

Abstract
Accumulating evidence suggests a direct role for cigarette smoke in pulmonary vascular remodeling, which contributes to the development of pulmonary hypertension. However, the molecular mechanisms underlying this process remain poorly understood. Platelet-derived growth factor (PDGF) is a potential mitogen and chemoattractant implicated in several biological processes, including cell survival, proliferation, and migration. In this study, we investigated the effect of cigarette smoke extract (CSE) on cell proliferation of rat pulmonary artery smooth muscle cells (rPASMCs). We found that stimulation of rPASMCs with CSE significantly increased cell proliferation and promoted cell cycle progression from G1 phase to the S and G2 phases. CSE treatment also significantly upregulated the mRNA and protein levels of PDGFB and PDGFRβ. Our study also revealed that Rottlerin, an inhibitor of PKCδ signaling, prevented CSE-induced cell proliferation, attenuated the increase of S and G2 phase populations induced by CSE treatment, and downregulated PDGFB and PDGFRβ mRNA and protein levels in rPASMCs exposed to CSE. Collectively, our data demonstrated that CSE-induced cell proliferation of rPASMCs involved upregulation of the PKCδ-PDGFB pathway.
1. Introduction
Pulmonary arterial hypertension (PAH) is a severe condition that frequently leads to right ventricular failure and death [1, 2]. Remodeling of small pulmonary arteries represents the main pathologic finding related to PAH. The remodeling process includes marked proliferation of pulmonary artery smooth muscle cells (PASMCs) that contribute to vascular wall hypertrophy [1, 3].
Studies in both animal models and in human patients have shown that cigarette smoke exerts a direct effect on pulmonary vascular structure, resulting in pulmonary vascular remodeling and pulmonary hypertension [4, 5]. However, the molecular mechanisms underlying this process remain unclear.
Platelet-derived growth factor (PDGF) is a potent mitogen and chemoattractant for pulmonary vascular smooth muscle cells [6]. Although there are several PDGF family members (A, B, C, and D), PDGFB is a prime candidate to study, because the PDGFB isoform is a potent inducer of VSMC proliferation and only acts through the beta-subunits of the PDGF receptor (PDGFR) [6–8]. The upregulated expression of PDGF and PDGFR has been correlated with PAH in various experimental animal models [9, 10] and in humans [11]. However, whether PDGF is also activated in rPASMCs by cigarette smoke extract (CSE) has not been explored.
Protein kinase C (PKC) is a crucial family of the serine-threonine kinases in the intracellular signal transduction pathway. It has been implicated in a variety of cellular functions in vascular smooth muscle cells, including proliferation [12], apoptosis [13], and differentiation [14]. Recent studies have shown that CSE activates PKC in monocytes [15], bronchial epithelial cells [16], and pulmonary artery endothelial cells [17]. It has also been reported that PKCδ plays an important role in hypoxia-induced proliferation of PASMCs [18]. Based on this background, we hypothesize that CSE may activate PKCδ to stimulate PDGFB and activate a cellular response in rPASMCs.
This study was designed to examine the impact of CSE on PKCδ expression to promote rPASMCs proliferation by upregulating the expression of PDGFB and PDGFRβ, which could contribute to pulmonary vascular remodeling.
2. Materials and Methods
2.1. Medium and Reagents
The rPASMCs were purchased from ATCC (Jing-Ke-Hong-Da Company, Beijing, China), and fetal bovine serum (FBS) and penicillin/streptomycin were obtained from Gibco Life Technologies Inc. (Rockville, MD, USA). Dulbecco's modified Eagle's medium (DMEM) was obtained from Clontech. The mouse polyclonal antibody against β-actin and the rabbit polyclonal antibodies against PKCδ, piPKCδ, and Rottlerin were all purchased from Sigma-Aldrich (St. Louis, MO, USA). Mouse monoclonal antibodies against PDGFB and PDGFRβ were purchased from Bioworld Technology Inc. (St. Louis Park, MN, USA). Fluorescein isothiocyanate- (FITC-) conjugated goat anti-rabbit IgG and horseradish peroxidase- (HRP-) conjugated goat anti-rabbit IgG were purchased from Pierce Biotechnology Co. Ltd (Rockford, IL, USA). Trizol reagent was obtained from Invitrogen (Carlsbad, CA, USA). All primers were synthesized by Aoke Bio-Engineering Co. (Beijing, China).
2.2. Cell Culture and Treatment
Cells were cultured at 37°C in DMEM supplemented with 10% FBS and antibiotics (penicillin and streptomycin) in a 5% CO2 incubator. Cells were treated with varying concentrations of CSE (0, 2.5%, 5%, 10%, and 20%) for 24 h, with or without a PKCδ inhibitor. For all experiments, cells were made quiescent in serum-free media for 24 h before exposure to CSE. Control untreated cells were maintained in serum-free media.
2.3. Preparation of CSE Solution
CSE was prepared as previously described by Oltmanns et al. with a few modifications [19]. Commercial filtered cigarettes (Daqianmen, Shanghai Cigarette Factory, China), which contain 13 mg of tar and 1.0 mg of nicotine/cigarette, were used. Briefly, cigarette smoke derived from one cigarette was drawn slowly into a 50 mL syringe and bubbled through 30 mL of DMEM at room temperature. A cigarette yielded 5 draws with a 50 mL syringe, with each individual draw taking approximately 10 seconds to complete. The resulting solution, which was considered 100% strength, was adjusted to pH 7.4 with concentrated NaOH and filtered through a 0.22 μm pore filter before diluting it into DMEM as a percentage of the total volume. Final concentrations of the solution are expressed as percent values ((CSE solution volume/total volume) × 100%). Solution concentrations ranging from 0 to 20% (0%, 2.5%, 5%, 10%, and 20%) were used to evaluate the effect of CSE on rPASMCs proliferation. The experiment was repeated at least twice for each biological sample.
2.4. Cell Proliferation Analysis (MTT Assay)
The effect of CSE treatment on cell viability and proliferation was examined by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) assay. Cells (1 × 104 per well) were seeded in 96-well plates in replicates of five. After 24 h, the absorbance at 490 nm was recorded.
2.5. Cell Cycle Analysis
Flow cytometric analyses of cells were conducted according to the manual provided with the propidium iodide (PI) flow kit (KeyGEN, Nanjing, China). Briefly, the cells were harvested and stained with PI solution for 15 min. Fluorescence was measured using a FACScalibur apparatus (Becton Dickinson, Franklin Lakes, NJ, USA). Data collection and analysis were performed using CellQuest software. The percentages of cells in different phases of the cell cycle were determined using the software program ModFit.
2.6. RT-PCR Assay
A total of 4 × 106 cells were lysed to isolate total RNA using Trizol reagent (Invitrogen), according to the manufacturer's instructions. Reverse transcription (RT) was performed as described by the company. Total RNA (5 μg) was RT to synthesize cDNA in a volume of 20 μL (Reverse Transcriptase M-MLV, Takara). For every 25 μL of PCR reaction, 1 μL of cDNA was used for 20–25 cycles. PCR products (10 μL) were loaded onto a 2% agarose gel, stained with ethidium bromide and photographed. The primer sequences used for the RT-PCR are as follows: PDGFB forward 5′-AAGTGAGCAGTGCCTCGGCG-3′, reverse 5′-TGGCCTCGGGGACCGTCAAA-3′; PDGFRβ forward 5′-GCCGCCAGCGCCCATTTTTC-3′, reverse 5′-AGGCGTTTTCCTCTGCCCGC-3′, and PKCδ forward 5′-AGCAGCGGGAGCCAGGACTA-3′; reverse 5′-GCGGTGCGGCATGTCGATGT-3′.
2.7. Immunoblot Analysis
Cells were lysed in ATM lysis buffer (containing 100 mM Tris-Cl, pH 7.5, 150 mM NaCl, 0.2 mM EDTA, 20% glycerol, 0.4% NP-40, 2% Tween-20, and 0.2 mM PMSF). The protein concentration of the supernatant was measured with a BCA Assay Kit (Calbiochem), and total protein (50 μg) was loaded onto 15% SDS-PAGE gels. After electrophoresis, proteins were transferred onto PVDF membranes (Amersham) and hybridized with primary antibodies at the following dilutions: PDGFRβ (Bio world; Cat No: BS1764; 1 : 2,000), PDGFB (Bio world; Cat No: BS1290 1 : 2,000), PKCδ (Sigma; 1 : 2,000), and phosphorylated PKCδ (Sigma; 1 : 2,000). HRP-labeled secondary antibodies (Zhongshan Golden Bridge) were all used at a dilution of 1 : 2,000. The ECL detection system (Amersham) was used to detect the signals on the membranes.
2.8. Statistical Analysis
All data were presented as mean ± standard deviation. Statistical analyses were carried out using one-way ANOVA (for multiple-group comparison) followed by the least significant difference (LSD) test with the computer software SPSS 12.0 (Chicago, USA). For all tests, groups were considered statistically significant when P < 0.05.
3. Results
3.1. Effect of CSE Treatment on rPASMCs Proliferation
To investigate the effect of CSE treatment on cell proliferation, rPASMCs were stimulated with 0%, 2.5%, 5%, 10%, and 20% CSE for 24 h. Cell proliferation was evaluated by MTT colorimetric assay. As shown in Figure 1(a), CSE treatment caused a significant increase in cell growth in a dose-dependent manner compared to the control group. The cell response reached its peak at the concentration of 20% CSE.
Figure 1.
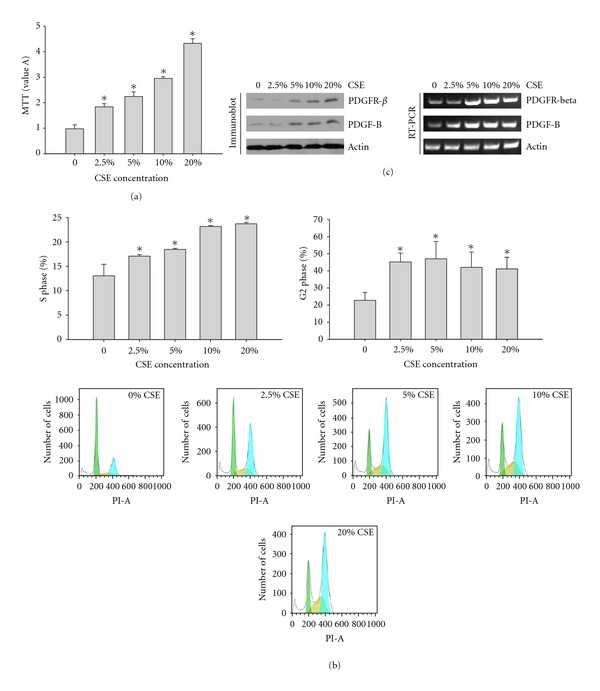
(a) CSE promoted cell proliferation of rPASMCs. The rPASMCs were seeded in a 96-well plate and treated with increasing concentrations of CSE for 24 h. Cell proliferation was analyzed by MTT assay. The data are shown as mean ± standard deviation from 6 experiments and were analyzed by ANOVA. *P < 0.05 compared with control. (b) CSE promoted cell cycle progression of rPASMCs. The rPASMCs were treated with increasing concentrations of CSE for 24 h. Cells were harvested and stained with PI solution for cell cycle assay. S phase and G2 phase cells were averaged. The data are shown as mean ± standard deviation from 6 experiments and were analyzed by ANOVA. *P < 0.05 compared with control. (c) The PDGF signaling pathway was activated after CSE treatment. The rPASMCs were treated with increasing concentrations of CSE for 24 h. Cells were lysed and submitted to RT-PCR or immunoblot assays, with the indicated primers or antibodies.
3.2. Effect of CSE Treatment on rPASMCs Cell Cycle Progression
Because cell proliferation is critically related to cell cycle changes, we further investigated the effect of CSE treatment on cell cycle progression. The rPASMCs were treated with 0%, 2.5%, 5%, 10%, and 20% CSE for 24 h, cell cycle was examined using the FACS assay with PI staining. According to the flow cytometry analysis, we found that there was an obvious increase in the S and G2 phases in CSE-treated cells compared to the nontreated control group. The data also indicated that there was a slight reduction in G2 phase cells when cells were treated with CSE at concentration of 10% (Figure 1(b)).
3.3. Effect of CSE on rPASMCs PDGFB and PDGFRβ Expression
We first investigated the effect of CSE treatment on the expression of PDGFB and PDGFRβ mRNA and protein levels. As shown in Figure 1(c), CSE significantly increased the mRNA and protein levels of PDGFB and PDGFRβ compared to the nontreated control rPASMCs.
According to the cell proliferation and cell cycle results, we selected the 10% CSE treatment to analyze the molecular mechanisms of CSE-induced cell proliferation. We further investigated the effect of PKCδ inhibition with Rottlerin (a PKCδ-specific inhibitor) on upregulated cell proliferation induced by CSE treatment. As shown in Figure 2(a), pre-incubation of rPASMCs with Rottlerin before CSE exposure significantly decreased the CSE-induced increased cell proliferation.
Figure 2.
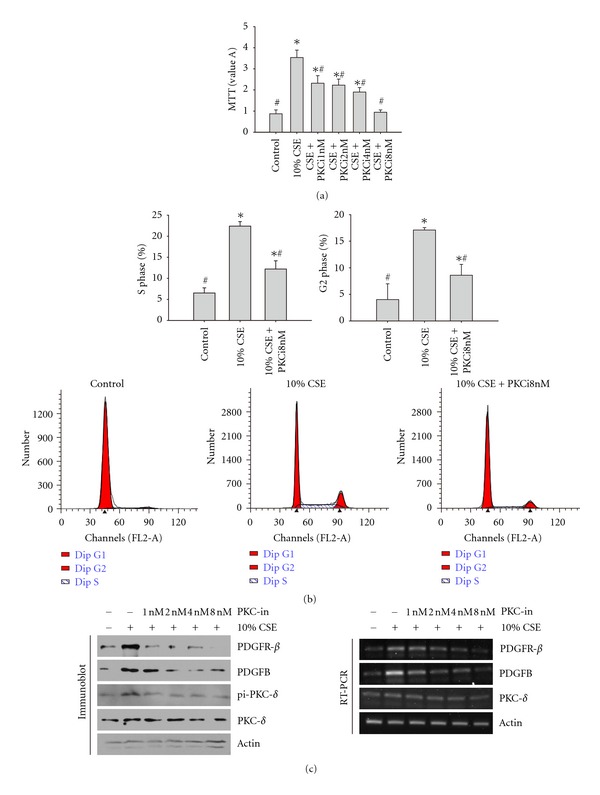
(a) The PKCδ-specific inhibitor Rottlerin inhibited 10% CSE-induced cell proliferation of rPASMCs. The rPASMCs were treated with different concentrations of Rottlerin for 1 h before exposure to 10% CSE. Cell proliferation was analyzed by MTT assay. The data are shown as mean ± standard deviation from 6 experiments and were analyzed by ANOVA. *P < 0.05 compared with control and #P < 0.05 compared with 10% CSE. (b) The PKCδ-specific inhibitor Rottlerin inhibited the CSE-induced cell cycle progression of rPASMCs. The rPASMCs were treated with different concentrations of Rottlerin for 1 h before exposure to 10% CSE. Cells were prepared and stained with propidium iodide solution for cell cycle assay. S phase and G2 phase cells were averaged. The data are shown as mean ± standard deviation from 6 experiments and were analyzed by ANOVA. *P < 0.05 compared with control and #P < 0.05 as compared with 10% CSE. (c) The PKCδ-specific inhibitor Rottlerin inhibited the CSE-induced upregulation of the mRNA and protein levels of PDGF signaling pathway. The rPASMCs were treated with different concentrations of Rottlerin for 1 h before exposure to 10% CSE. Cell extracts were prepared and submitted to RT-PCR or immunoblot assay with primers or antibodies as indicated.
Since cell proliferation was reduced with Rottlerin administration, we determined whether Rottlerin affected the cell cycle alterations with CSE treatment. As illustrated in Figure 2(b), preincubation of rPASMCs with Rottlerin before CSE exposure attenuated the increase of S and G2 phase populations induced by CSE treatment.
We also investigated the changes of mRNA and protein levels of PKCδ after CSE treatment. We found that 10% CSE treatment had no obvious effect on total PKCδ mRNA and protein levels. PKCδ has many different modifications that affect its activity, including phosphorylation to generate the active form. Therefore, we explored whether CSE treatment affected the phosphorylation of PKCδ. We measured the phosphorylation level of PKCδ and found that CSE treatment indeed promoted the phosphorylation of PKCδ (Figure 2(c)).
We further investigated the effect of Rottlerin on the level of PDGFB, PDGFRβ, and phosphorylated PKCδ. Cells were incubated with different concentrations of Rottlerin prior to 10% CSE treatment, harvested, lysed, and subjected to RT-PCR or immunoblot assay. As shown in Figure 2(c), Rottlerin treatment significantly reduced the increase in PDGFB, PDGFRβ, and phosphorylated PKCδ levels induced by CSE treatment. These changes occurred at both the mRNA and protein level.
4. Discussion
Cigarette smoking is a major public health problem associated with a multitude of diseases, including vascular diseases such as atherosclerosis and pulmonary hypertension. Proliferation of vascular smooth muscle cells plays an important role in the development of these vascular diseases. The relationship between cigarette smoking and cell function has been explored by stimulating isolated cells with CSE to determine direct causes [20]. In this study, we demonstrated that CSE induced rPASMCs proliferation by upregulating phosphorylating PKCδ and PDGFB. This stimulation was suppressed with a PKCδ inhibitor Rottlerin, which implicate PKCδ and PDGFB as therapeutic targets to block abnormal smooth muscle cell proliferation induced by CSE.
Abnormal vascular smooth muscle cell proliferation leads to medial hypertrophy of the vessel wall, vascular remodeling, and vascular lumen narrowing, all of which contribute to the development of pulmonary hypertension [21, 22]. In our in vitro study, CSE significantly promoted the proliferation of rPASMCs. Because cell proliferation is critically related to cell cycle changes, we further analyzed the effect of CSE on cell cycle distribution. Our study showed that CSE promoted cell cycle progression from the G1 phase to the S phase, indicating that CSE promoted cell proliferation. It is important to note that the results of the current study are consistent with those obtained by Luppi et al. [23], but contradictory to other investigations. In particular, a report by Ambalavanan et al. found that 20% CSE did not stimulate the proliferation of neonatal porcine vascular smooth muscle cells. Instead, CSE induced cell necrosis in the study [24]. A major difference between that study and ours is that neonatal porcine cells were used in the former while our study used adult rat cells. The different CSE preparation methods between the two studies could be another possible explanation for these discrepancies.
Many different cell types synthesize PDGF, including smooth muscle cells, endothelial cells, and macrophages [25]. PDGF has the ability to induce the proliferation and migration of smooth muscle cells and fibroblasts and has been proposed as a key mediator in the progression of several fibroproliferative disorders such as atherosclerosis, lung fibrosis, and PAH [11, 25, 26]. There is much evidence to support the hypothesis that PDGF pathways play an important role in the pulmonary vascular remodeling process responsible in the progression of PAH [10]. Indeed, PDGF is known to induce proliferation of smooth muscle cells of different origins such as the aorta and carotid [27, 28]. Our results showed that after rPASMCs were exposed to CSE, the expression of PDGFB and PDGFRβ mRNA and protein levels were markedly increased. In the current study, we extended the previous findings and demonstrated that PDGF was involved in the proliferation of rPASMCs induced by CSE.
Moreover, we investigated the molecular mechanisms by which PDGF stimulated proliferation of rPASMCs. PKC is a critical regulator in the intracellular signal transduction pathway and plays an important role in PASMCs proliferation [29]. Currently, 10 members in the PKC family have been identified, and each isozyme has distinct cellular functions [30]. There are at least four isozymes in smooth muscle cells, including PKCδ, PKCα, PKCε, and PKCζ [31]. This study focused on the role of the PKCδ isoform in CSE-induced rPASMCs proliferation. Although it has been shown that PKCδ activation can induce smooth muscle cell proliferation [32], Sasaguri and colleagues found that activation of PKCα and PKCε blunted smooth muscle cell proliferation [33]. In our study, preincubation with Rottlerin, a specific PKCδ inhibitor, inhibited cell proliferation, caused cell cycle arrest at the G1 phase, and downregulated PDGFB and PDGFRβ mRNA and protein levels in rPASMCs exposed to CSE. These results suggest that CSE induces rPASMCs proliferation by upregulating PDGFB and PDGFRβ expression mediated by the PKCδ signaling pathway.
Although the specific PKCδ inhibitor Rottlerin significantly suppressed PDGFB and PDGFRβ expression and CSE-induced rPASMCs proliferation, the upregulation was not completely inhibited. This result indicates that other mediators might also be involved in the process, such as EAK signaling, which plays an important role in mediating growth factor functions. The EAK signaling pathway has been shown to mediate cell proliferation in other cell types [34]. Future studies evaluating the roles of additional signaling cascades on cell proliferation in rPASMCs are warranted.
In conclusion, we determined that the effect of CSE on rPASMCs proliferation occurs through PKCδ and PDGFB activation. This study provides a foundation on which to further examine the precise roles of PDGF in pulmonary vascular remodeling that leads to PAH. Understanding the intracellular mechanisms responsible for rPASMCs growth provides therapeutic targets to prevent or limit rPASMCs proliferation and pulmonary vascular remodeling.
5. Conflict of Interests
The authors affirm that they have no conflict of interests.
Acknowledgment
This work was supported by Shanxi Scholarship Council of China (no. 2011-094).
References
- 1.Mandegar M, Fung YCB, Huang W, Remillard CV, Rubin LJ, Yuan JXJ. Cellular and molecular mechanisms of pulmonary vascular remodeling: role in the development of pulmonary hypertension. Microvascular Research. 2004;68(2):75–103. doi: 10.1016/j.mvr.2004.06.001. [DOI] [PubMed] [Google Scholar]
- 2.Pidgeon GP, Tamosiuniene R, Chen G, et al. Intravascular thrombosis after hypoxia-induced pulmonary hypertension: regulation by cyclooxygenase-2. Circulation. 2004;110(17):2701–2707. doi: 10.1161/01.CIR.0000145613.01188.0B. [DOI] [PubMed] [Google Scholar]
- 3.Barberà JA, Peinado VI, Santos S. Pulmonary hypertension in chronic obstructive pulmonary disease. European Respiratory Journal. 2003;21(5):892–905. doi: 10.1183/09031936.03.00115402. [DOI] [PubMed] [Google Scholar]
- 4.Wright JL, Levy RD, Churg A. Pulmonary hypertension in chronic obstructive pulmonary disease: current theories of pathogenesis and their implications for treatment. Thorax. 2005;60(7):605–609. doi: 10.1136/thx.2005.042994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Peinado VI, Pizarro S, Barberà JA. Pulmonary vascular involvement in COPD. Chest. 2008;134(4):808–814. doi: 10.1378/chest.08-0820. [DOI] [PubMed] [Google Scholar]
- 6.Jawien A, Bowen-Pope DF, Lindner V, Schwartz SM, Clowes AW. Platelet-derived growth factor promotes smooth muscle migration and intimal thickening in a rat model of balloon angioplasty. Journal of Clinical Investigation. 1992;89(2):507–511. doi: 10.1172/JCI115613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sachinidis A, Locher R, Hoppe J, Vetter W. The platelet-derived growth factor isomers, PDGF-AA, PDGF-AB and PDGF-BB, induce contraction of vascular smooth muscle cells by different intracellular mechanisms. FEBS Letters. 1990;275(1-2):95–98. doi: 10.1016/0014-5793(90)81447-v. [DOI] [PubMed] [Google Scholar]
- 8.Sakao S, Tatsumi K, Voelkel NF. Reversible or irreversible remodeling in pulmonary arterial hypertension. American Journal of Respiratory Cell and Molecular Biology. 2010;43(6):629–634. doi: 10.1165/rcmb.2009-0389TR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Humbert M, Monti G, Fartoukh M, et al. Platelet-derived growth factor expression in primary pulmonary hypertension: comparison of HIV seropositive and HIV seronegative patients. European Respiratory Journal. 1998;11(3):554–559. [PubMed] [Google Scholar]
- 10.Balasubramaniam V, Le Cras TD, Ivy DD, Grover TR, Kinsella JP, Abman SH. Role of platelet-derived growth factor in vascular remodeling during pulmonary hypertension in the ovine fetus. American Journal of Physiology. 2003;284(5):L826–L833. doi: 10.1152/ajplung.00199.2002. [DOI] [PubMed] [Google Scholar]
- 11.Schermuly RT, Dony E, Ghofrani HA, et al. Reversal of experimental pulmonary hypertension by PDGF inhibition. Journal of Clinical Investigation. 2005;115(10):2811–2821. doi: 10.1172/JCI24838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yamamoto M, Acevedo-Duncan M, Chalfant CE, Patel NA, Watson JE, Cooper DR. The roles of protein kinase C βI and βII in vascular smooth muscle cell proliferation. Experimental Cell Research. 1998;240(2):349–358. doi: 10.1006/excr.1998.3999. [DOI] [PubMed] [Google Scholar]
- 13.Li PF, Maasch C, Haller H, Dietz R, von Harsdorf R. Requirement for protein kinase C in reactive oxygen species-induced apoptosis of vascular smooth muscle cells. Circulation. 1999;100(9):967–973. doi: 10.1161/01.cir.100.9.967. [DOI] [PubMed] [Google Scholar]
- 14.Haller H, Lindschau C, Quass P, Distler A, Luft FC. Differentiation of vascular smooth muscle cells and the regulation of protein kinase C-α. Circulation Research. 1995;76(1):21–29. doi: 10.1161/01.res.76.1.21. [DOI] [PubMed] [Google Scholar]
- 15.Kalra VK, Ying Y, Deemer K, Natarajan R, Nadler JL, Coates TD. Mechanism of cigarette smoke condensate induced adhesion of human monocytes to cultured endothelial cells. Journal of Cellular Physiology. 1994;160(1):154–162. doi: 10.1002/jcp.1041600118. [DOI] [PubMed] [Google Scholar]
- 16.Kashyap R, Floreani AA, Heires AJ, Sanderson SD, Wyatt TA. Protein kinase c-α mediates cigarette smoke extract- and complement factor 5a-stimulated interleukin-8 release in human bronchial epithelial cells. Journal of Investigative Medicine. 2002;50(1):46–53. doi: 10.2310/6650.2002.33517. [DOI] [PubMed] [Google Scholar]
- 17.Lee SD, Lee DS, Chun YG, et al. Cigarette smoke extract induces endothelin-1 via protein kinase C in pulmonary artery endothelial cells. American Journal of Physiology. 2001;281(2):L403–L411. doi: 10.1152/ajplung.2001.281.2.L403. [DOI] [PubMed] [Google Scholar]
- 18.Leszczynski D, Joenväärä S, Foegh ML. Protein kinase C-α regulates proliferation but not apoptosis in rat coronary vascular smooth muscle cells. Life Sciences. 1996;58(7):599–606. doi: 10.1016/0024-3205(95)02329-1. [DOI] [PubMed] [Google Scholar]
- 19.Oltmanns U, Chung KF, Walters M, John M, Mitchell JA. Cigarette smoke induces IL-8, but inhibits eotaxin and RANTES release from airway smooth muscle. Respiratory Research. 2005;6:10–19. doi: 10.1186/1465-9921-6-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shapira SD. Smoke gets in your cells. American Journal of Respiratory Cell and Molecular Biology. 2004;31(5):481–484. doi: 10.1165/rcmb.F285. [DOI] [PubMed] [Google Scholar]
- 21.Pietra GG, Capron F, Stewart S, et al. Pathologic assessment of vasculopathies in pulmonary hypertension. Journal of the American College of Cardiology. 2004;43(12):25S–32S. doi: 10.1016/j.jacc.2004.02.033. [DOI] [PubMed] [Google Scholar]
- 22.Rabinovitch M. The mouse through the looking glass: a new door into the pathophysiology of pulmonary hypertension. Circulation Research. 2004;94(8):1001–1004. doi: 10.1161/01.RES.0000128079.89263.68. [DOI] [PubMed] [Google Scholar]
- 23.Luppi F, Aarbiou J, van Wetering S, et al. Effects of cigarette smoke condensate on proliferation and wound closure of bronchial epithelial cells in vitro: role of glutathione. Respiratory Research. 2005;6:140–151. doi: 10.1186/1465-9921-6-140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ambalavanan N, Carlo WF, Bulger A, Shi J, Philips JB. Effect of cigarette smoke extract on neonatal porcine vascular smooth muscle cells. Toxicology and Applied Pharmacology. 2001;170(2):130–136. doi: 10.1006/taap.2000.9094. [DOI] [PubMed] [Google Scholar]
- 25.Heldin CH, Westermark B. Mechanism of action and in vivo role of platelet-derived growth factor. Physiological Reviews. 1999;79(4):1283–1316. doi: 10.1152/physrev.1999.79.4.1283. [DOI] [PubMed] [Google Scholar]
- 26.Reigstad LJ, Varhaug JE, Lillehaug JR. Structural and functional specificities of PDGF-C and PDGF-D, the novel members of the platelet-derived growth factors family. The FEBS Journal. 2005;272(22):5723–5741. doi: 10.1111/j.1742-4658.2005.04989.x. [DOI] [PubMed] [Google Scholar]
- 27.Yahiaoui L, Villeneuve A, Valderrama-Carvajal H, Burke F, Fixman ED. Endothelin-1 regulates proliferative responses, both alone and synergistically with PDGF, in rat tracheal smooth muscle cells. Cellular Physiology and Biochemistry. 2006;17(1-2):37–46. doi: 10.1159/000091462. [DOI] [PubMed] [Google Scholar]
- 28.Deng DXF, Spin JM, Tsalenko A, et al. Molecular signatures determining coronary artery and saphenous vein smooth muscle cell phenotypes: distinct responses to stimuli. Arteriosclerosis, Thrombosis, and Vascular Biology. 2006;26(5):1058–1065. doi: 10.1161/01.ATV.0000208185.16371.97. [DOI] [PubMed] [Google Scholar]
- 29.Barnett ME, Madgwick DK, Takemoto DJ. Protein kinase C as a stress sensor. Cellular Signalling. 2007;19(9):1820–1829. doi: 10.1016/j.cellsig.2007.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dempsey EC, Cool CD, Littler CM. Lung disease and PKCs. Pharmacological Research. 2007;55(6):545–559. doi: 10.1016/j.phrs.2007.04.010. [DOI] [PubMed] [Google Scholar]
- 31.Assenderab JW, Kontny E, Bertil B. Expression of protein kinase C isoforms in smooth muscle cells in various states of differentiation. FEBS Letters. 1994;55(1):76–80. doi: 10.1016/0014-5793(94)80588-1. [DOI] [PubMed] [Google Scholar]
- 32.Leszczynski D, Joenväärä S, Foegh ML. Protein kinase C-α regulates proliferation but not apoptosis in rat coronary vascular smooth muscle cells. Life Sciences. 1996;58(7):599–606. doi: 10.1016/0024-3205(95)02329-1. [DOI] [PubMed] [Google Scholar]
- 33.Sasaguri T, Kosaka C, Zen K, Masuda J, Shimokado K, Ogata J. Protein kinase C isoforms that may mediate G1/S inhibition in cultured vascular smooth muscle cells. Annals of the New York Academy of Sciences. 1995;748:590–591. doi: 10.1111/j.1749-6632.1994.tb17367.x. [DOI] [PubMed] [Google Scholar]
- 34.Pukac L, Huangpu J, Karnovsky MJ. Platelet-derived growth factor-BB, insulin-like growth factor-I, and phorbol ester activate different signaling pathways for stimulation of vascular smooth muscle cell migration. Experimental Cell Research. 1998;242(2):548–560. doi: 10.1006/excr.1998.4138. [DOI] [PubMed] [Google Scholar]