S, Pfeiffer P, Gassenhuber H, Beimfohr C, Rabes HM. RET rearrangements in radiation-induced papillary thyroid carcinomas: high prevalence of topoisomerase I sites at breakpoints and microhomology-mediated end joining in ELE1 and RET chimeric genes. Genomics. 2001;73(2):149–160. doi: 10.1006/geno.2000.6434. [DOI] [PubMed] [Google Scholar]
- 74.Télez M, Ortiz-Lastra E, Gonzalez AJ, et al. Assessment of the genotoxicity of atenolol in human peripheral blood lymphocytes: correlation between chromosomal fragility and content of micronuclei. Mutation Research. 2010;695(1-2):46–54. doi: 10.1016/j.mrgentox.2009.02.015. [DOI] [PubMed] [Google Scholar]
- 75.Yunis JJ, Soreng AL, Bowe AE. Fragile sites are targets of diverse mutagens and carcinogens. Oncogene. 1987;1(1):59–69. [PubMed] [Google Scholar]
- 76.Glover TW, Coyle-Morris J, Morgan R. Fragile sites: overview, occurrence in acute nonlymphocytic leukemia and effects of caffeine on expression. Cancer Genetics and Cytogenetics. 1986;19(1-2):141–150. doi: 10.1016/0165-4608(86)90381-x. [DOI] [PubMed] [Google Scholar]
- 77.Yunis JJ, Soreng AL. Constitutive fragile sites and cancer. Science. 1984;226(4679):1199–1204. doi: 10.1126/science.6239375. [DOI] [PubMed] [Google Scholar]
- 78.Kao-Shan C-S, Fine RL, Whang-Peng J, Lee EC, Chabner BA. Increased fragile sites and sister chromatid exchanges in bone marrow and peripheral blood of young cigarette smokers. Cancer Research. 1987;47(23):6278–6282. [PubMed] [Google Scholar]
- 79.Stein CK, Glover TW, Palmer JL, Glisson BS. Direct correlation between FRA3B expression and cigarette smoking. Genes Chromosomes and Cancer. 2002;34(3):333–340. doi: 10.1002/gcc.10061. [DOI] [PubMed] [Google Scholar]
- 80.Kuwano A, Kajii T. Synergistic effect of aphidicolin and ethanol on the induction of common fragile sites. Human Genetics. 1987;75(1):75–78. doi: 10.1007/BF00273845. [DOI] [PubMed] [Google Scholar]
- 81.Demirhan O, Taştemir D. Cytogenetic effects of ethanol on chronic alcohol users. Alcohol and Alcoholism. 2008;43(2):127–136. doi: 10.1093/alcalc/agm155. [DOI] [PubMed] [Google Scholar]
- 82.Coquelle A, Toledo F, Stern S, Bieth A, Debatisse M. A new role for hypoxia in tumor progression: induction of fragile site triggering genomic rearrangements and formation of complex DMs and HSRs. Molecular Cell. 1998;2(2):259–265. doi: 10.1016/s1097-2765(00)80137-9. [DOI] [PubMed] [Google Scholar]
- 83.Musio A, Sbrana I. Aphidicolin-Sensitive specific common fragile sites: a biomarker of exposure to pesticides. Environmental and Molecular Mutagenesis. 1997;29(3):250–255. doi: 10.1002/(sici)1098-2280(1997)29:3<250::aid-em4>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- 84.Sbrana I, Musio A. Enhanced expression of common fragile site with occupational exposure to pesticides. Cancer Genetics and Cytogenetics. 1995;82(2):123–127. doi: 10.1016/0165-4608(95)00020-p. [DOI] [PubMed] [Google Scholar]
- 85.Webster LR, McKenzie GH, Moriarty HT. Organophosphate-based pesticides and genetic damage implicated in bladder cancer. Cancer Genetics and Cytogenetics. 2002;133(2):112–117. doi: 10.1016/s0165-4608(01)00576-3. [DOI] [PubMed] [Google Scholar]
- 86.Sbrana I, Zavattari P, Barale R, Musio A. Common fragile sites on human chromosomes represent transcriptionally active regions: evidence from camptothecin. Human Genetics. 1998;102(4):409–414. doi: 10.1007/s004390050713. [DOI] [PubMed] [Google Scholar]
- 87.Craig-Holmes AP, Strong LC, Goodacre A, Pathak S. Variation in the expression of aphidicolin-induced fragile sites in human lymphocyte cultures. Human Genetics. 1987;76(2):134–137. doi: 10.1007/BF00284909. [DOI] [PubMed] [Google Scholar]
- 88.Tunca B, Egeli Ü, Zorluoglu A, Yilmazlar T, Yerci Ö, Kizil A. The expression of fragile sites in lymphocytes of patients with rectum cancer and their first-degree relatives. Cancer Letters. 2000;152(2):201–209. doi: 10.1016/s0304-3835(00)00334-7. [DOI] [PubMed] [Google Scholar]
- 89.Blair A, Zahm SH. Cancer among farmers. Occupational Medicine. 1991;6(3):335–354. [PubMed] [Google Scholar]
- 90.Morris Brown L, Blair A, Gibson R, et al. Pesticide exposures and other agricultural risk factors for leukemia among men in Iowa and Minnesota. Cancer Research. 1990;50(20):6585–6591. [PubMed] [Google Scholar]
- 91.NTP. Report on Carcinogens. 12th edition. Durham, NC, USA: U.S. Department of Health and Human Services, Public Health Service, National Toxicology Program: Research Triangle Park; 2011. [Google Scholar]
- 92.Pellegriti G, De Vathaire F, Scollo C, et al. Papillary thyroid cancer incidence in the volcanic area of sicily. Journal of the National Cancer Institute. 2009;101(22):1575–1583. doi: 10.1093/jnci/djp354. [DOI] [PubMed] [Google Scholar]
- 93.Pecoraino G, Scalici L, Avellone G, et al. Distribution of volatile organic compounds in Sicilian groundwaters analysed by head space-solid phase micro extraction coupled with gas chromatography mass spectrometry (SPME/GC/MS) Water Research. 2008;42(14):3563–3577. doi: 10.1016/j.watres.2008.07.022. [DOI] [PubMed] [Google Scholar]
- 94.Jordan A. Volcanic formation of halogenated organic compounds. In: Gribble GW, editor. The Handbook of Environmental Chemistry. Vol. 3. Berlin, Germany: Springer; 2003. pp. 121–139. [Google Scholar]
- 95.Télez M, Martínez B, Criado B, et al. In vitro and in vivo evaluation of the antihypertensive drug atenolol in cultured human lymphocytes: effects of long-term therapy. Mutagenesis. 2000;15(3):195–202. doi: 10.1093/mutage/15.3.195. [DOI] [PubMed] [Google Scholar]
- 96.Brambilla G, Martelli A. Genotoxicity and carcinogenicity studies of antihypertensive agents. Mutation Research. 2006;612(2):115–149. doi: 10.1016/j.mrrev.2005.12.002. [DOI] [PubMed] [Google Scholar]
- 97.Ozkaynak MF, Avramis VI, Carcich S, Ortega JA. Pharmacology of cytarabine given as a continuous infusion followed by mitoxantrone with and without amsacrine/etoposide as reinduction chemotherapy for relapsed or refractory pediatric acute myeloid leukemia. Medical and Pediatric Oncology. 1998;31(6):475–482. doi: 10.1002/(sici)1096-911x(199812)31:6<475::aid-mpo3>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 98.Allan JM, Travis LB. Mechanisms of therapy-related carcinogenesis. Nature Reviews Cancer. 2005;5(12):943–955. doi: 10.1038/nrc1749. [DOI] [PubMed] [Google Scholar]
- 99.Boffetta P, Kaldor JM. Secondary malignancies following cancer chemotherapy. Acta Oncologica. 1994;33(6):591–598. doi: 10.3109/02841869409121767. [DOI] [PubMed] [Google Scholar]
- 100.Swerdlow AJ, Douglas AJ, Vaughan Hudson G, Bennett MH, MacLennan KA. Risk of second primary cancers after Hodgkin’s disease by type of treatment: analysis of 2846 patients in the British National Lymphoma Investigation. British Medical Journal. 1992;304(6835):1137–1143. doi: 10.1136/bmj.304.6835.1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Travis LB, Curtis RE, Storm H, et al. Risk of second malignant neoplasms among long-term survivors of testicular cancer. Journal of the National Cancer Institute. 1997;89(19):1429–1439. doi: 10.1093/jnci/89.19.1429. [DOI] [PubMed] [Google Scholar]
- 102.Jiménez M, León P, Castro L, Azcona C, Sierrasesúmaga L. Second tumors in pediatric oncologic patients. Report of 5 cases. Revista de medicina de la Universidad de Navarra. 1995;40(2):72–77. [PubMed] [Google Scholar]
- 103.Tsuchiya H, Tomita K, Ohno M, Inaoki M, Kawashima A. Werner’s syndrome combined with quintuplicate malignant tumors: a case report and review of literature data. Japanese Journal of Clinical Oncology. 1991;21(2):135–142. doi: 10.1093/oxfordjournals.jjco.a039443. [DOI] [PubMed] [Google Scholar]
- 104.Verneris M, Ross McDougall I, Becton D, Link MP. Thyroid carcinoma after successful treatment of osteosarcoma: a report of three patients. Journal of Pediatric Hematology/Oncology. 2001;23(5):312–315. doi: 10.1097/00043426-200106000-00016. [DOI] [PubMed] [Google Scholar]
- 105.Yen BC, Kahn H, Schiller AL, Klein MJ, Phelps RG, Lebwohl MG. Multiple hamartoma syndrome with osteosarcoma. Archives of Pathology and Laboratory Medicine. 1993;117(12):1252–1254. [PubMed] [Google Scholar]
- 106.Min SK, Yoon SS, Lee SY, Jeon DG. Secondary thyroid papillary carcinoma in osteosarcoma patients: report of two cases. Journal of Korean Medical Science. 2008;23(1):149–152. doi: 10.3346/jkms.2008.23.1.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Venkitaraman R, Affolter A, Ahmed M, et al. Childhood papillary thyroid cancer as second malignancy after successful treatment of rhabdomyosarcoma. Acta Oncologica. 2008;47(3):469–472. doi: 10.1080/02841860701864676. [DOI] [PubMed] [Google Scholar]
- 108.De Vathaire F, Hawkins M, Campbell S, et al. Second malignant neoplasms after a first cancer in childhood: temporal pattern of risk according to type of treatment. British Journal of Cancer. 1999;79(11-12):1884–1893. doi: 10.1038/sj.bjc.6690300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Gow KW, Lensing S, Hill DA, et al. Thyroid carcinoma presenting in childhood or after treatment of childhood malignancies: an institutional experience and review of the literature. Journal of Pediatric Surgery. 2003;38(11):1574–1580. doi: 10.1016/s0022-3468(03)00563-3. [DOI] [PubMed] [Google Scholar]
- 110.Vane D, King DR, Boles ET. Secondary thyroid neoplasms in pediatric cancer patients: increased risk with improved survival. Journal of Pediatric Surgery. 1984;19(6):855–860. doi: 10.1016/s0022-3468(84)80384-x. [DOI] [PubMed] [Google Scholar]
- 111.Smith MB, Xue H, Strong L, et al. Forty-year experience with second malignancies after treatment of childhood cancer: analysis of outcome following the development of the second malignancy. Journal of Pediatric Surgery. 1993;28(10):1342–1349. doi: 10.1016/s0022-3468(05)80325-2. [DOI] [PubMed] [Google Scholar]
- 112.Arlt MF, Glover TW. Inhibition of topoisomerase I prevents chromosome breakage at common fragile sites. DNA Repair. 2010;9(6):678–689. doi: 10.1016/j.dnarep.2010.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Tuduri S, Crabbé L, Conti C, et al. Topoisomerase I suppresses genomic instability by preventing interference between replication and transcription. Nature Cell Biology. 2009;11(11):1315–1324. doi: 10.1038/ncb1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.McQuarrie HG, Scott CD, Ellsworth HS, Harris JW, Stone RA. Cytogenetic studies on women using oral contraceptives and their progeny. American Journal of Obstetrics and Gynecology. 1970;108(4):659–665. doi: 10.1016/0002-9378(70)90249-8. [DOI] [PubMed] [Google Scholar]
- 115.Biri A, Civelek E, Karahalil B, Şardaş S. Assessment of DNA damage in women using oral contraceptives. Mutation Research. 2002;521(1-2):113–119. doi: 10.1016/s1383-5718(02)00217-6. [DOI] [PubMed] [Google Scholar]
- 116.Dhillon VS, Dhillon IK. Genotoxicity evaluation of estradiol. Mutation Research. 1995;345(1-2):87–95. doi: 10.1016/0165-1218(95)90073-x. [DOI] [PubMed] [Google Scholar]
- 117.Hundal BS, Dhillon VS, Sidhu IS. Genotoxic potential of estrogens. Mutation Research. 1997;389(2-3):173–181. doi: 10.1016/s1383-5718(96)00144-1. [DOI] [PubMed] [Google Scholar]
- 118.La Vecchia C, Ron E, Franceschi S, et al. A pooled analysis of case-control studies of thyroid cancer. III. Oral contraceptives, menopausal replacement therapy and other female hormones. Cancer Causes and Control. 1999;10(2):157–166. doi: 10.1023/a:1008832513932. [DOI] [PubMed] [Google Scholar]
- 119.Furuya T, Hagiwara J, Ochi H, et al. Changes in common fragile sites on chromosomes according to the menstrual cycle. Human Genetics. 1991;86(5):471–474. doi: 10.1007/BF00194635. [DOI] [PubMed] [Google Scholar]
- 120.Mosher WD, Jones J. Use of contraception in the United States: 1982–2008. Vital and health statistics. Series 23. 2010;(29):1–44. [PubMed] [Google Scholar]
- 121.Ehrlich E, Gibson TB, Mark TL. Trends in prescriptions for oral contraceptives among U.S. teenagers. Thomson Reuters, March 2011.
- 122.BLS. Spotlight on statistics: women at work. Department of Labor, Bureau of Labor Statistics. March 2011, http://www.bls.gov/spotlight/2011/women.
- 123.Kim S, Vermeulen R, Waidyanatha S, et al. Modeling human metabolism of benzene following occupational and environmental exposures. Cancer Epidemiology Biomarkers and Prevention. 2006;15(11):2246–2252. doi: 10.1158/1055-9965.EPI-06-0262. [DOI] [PubMed] [Google Scholar]
- 124.Yamane GK. Cancer incidence in the U.S. Air Force: 1989–2002. Aviation Space and Environmental Medicine. 2006;77(8):789–794. [PubMed] [Google Scholar]
- 125.Health, United States, 2010: With Special Feature in Death and Dying. Hyattsville, Md, USA: National Center for Health Statistics (U.S.) and National Center for Health Services Research; 2011. [PubMed] [Google Scholar]
- 126.CDC. Alcohol use among pregnant and nonpregnant women of childbearing age—United States, 1991–2005. Morbidity and Mortality Weekly Report. 2009;58(19):529–532. [PubMed] [Google Scholar]
- 127.Eaton DK, Kann L, Kinchen S, et al. Youth risk behavior surveillance—United States, 2009. Morbidity and Mortality Weekly Report. 2010;59(SS-5):1–142. [PubMed] [Google Scholar]
- 128.Grucza RA, Norberg KE, Bierut LJ. Binge drinking among youths and young adults in the united states: 1979–2006. Journal of the American Academy of Child and Adolescent Psychiatry. 2009;48(7):692–702. doi: 10.1097/CHI.0b013e3181a2b32f. [DOI] [PMC free article] [PubMed] [Google Scholar]