

Abstract
Primary amenorrhea refers to absence of spontaneous menarche even after the age of 16. Cytogenetic analysis in two cases with primary amenorrhea, short stature, poorly developed secondary sexual characteristics, and growth retardation were studied. Routine GTG-band analysis of metaphases from peripheral blood leucocytes revealed female karyotype with a 15(ps+) and an isochromosome of X, i(Xq), in one patient and 46,X, i(Xq), in another patient. Ascertainment of the karyotype aided in confirmation of the provisional diagnosis, a better phenotype–genotype correlation to understand clinical heterogeneity in genetic counseling.
Keywords: Amenorrhea, isochromosome X, short stature, turner syndrome
Introduction
Primary amenorrhea (PA) refers to absence of spontaneous menarche by age 14 years with the absence of growth or development of secondary sexual characteristics or as absence of menses by age 16 years with normal development of secondary sexual characteristics.[1] Genetic causes of amenorrhea account for about 45% of the cases in which ovarian failure due to chromosomal abnormalities include primarily Turner syndrome (TS) and its variants and gonadal dysgenesis. The importance of chromosomal analysis in the evaluation of women with absence of menstruation for better management and counseling has been emphasized by several investigators. The frequency of abnormal karyotypes has been reported to vary between 20% and 31% among women experiencing PA.[2,3] In a recent survey on a large sample size (n = 944), Jyothy et al.,[4] identified 21.5% of women with PA and 4.42% women presenting secondary amenorrhea (SA) to have an abnormal karyotype. In the present study, the cytogenetic profile of two patients referred with complaints of amenorrhea, short stature, poorly developed secondary sexual characters, and growth retardation to correlate the karyotype with the phenotypic features.
Case Report
Two patients (14 and 15 years old girls) presented to us with PA, short stature, poorly developed secondary sexual characteristics, and growth retardation. A detailed work up was planned for investigating the patient objectively for diagnosis. The routine diagnostic tests such as complete blood picture, biochemical parameters (T3, T4, and TSH), estimation of calcium levels in the serum, ultrasound whole abdomen, chromosomal analysis, and genetic counseling were done. The chromosomal analysis was performed in the Molecular Biology and Cytogenetics Lab, Apollo Hospitals, Hyderabad.
Blood samples from the two patients were drawn in a heparinized vacutainers for cytogenetic analysis and other appropriate vacutainers for other investigations. The lymphocyte cultures were set up in duplicates.[5] Two set of slides were prepared from each culture. On these slides, Giemsa banding (GTG banding) was performed.[6] At least 30 metaphases were scored for each patient. Three cells were karyotyped according to International System for Human Cytogenetic Nomenclature (ISCN) criteria.[7] Usually, the total chromosome count was determined in 30 cells, but if mosaicism was suspected then 50 or more cell counts were undertaken.[8]
Result
The following are the results of the investigations performed in the two patients
Patient A: 14-year-old patient
Complete blood picture showed normocytic and normochromic, with total and differential counts are within normal limits, platelets adequate on the smear.
The biochemical parameters such as TSH was elevated with the value being 5.25* (Girls - 14–15 years: 0.77–4.3) IU/ Ml and calcium serum was also abnormal with value being 9.1* (Girls - 13–15 years: 9.3-10.7) mg/dL. Growth hormone was decreased with the value being 0.10* (0.14–9.9) ng/Ml.
The whole abdomen scan showed infantile small uterus with nonvisualized ovaries and endometrium seen as a thin line. Menstrual cycle was not yet started.
The cytogenetic analysis with GTG banding showed female karyotype with increase in the length of the satellite on the short arm of chromosome 15 and isochromosome of one X chromosome detectable at the level of banding resolution (ISCN 400) (46, XX, 15ps+, i(Xq) (Figures 1a and 1b)].
Figure 1.
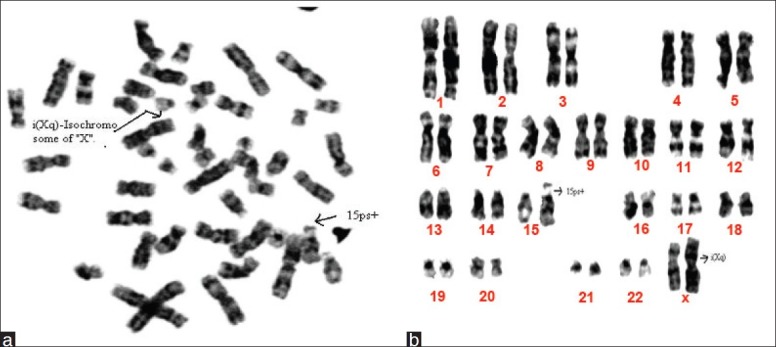
(a) Metaphase of a 14-year-old girl showing 46, XX, 15ps+, i(Xq). (b) Karyotype of a 14-year-old girl showing 46, XX, 15ps+,i(Xq)
Patient B: 15-year-old patient
Complete blood report showed normocytic and normochromic, leukopenia, with platelets adequate in smear with decrease in total leukocyte count TLC - 3.6* %. The whole abdomen scan showed hypoplastic uterus and not visualized, pelvic u/s ovaries 17Xcm and uterus 35 × 9 cm, Left breast is absent. The height of the patient is (Ht) – 126 cm, weight (Wt- 22) kg.
No evidence of any free fluid in POD, 2D ECHO normal. The biochemical parameters such as FSH is elevated with value being 144* (1.78–11.5) MIU/ml; TSH also elevated with value being 3.09* (0.52–2.9) IU/ml.
The cytogenetics analysis with GTG banding revealed a female karyotype with isochromosome of one X chromosome and another normal X chromosome detectable at the level of banding resolution (ISCN 400) (46, X, i(Xq) (Figures 2a and 2b)].
Figure 2.
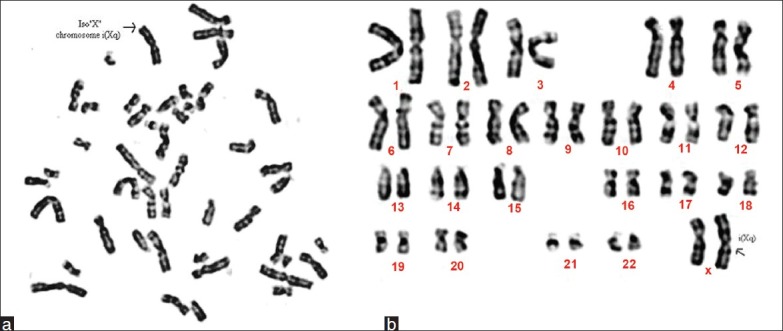
(a) Metaphase of a 15-year-old girl showing 46, X, i(Xq). (b) Karyotype of a 15-year-old girl showing 46, X, i(Xq)
Discussion
Amenorrhea is one of the major problems encountered by women. This problem may lead to anxiety, depression, and suicidal tendencies in the affected individuals. Since the psychological and social impact is high, patients seeking genetic evaluation are a small fraction of affected women, and hence. the exact incidence or prevalence of amenorrhea is not known. The World Health Organization has estimated 15% of the human population as being infertile and amenorrhea as the sixth largest major cause of female infertility. Among the general population, amenorrhea seemed to have affected 2%–5% of all women of childbearing age.[9] The etiology of amenorrhea has been compartmentalized as disorders of the outflow tract/ovary/anterior pituitary/CNS factors, and with genetic basis.[10] Genetic factors are considered to be “the causal factors” for most of the conditions in the field of Medicine. Likewise, whatever be the etiology of amenorrhea, the “genetic basis” needs to be emphasized. Genetic factors could be single gene disorders/chromosomal, or multifactorial. Among them, mostly it is chromosomes and their abnormalities, contributing to the constitutional etiology of amenorrhea. The incidence of the chromosomal abnormality (CA) in live births is around 90 per 10,000. Included in the incidence are the numerical (monosomy/trisomy/mosaicism) as well as the structural (translocation/ isochromosome/ deletion/duplication/ring) chromosomal anomalies.[11] The reported incidence of CA in PA is 20%–40%. Structural abnormalities leading to PA mostly are isochromosome of the long arm of X [i(Xq)].[12] The structurally abnormal X chromosome may be inactivated, thereby may minimize the disturbance of cellular function. The same is seen in our study where the PA reported in these two patients could be attributed to isochromosome of (Xq). Moreover, the phenotype may be indirectly influenced as per the size, and the loss/gain/altered genetic function, in the deleted or duplicated segments in X. Similar observation is seen in our study as well where both our patients had isochromosome of Xq.
A large number of surveys undertaken worldwide to ascertain the frequency of sex chromosomal anomalies in patients present with primary or SA have shown a wide variation in the incidence of chromosomal anomalies.[13,14] A retrospective survey of 620 women with PA and 245 with SA revealed chromosomal abnormalities in 26.13% and 16.33% of the patients, respectively.[15] Besides numerical abnormalities of X chromosome, the structural X chromosomal anomalies constituted isochromosome for the long arm, marker, ring, and X autosome translocations. All these aberrations were seen in both pure and mosaic forms. Robertsonian and reciprocal translocations were also documented. Suri et al.,[16] observed 45,X to be the most frequently occurring karyotype (44.4%) followed by 45,X/46,XX mosaicism (24.4%) in their analysis of 45 cases of Turner syndrome. Many of the chromosomal aberrations especially related to the heterochromatin region, namely, p+/q+ are considered as normal variants, which may or may not lead to mental retardation. In the present study also the patient whose chromosomal analysis showed 46, XX, 15ps+, i(Xq) did not show any symptoms of mental retardation other than PA, short stature and poorly developed secondary sexual characters which could have been contributed to i(Xq). The abnormal levels in the biochemical parameters can also be attributed to the isochromosome of X. Knowledge of the critical regions in chromosomes is very useful in correlating the genotype and the phenotype.
Karyotyping determines these critical regions and thus various congenital malformations can be observed for the chromosomal aberrations. The positioning of the genes responsible for human malformations could be gained from the studies of phenotypic effects of human chromosomal aberrations. Therefore, further molecular mutational studies are necessary to define the abnormality. Genetic diagnosis by cytogenetic screening thus proved to be crucial in counseling of parents, and special education and management of these children. In cases such as the present study, where a family has significant emotional and social issues to grapple with the conflict as to whether the child would ever get married and lead a normal life or not, the chromosomal analysis can answer if not all but a significant number of queries of the anxious parents. Given these concerns, a thorough explanation of the risk-benefit ratio should be considered whenever diagnostic tests are considered, in patients along with a thorough discussion with parents regarding the potential risks involved.
Conclusion
The karyotype of parents with chromosomally abnormal children could help to establish the inheritance or recurrence risk in the family, and proved significant in prevention and genetic counseling. At the time of counseling, it should be informed that in women with sex chromosome anomalies, especially having X mosaicism, pregnancy cannot be ruled out. Nowadays, even in 45, X condition with hormonally prepared milieu, assisted reproductive strategy has become successful. The educational/cultural/regional/religious and the psychological background of the women with amenorrhea and their family should be kept in mind. Also, during counseling the following information also thought to be provided: mechanism of origin of the chromosomal anomalies, the almost negligible recurrence risk in sex chromosomal anomalies, the hormonal therapy, education/hobby/career, and marriage/reproductive options. Hence, it is recommended that patients with any symptoms of mental retardation, PA, short stature and poorly developed secondary sexual characters which could have been contributed to should be subjected for karyotyping. Karyotyping determines these critical regions and thus various congenital malformations can be observed for the chromosomal aberrations. Knowledge of the critical regions in chromosomes are very useful in correlating the genotype and the phenotype.
Acknowledgments
The authors acknowledge the two patients and their families for accepting to give the samples and management of Apollo Hospitals for their support.
Footnotes
Source of Support: Nil
Conflict of Interest: None declared.
References
- 1.Doody KM, Carr BR. Amenorrhea. Obstet Gynecol Clin North Am. 1990;17:361–87. [PubMed] [Google Scholar]
- 2.Opitz O, Zoll B, Hansmann I, Hinney B. Cytogenetic investigation of 103 patients with primary or secondary amenorrhea. Hum Genet. 1982;65:46–7. doi: 10.1007/BF00285026. [DOI] [PubMed] [Google Scholar]
- 3.Anglani F, Baccichetti C, Artifoni L, Lenzini E, Tenconi R. Frequency of abnormal karyotypes in relation to the ascertained method in females referred for suspected sex chromosome abnormality. Clin Genet. 1984;25:242–7. doi: 10.1111/j.1399-0004.1984.tb01984.x. [DOI] [PubMed] [Google Scholar]
- 4.Jyothy A, Kumar KS, Swarna M, Sekhar M Raja, Devi B Uma, Reddy PP. Cytogenetic investigations in 1843 referral cases of disordered sexual development from Andhra Pradesh, India. Int J Hum Genet. 2002;2:55–9. [Google Scholar]
- 5.Moorhead PS, Nowell PC, Mellman WJ, Battips DM, Hungerford D. Chromosome preparations of leucocytes cultured from human peripheral blood. Exp Cell Res. 1960;20:613–6. doi: 10.1016/0014-4827(60)90138-5. [DOI] [PubMed] [Google Scholar]
- 6.Seabright M. A rapid banding technique for human chromosome. Lancet. 1971;2:971–2. doi: 10.1016/s0140-6736(71)90287-x. [DOI] [PubMed] [Google Scholar]
- 7.Kingston MH. Chromosomal analysis. In: Kingston MH, editor. ABC of Clinical Genetics. London: BMJ Publishing Group; 1994. pp. 21–5. [Google Scholar]
- 8.Mitelman F. ISCN. An International System for Human Cytogenetic Nomenclature. Basel: Karger; 2005. pp. 1–115. [Google Scholar]
- 9.14th Annual report. Geneva: 1985. World Health Organization, Special Programme of Research Development and Research training in Human Reproduction; pp. 114–9. [Google Scholar]
- 10.Speroff L, Glass RH, Kase NG, editors. In: Clinical Gynecologic Endocrinology and Infertility. 6th ed. USA: Lippincott Williams and Wilkins; 1999. Amenorrhea; pp. 421–76. [Google Scholar]
- 11.Mueller RF, Young ID, editors. Emery's elements of medical genetics. 11th ed. Edinburgh, UK: Churchill Livingstone; 2001. p. 249. [Google Scholar]
- 12.Opitz O, Zoll B, Hansmann I, Hinney B. Cytogenetic investigation of 103 patients with primary or secondary amenorrhea. Hum Genet. 1982;65:46–7. doi: 10.1007/BF00285026. [DOI] [PubMed] [Google Scholar]
- 13.Ten SK, Chin YM, Noor PJ, Hassan K. Cytogenetic studies in women with primary amenorrhea. Singapore Med J. 1990;31:355–9. [PubMed] [Google Scholar]
- 14.Park YS, Kang KC. Cytogenetic study of primary amenorrhea. Korean J Obstet Gynecol. 1999;42:814–20. [Google Scholar]
- 15.Sayee R, Leelavathy N. Cytogenetic studies in amenorrhea. Saudi Med J. 2007;28:187–92. [PubMed] [Google Scholar]
- 16.Suri M, Kabra M, Jain U, Sanders V, Saxena R, Shukla A, et al. A clinical and cytogenetic study of Turner syndrome. Indian Pediatr. 1995;32:433–42. [PubMed] [Google Scholar]