

Abstract
Compared with HLA-DR molecules, the specificities of HLA-DP and HLA-DQ molecules have only been studied to a limited extent. The description of the binding motifs has been mostly anecdotal and does not provide a quantitative measure of the importance of each position in the binding core and the relative weight of different amino acids at a given position. The recent publication of larger data sets of peptide-binding to DP and DQ molecules opens the possibility of using data-driven bioinformatics methods to accurately define the binding motifs of these molecules. Using the neural network-based method NNAlign, we characterized the binding specificities of five HLA-DP and six HLA-DQ among the most frequent in the human population. The identified binding motifs showed an overall concurrence with earlier studies but revealed subtle differences. The DP molecules revealed a large overlap in the pattern of amino acid preferences at core positions, with conserved hydrophobic/aromatic anchors at P1 and P6, and an additional hydrophobic anchor at P9 in some variants. These results confirm the existence of a previously hypothesized supertype encompassing the most common DP alleles. Conversely, the binding motifs for DQ molecules appear more divergent, displaying unconventional anchor positions and in some cases rather unspecific amino acid preferences.
Keywords: binding motif, HLA-DP, HLA-DQ, immunoinformatics, MHC class II
Introduction
The MHC performs an essential role in the cellular immune system, and regulates immune responses through presentation of processed antigens to T lymphocytes. The MHC is also widely studied because of its association with many autoimmune and inflammatory diseases, including type I diabetes, rheumatoid arthritis, multiple sclerosis and Crohn’s disease, and certain MHC alleles have been linked to susceptibility to infectious diseases such as malaria and HIV (reviewed in ref. 1).
Unlike MHC class I, which samples peptides from cytosolic proteins, MHC class II molecules present short peptide sequences derived from extracellular proteins. Human MHC class II molecules are heterodimers consisting of an α-chain and a β-chain encoded on chromosome 6 in one of three HLA loci: DR, DP and DQ. Compared with DR molecules, the specificities of DP and DQ molecules have only been studied to a limited extent, and their binding motifs are poorly characterized and understood. The scarcity of binding data for DP and DQ molecules is mainly the result of the relative difficulty, compared with HLA-DR, of obtaining experimental binding data for these molecules, but the common assumption that DR molecules are more important in mediating immune responses has exacerbated the lack of information on DP and DQ. However, a growing number of reports associate certain DP and DQ alleles with several diseases, such as type I diabetes and coeliac disease,1–3 as well as in cancer.4–6
This gap in knowledge between DR and the other class II molecules has only recently begun to be filled, with the publication of larger sets of binding data for HLA DP and DQ molecules. In particular, a recent study by Wang et al.7 describes the release of an unprecedentedly large set of measured MHC class II binding affinities covering 26 allelic variants, including a total of about 17 000 affinity measurements for five DP and six DQ molecules. The same study also compared the predictive performance of some of the best available bioinformatics methods on these data, and found that it was possible to obtain reliable binding predictions for DP and DQ at levels comparable to those for DR molecules. The same group, in two additional publications8,9 attempted to characterize the binding specificities of a number of DP and DQ molecules using a matrix method called ARB (average relative binding).10 However, this method has been shown to perform significantly worse than other comparable approaches for MHC class II binding prediction, such as the NN-align method.11 In this report, we applied the latest version of the NN-align algorithm, implemented as the NNAlign web-server,12 to exploit the newly available large data sets of peptide binding affinity to DP and DQ molecules and finely characterize the binding specificities of 11 DP and DQ molecules.
Materials and methods
NNAlign is a neural network-based method specifically designed to identify short linear motifs contained in large peptide data sets. As a direct result of the method, it identifies a core of consecutive amino acids within the peptide sequences that constitutes an informative motif. The method has been shown to perform significantly better than any other publicly available method for MHC class II binding prediction, including HLA-DP and HLA-DQ molecules.7 One of the strengths of this approach is the use of multiple neural networks, trained with different architectures and initial conditions, to reduce stochastic factors and at the same time combine information from the different networks in the ensemble to obtain a prediction that is better than what can be obtained from the individual networks. Although this ensemble approach has earlier proved to be highly effective in terms of improving the accuracy for binding affinity predictions,11 it has been demonstrated that the use of network ensembles could lead to a loss in accuracy when it comes to identification of the motif binding core.12 However, using an offset correction algorithm implemented in NNAlign, this problem is resolved allowing not only improved predictive performance for network ensembles but also a more accurate representation of the identified sequence motif.
In this report, we applied NNAlign to peptide–MHC class II binding data for five HLA-DP and six HLA-DQ molecules to characterize their specificities and binding motifs. The binding data were obtained from the publication by Wang et al.7 They comprise a total of 17 092 measured peptide–MHC affinities, with an average of over 1500 measurements per allelic variant. Each data set was split in five random subsets and, each time excluding one subset, a network was trained on the remaining four subsets. We set the motif length to nine amino acids, and for all the remaining parameters we used the default values of the NNAlign web server: sequences were presented to the networks using Blosum encoding,13 hidden layers were composed of three neurons, training lasted 500 iterations per training example, starting from five different initial configurations for each cross-validation fold, subsets for cross-validation were created using a homology clustering at 80% to reduce similarity between subsets, using the best four networks for each cross-validation step.
The resulting 20 networks in each ensemble, trained on different subsets of the data and from alternative initial conditions, capture motifs that can be different from each other to some extent. They often place the alignment core in a different register, and might disagree on the exact boundaries of the motif. The offset correction algorithm described by Andreatta et al.12 proved extremely efficient in correcting for this disagreement, allowing re-alignment of different networks to a common core. This alignment procedure creates a position-specific scoring matrix (PSSM) representation of the motif of each network, and then aligns the matrices to maximize the information content of the combined core. We used a slightly modified version of the algorithm described in detail in a previous publication,12 where PSSMs are extended at both ends with background frequencies before alignment, so allowing the PSSMs to be aligned on a window of the same length as the matrices. This process assigns to each PSSM, and its relative network, an offset value that quantifies the shift distance from other networks. Note that the alignment procedure does not guarantee that the final combined register corresponds to the biologically correct register (in the case of peptide–MHC binding, the nine-amino-acid stretch bound in the MHC binding cleft), but rather to the window with the maximum information content. In most of the cases informative positions are also biologically important positions, so the core register would be in the correct place. However, if either terminal of the core has very weak information content (i.e. no particular amino acid preference at terminal positions), the sequences might possibly, although aligned correctly, all be shifted by one or more positions with respect to the biologically correct core register. This is an aspect to keep in mind when interpreting the results, and possibly adjust the register based on previous knowledge about the location of the motif anchors.
An effective way of visualizing the receptor-binding motif is by using sequence logos. Sequence logos were introduced by Schneider and Stephens14 to graphically represent the sequence motif contained in a set of aligned sequences, where at each position, the frequency of all amino acids is displayed as a stack of letters. The height of a column in the logo is given as the information content in bits of the alignment at that particular position, and the relative height of individual letters is proportional to the frequency of the corresponding amino acid at that position. In this paper, we use such sequence logos to display the HLA-DP and HLA-DQ binding specificities identified by NNAlign.
Results and discussion
HLA-DP
The five HLA-DP allelic variants were chosen by Wang et al.7 to cover a high percentage of the human population. Only considering the β-chain, more polymorphic than the α-chain and the main determinant for HLA-DP binding,15,16 the allele choice provides coverage of about 92% of the average population at the DPB1 locus.9
The sequence motifs identified by NNAlign for the five HLA-DP molecules are shown in Fig. 1. In general, all variants share a common pattern characterized by anchors at positions P1 and P6, with strong preferences for phenylalanine (F) and other aromatic or hydrophobic amino acids. Additionally, some molecules appear to have a hydrophobic preference at P9 especially for leucine (L). This P9 anchor was previously described for DPB1*04:02,17 but here we observe it also for other variants such as DPA1*02:01-DPB1*01:01 and DPA1*02:01-DPB1*05:01. In some instances, and notably for DPA1*03:01-DPB1*04:02, the residues at position P7 appear to have influence on the binding specificity of the molecule. This has not been described in previous reports. Another small exception to the P1–P6 hydrophobic/aromatic pattern is observed in the allelic variant DPA1*02:01-DPB1*05:01, where the positively charged amino acids R and K are moderately preferred at P1 together with hydrophobic ones, as was also previously noted.9
Figure 1.
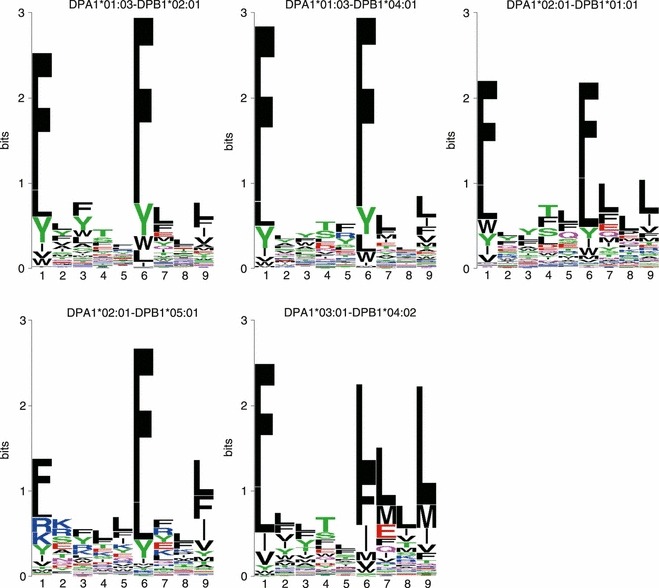
Sequence logos for five HLA-DP molecules. Hydrophobic amino acids are shown as black, acidic amino acids as red, basic amino acids as blue, neutral and polar amino acids as green and pink. All variants appear to share two main hydrophobic/aromatic anchors at P1 and P6, with an additional P9 anchor for some variants.
Taken as a whole, there appears to be a large overlap in the peptide-binding specificities of the five DP molecules, characterized by strong hydrophobic/aromatic anchors at P1 and P6, with the few exceptions noted above. Consistent with these observations, previous studies have found considerable overlaps in the peptide repertoires that can bind different DP alleles, and suggested the existence of a DP supertype encompassing the most common variants.9,17 Greenbaum et al.,18 on the basis of shared binding repertoires, suggested the presence of two DP supertypes: a ‘main DP’ supertype (composed of DPB1*01:01, 05:01 and 04:02) and a DP2 supertype (DPB1*02:01 and 04:01). These two subgroups correspond, in our analysis, to molecules with a strong P9 anchor (main DP) as opposed to molecules with weak or no P9 hydrophobic preference (DP2).
HLA-DQ
Most efforts in characterizing HLA-DQ binding specificities have been directed towards a few selected molecules, such as DQA1*05:01-DQB1*02:01 (also known as DQ2) or DQA1*03:01-DQB1*03:02 (DQ8) because of their association with disease.19–21 The data published by Wang et al.7 aim to be more comprehensive in terms of human population coverage, and they include binding data for the six most common allelic variants across different ethnicities.
The HLA-DQ sequence motifs identified by NNAlign are shown in Fig. 2. In contrast to the DP variants, which appear to share a common supertypical pattern, the DQ molecules show very little overlap in specificity. There do not appear to be common amino acid preferences, and the anchors are found at different positions within the 9-mer core. In particular, DQA1*01:01-DQB1*05:01 shows a strong preference for aromatic residues (F, W, Y) at P5, and secondary anchors at P6 and P7. The only previous report addressing the binding motif of this molecule8 also found a dominant anchor characterized by a preference for W and F, but placed this anchor at P4, and is generally in disagreement with our findings on other positions. The binding motif for DQA1*01:02-DQB1*06:02 appears loose, with several amino acids allowed at most positions. Previous reports22,23 identified mainly a P4–P6–P9 anchor spacing, with small and hydrophobic residues at P4, hydrophobic/aliphatic amino acids such as I, L, M, V at P6, and small residues like A and S at P9. Similar amino acid preferences are reflected in the binding motif detected by NNAlign, with additional anchors at P3 and P7. The only pair of molecules that appear to have a somewhat similar specificity is composed of DQA1*03:01-DQB1*03:02 and DQA1*04:01-DQB1*04:02. Both show a dominant anchor at P9, with preference for the acidic residues E and D. Additionally, they both show a preference for hydrophobic amino acids at P6, and mainly for A or E at P8. The strong acidic anchor at P9 was observed before.19,24 In the case of DQA1*05:01-DQB1*02:01, previous studies describe a motif with P1 and P9 binding pockets with hydrophobic/aromatic preferences, and acidic residues in the centre of the core, particularly at P4, P6 and P7.8,24–28 Besides the hydrophobic/aromatic P1–P9, NNAlign places the strongest anchor at P7, but with preferences for glutamic acid (E) also at P6 and P8. Finally, the somewhat peculiar sequence motif of DQA1*05:01-DQB1*03:01 seems to just prefer small amino acids such as A, G and S, especially on the central positions of the core, in agreement with the motif previously suggested for this molecule.8
Figure 2.
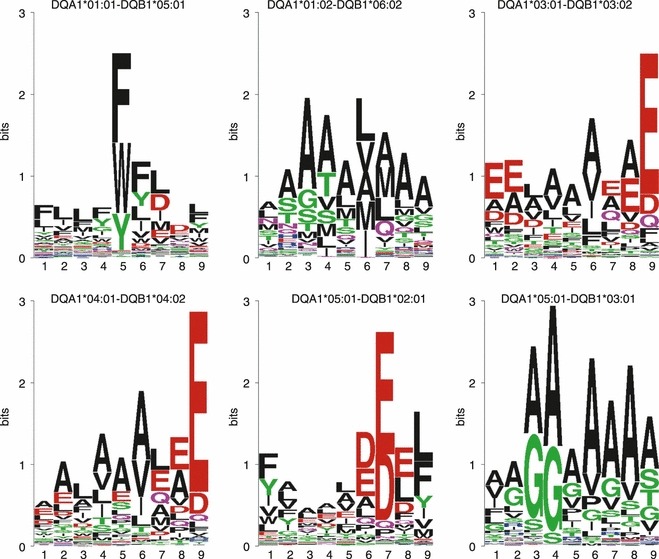
Sequence logos for six HLA-DQ molecules. Most of the variants display unique anchor positions and spacing, and very diverse amino acid preferences.
It is evident that the peptide-binding specificities for HLA-DQ variants are much more diverse than for HLA-DP variants. In particular, the strong hydrophobic/aromatic P1 anchor that generally characterizes all known HLA-DR and DP molecules is not observed here. There appears to be no general pattern in the spacing of the anchors, as well as in the kinds of permitted amino acids. In particular, we find a preference for acidic amino acids close to or at the C-terminal of the binding motif for three of the six molecules, and generally, the motifs seem rather promiscuous, with several residues allowed in the binding groove of the MHC.
Conclusions
In this report, we applied a state-of-the-art neural network-based method, NNAlign, to characterize the binding specificities of five HLA-DP and six HLA-DQ molecules. The allelic variants are among the most common human MHC class II molecules at the two HLA loci DP and DQ, covering a large percentage of the human population.8,9
For what concerns HLA-DP, there appears to be a common pattern in all the five variants under consideration, with primary anchor positions at P1 and P6 with preference for hydrophobic and aromatic residues. Some variants show an additional hydrophobic anchor at P9 and other minor differences, but in general there appears to be a consistent overlap in the binding specificities of all five molecules. The same cannot be said for HLA-DQ, where most of the molecules have very different anchor positions, anchor spacing and amino acid preferences. Hence, there does not seem to be a supertypical mode of binding for DQ, and each variant appears to be characterized by a distinct binding specificity. The most striking observation for the DQ loci binding motifs is the preference for acidic amino acids close to or at the C-terminal of the binding groove. Such an amino acid preference has not, to the best of our knowledge, previously been described for any HLA class I molecules, and has only sporadically been reported for HLA class II molecules. Binding predictions (including identification of the binding core) for any peptide sequence to all the alleles described in this report can be obtained at the NetMHCII server (http://www.cbs.dtu.dk/services/NetMHCII).
The binding motifs described in this work confirm most of the observations brought up by previous studies, but also highlight some interesting differences. Importantly, the sequence logo representation provides a quantitative measure of the relevance of each position in the binding core, and the relative importance of each amino acid, in determining the specificities of a given molecule, a differentiation that was not obtained in previous studies. The study first and foremost demonstrates the power of the NNalign method to, in a fully automated manner, identify and characterize the receptor-binding motif from a set of peptide-binding data. Second, it underlines the importance of generating such peptide data sets to carry out receptor-binding motif characterizations, gain insights into the peptide-binding repertoire of MHC molecules and reveal details about which amino acids and amino acid positions are critical for binding and, potentially, for peptide immunity.
Acknowledgments
The research leading to these results received funding from National Institute of Allergy and Infectious Diseases under contract number HHSN 272 2009 00045C.
Disclosures
The authors declare no conflict of interest.
References
- 1.Jones EY, Fugger L, Strominger JL, Siebold C. MHC class II proteins and disease: a structural perspective. Nat Rev Immunol. 2006;6:271–82. doi: 10.1038/nri1805. [DOI] [PubMed] [Google Scholar]
- 2.Fernando MMA, Stevens CR, Walsh EC, De Jager PL, Goyette P, Plenge RM, Vyse TJ, Rioux JD. Defining the role of the MHC in autoimmunity: a review and pooled analysis. PLoS Genet. 2008;4:e1000024. doi: 10.1371/journal.pgen.1000024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fallang LE, Bergseng E, Hotta K, Berg-Larsen A, Kim CY, Sollid LM. Differences in the risk of celiac disease associated with HLA-DQ2.5 or HLA-DQ2.2 are related to sustained gluten antigen presentation. Nat Immunol. 2009;10:1096–102. doi: 10.1038/ni.1780. [DOI] [PubMed] [Google Scholar]
- 4.Mandic M, Castelli F, Janjic B, et al. One NY-ESO-1-derived epitope that promiscuously binds to multiple HLA-DR and HLA-DP4 molecules and stimulates autologous CD4+ T cells from patients with NY-ESO-1-expressing melanoma. J Immunol. 2005;174:1751–9. doi: 10.4049/jimmunol.174.3.1751. [DOI] [PubMed] [Google Scholar]
- 5.Qian F, Gnjatic S, Jäger E, et al. Th1/Th2 CD4+ T cell responses against NY-ESO-1 in HLA-DPB1*0401/0402 patients with epithelial ovarian cancer. Cancer Immun. 2004;4:12. [PubMed] [Google Scholar]
- 6.Kamatani Y, Wattanapokayakit S, Ochi H, et al. A genome-wide association study identifies variants in the HLA-DP locus associated with chronic hepatitis B in Asians. Nat Genet. 2009;41:591–5. doi: 10.1038/ng.348. [DOI] [PubMed] [Google Scholar]
- 7.Wang P, Sidney J, Kim Y, Sette A, Lund O, Nielsen M, Peters B. Peptide binding predictions for HLA DR, DP and DQ molecules. BMC Bioinformatics. 2010;11:568. doi: 10.1186/1471-2105-11-568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sidney J, Steen A, Moore C, Ngo S, Chung J, Peters B, Sette A. Divergent motifs but overlapping binding repertoires of six HLA-DQ molecules frequently expressed in the worldwide human population. J Immunol. 2010;185:4189–98. doi: 10.4049/jimmunol.1001006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sidney J, Steen A, Moore C, Ngo S, Chung J, Peters B, Sette A. Five HLA-DP molecules frequently expressed in the worldwide human population share a common HLA supertypic binding specificity. J Immunol. 2010;184:2492–503. doi: 10.4049/jimmunol.0903655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bui HH, Sidney J, Peters B, et al. Automated generation and evaluation of specific MHC binding predictive tools: ARB matrix applications. Immunogenetics. 2005;57:304–14. doi: 10.1007/s00251-005-0798-y. [DOI] [PubMed] [Google Scholar]
- 11.Nielsen M, Lund O. NN-align. An artificial neural network-based alignment algorithm for MHC class II peptide binding prediction. BMC Bioinformatics. 2009;10:296. doi: 10.1186/1471-2105-10-296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Andreatta M, Schafer-Nielsen C, Lund O, Buus S, Nielsen M. NNAlign: a web-based prediction method allowing non-expert end-user discovery of sequence motifs in quantitative peptide data. PLoS ONE. 2011;6:e26781. doi: 10.1371/journal.pone.0026781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Henikoff S, Henikoff JG. Amino acid substitution matrices from protein blocks. Proc Natl Acad Sci U S A. 1992;89:10915–9. doi: 10.1073/pnas.89.22.10915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schneider TD, Stephens RM. Sequence logos: a new way to display consensus sequences. Nucleic Acids Res. 1990;18:6097–100. doi: 10.1093/nar/18.20.6097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bugawan TL, Horn GT, Long CM, Mickelson E, Hansen JA, Ferrara GB, Angelini G, Erlich HA. Analysis of HLA-DP allelic sequence polymorphism using the in vitro enzymatic DNA amplification of DP-α and DP-β loci. J Immunol. 1988;141:4024–30. [PubMed] [Google Scholar]
- 16.Diaz G, Amicosante M, Jaraquemada D, Butler RH, Guillen MV, Sanchez M, Nombela C, Arroyo J. Functional analysis of HLA-DP polymorphism: a crucial role for DPβ residues 9, 11, 35, 55, 56, 69 and 84–87 in T cell allorecognition and peptide binding. Int Immunol. 2003;15:565–76. doi: 10.1093/intimm/dxg057. [DOI] [PubMed] [Google Scholar]
- 17.Castelli FA, Buhot C, Sanson A, et al. HLA-DP4, the most frequent HLA II molecule, defines a new supertype of peptide-binding specificity. J Immunol. 2002;169:6928–34. doi: 10.4049/jimmunol.169.12.6928. [DOI] [PubMed] [Google Scholar]
- 18.Greenbaum J, Sidney J, Chung J, Brander C, Peters B, Sette A. Functional classification of class II human leukocyte antigen (HLA) molecules reveal seven different supertypes and a surprising degree of repertoire sharing across supertypes. Immunogenetics. 2010;63:325–35. doi: 10.1007/s00251-011-0513-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee KH, Wucherpfenning KW, Wiley DC. Structure of a human insulin peptide-HLA-DQ8 complex and susceptibility to type I diabetes. Nat Immunol. 2001;2:501–7. doi: 10.1038/88694. [DOI] [PubMed] [Google Scholar]
- 20.Megiorni F, Mora B, Bonamico M, Barbato M, Nenna R, Maiella G, Lulli P, Mazzilli MC. HLA-DQ and risk gradient for celiac disease. Hum Immunol. 2009;70:55–9. doi: 10.1016/j.humimm.2008.10.018. [DOI] [PubMed] [Google Scholar]
- 21.Hovhannisyan Z, Weiss A, Martin A, et al. The role of HLA-DQ8 β57 polymorphism in the anti-gluten T-cell response in coeliac disease. Nature. 2008;456:534–8. doi: 10.1038/nature07524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Siebold C, Hansen BE, Wyer JR, et al. Crystal structure of HLA-DQ0602 that protects against type I diabetes and confers strong susceptibility to narcolepsy. Proc Natl Acad Sci USA. 2004;101:1999–2004. doi: 10.1073/pnas.0308458100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ettinger RA, Kwok WW. A peptide binding motif for HLA-DQA1*0102/DQB1*0602, the class II MHC molecule associated with dominant protection in insulin-dependent diabetes mellitus. J Immunol. 1998;160:2365–73. [PubMed] [Google Scholar]
- 24.Sidney J, del Guercio MF, Southwood S, Sette A. The HLA molecules DQA1*0501/B1*0201 and DQA1*0301/B1*0302 share an extensive overlap in peptide binding specificity. J Immunol. 2002;169:5098–108. doi: 10.4049/jimmunol.169.9.5098. [DOI] [PubMed] [Google Scholar]
- 25.van de Wal Y, Kooy YMC, Drijfhout JW, Amons R, Papadopoulos GK, Koning F. Unique peptide binding characteristics of the disease-associated DQ(α1*0501, β1*0201) vs the non-disease-associated DQ(α1*0201, β1*0202) molecule. Immunogenetics. 1997;46:484–92. doi: 10.1007/s002510050309. [DOI] [PubMed] [Google Scholar]
- 26.Quarsten H, Paulsen G, Johansen BH, Thorpe CJ, Holm A, Buus S, Sollid LM. The P9 pocket of HLA-DQ2 (non-Aspβ57) has no particular preference for negatively charged anchor residues found in other type 1 diabetes-predisposing non-Aspβ57 MHC class II molecules. Int Immunol. 1998;10:1229–36. doi: 10.1093/intimm/10.8.1229. [DOI] [PubMed] [Google Scholar]
- 27.Stepniak D, Wiesner M, de Ru AH, Moustakas AK, Drijfhout JW, Papadopoulos GK, van Veelen PA, Koning F. Large-scale characterization of natural ligands explains the unique gluten-binding properties of HLA-DQ2. J Immunol. 2008;180:3268–78. doi: 10.4049/jimmunol.180.5.3268. [DOI] [PubMed] [Google Scholar]
- 28.Vartdal F, Johansen BH, Friede T, et al. The peptide binding motif of the disease associated HLA-DQ (α1*0501, β1*0201) molecule. Eur J Immunol. 1996;26:2764–72. doi: 10.1002/eji.1830261132. [DOI] [PubMed] [Google Scholar]