

Abstract
Periodontal diseases are inflammatory diseases of supporting structures of the tooth. It results in the destruction of the supporting structures and most of the destructive processes involved are host derived. The processes leading to destruction and regeneration of the destroyed tissues are of great interest to both researchers and clinicians. The selective susceptibility of subjects for periodontitis has remained an enigma and wide varieties of risk factors have been implicated for the manifestation and progression of periodontitis. Genetic factors have been a new addition to the list of risk factors for periodontal diseases. With the availability of human genome sequence and the knowledge of the complement of the genes, it should be possible to identify the metabolic pathways involved in periodontal destruction and regeneration. Most forms of periodontitis represent a life-long account of interactions between the genome, behaviour, and environment. The current practical utility of genetic knowledge in periodontitis is limited. The information contained within the human genome can potentially lead to a better understanding of the control mechanisms modulating the production of inflammatory mediators as well as provides potential therapeutic targets for periodontal disease. Allelic variants at multiple gene loci probably influence periodontitis susceptibility.
Keywords: Genes, periodontal disease, polymorphisms
Introduction
Periodontal disease is an infectious disease of the structures surrounding the tooth, which results from complex interactions between plaque microorganisms and host immune system. It is one of the most prevalent diseases affecting the mankind. Yet surprisingly a small percentage of people develop periodontitis, which is characterised by irreversible destruction of periodontal tissues. The selective susceptibility of subjects for periodontitis has remained an enigma and wide varieties of risk factors have been implicated for the manifestation and progression of periodontitis. These include microbial composition of plaque tooth-associated factors subject characteristics, social and behavioural factors, systemic and genetic factors.
In the recent years, the greatest of feats in biological sciences that is deciphering of human genome has given a new dimension to our understanding of various chronic immuno-inflammatory diseases, including periodontal disease. This has opened up new vistas of research in the field of medicine and dentistry. There is an exponential increase in the number of reports implicating an association between genes, genetic polymorphisms, and progression of periodontal disease.
Genetic factors are known to influence inflammatory and immune responses in periodontitis. As the immune system plays a crucial role in the pathogenesis of periodontitis, researchers have concentrated on the identification of genetic polymorphisms in several aspects of immunity. Allelic variants at multiple gene loci probably influence periodontitis susceptibility. The effects of some of these genetic variants may be large and of clinical significance, while the effects of others are probably minor and not clinically significant.
Genetic Disease Paradigms
Most human diseases have a genetic component to the etiology. The manner and extent to which genetic factors contribute to disease have important implications for identifying the genetic basis of aetiology and for utilizing this information for the diagnosis and treatment of the disease. Genetic diseases have been broadly classified into two groups: simple mendelian diseases and complex diseases.[1]
Simple mendelian diseases (monogenic disorders)
These are caused by a mutation in a single gene and are referred to as single gene (major gene effect) disorders. Inheritance patterns are autosomal dominant or of the autosomal recessive type. Severe periodontitis presents as part of the clinical manifestations of several monogenetic syndromes and the gene mutation and biochemical defect are known for many of these conditions. Various syndromes, which have periodontal disease manifestations as part of syndromic manifestations are Papillon Lefevre syndrome, Chediak-Higashi syndrome, Ehler-Danlos syndrome, cyclic neutropenia, and leukocyte adhesion deficiency.[1] Hereditary gingival fibromatosis is found to be associated with a mutation in the SOS (Son of Sevenless) gene.[2] The significance of these conditions is that they clearly demonstrate that a genetic mutation at a single locus can impart susceptibility to periodontitis.
Complex genetic diseases (polygenic disorders)
Genetically complex diseases are much more prevalent and usually occur with the frequency of greater than 1% of the population. Complex traits do not fit the typical inheritance patterns within families. They are a result of the interaction of multiple different gene loci, environmental, and behavioural factors. Many of the diagnostic features of these complex diseases also called quantitative trait disorders are regulated by several genes. Periodontitis, which is the most common of the periodontal diseases, is considered to be a complex disease. Common features of complex human diseases (e.g., Alzheimer's disease, Crohn's disease, and cardiovascular disease) are that these conditions present mostly with a relatively mild phenotype and are slowly progressive and chronic in nature.[3] Various biological pathways leading to similar clinical phenomena characterise the patho-physiology of complex diseases. Complex diseases are associated with variations in multiple genes, each having a small overall contribution and relative risk for the disease process. The clinical condition may not be evident unless two different genetic factors are present.
Inheritance of monogenic and complex (multifactorial) disorders
In monogenic diseases, mutations in a single gene are both necessary and sufficient to produce the clinical phenotype and to cause the disease. The impact of the gene on genetic risk for the disease is same in all families. In complex disorders with multiple causes, variations in a number of genes encoding different proteins result in a genetic predisposition to a clinical phenotype. Pedigrees reveal no Mendelian inheritance pattern, and gene mutations are often neither sufficient nor necessary to explain the disease phenotype. Environment and life-style are major contributors to the pathogenesis of complex diseases. In a given population, epidemiological studies expose the relative impact of individual genes on the disease phenotype.[4] However, between families the impact of these genes might be totally different. In one family, a rare gene C (family 3) might have a large impact on genetic predisposition to a disease. However, because of its rarity in the general population, the overall population effect of this gene would be small. Some genes that predispose individuals to disease might have minuscule effects in some families (gene D, Family 3) [Figure 1].
Figure 1.
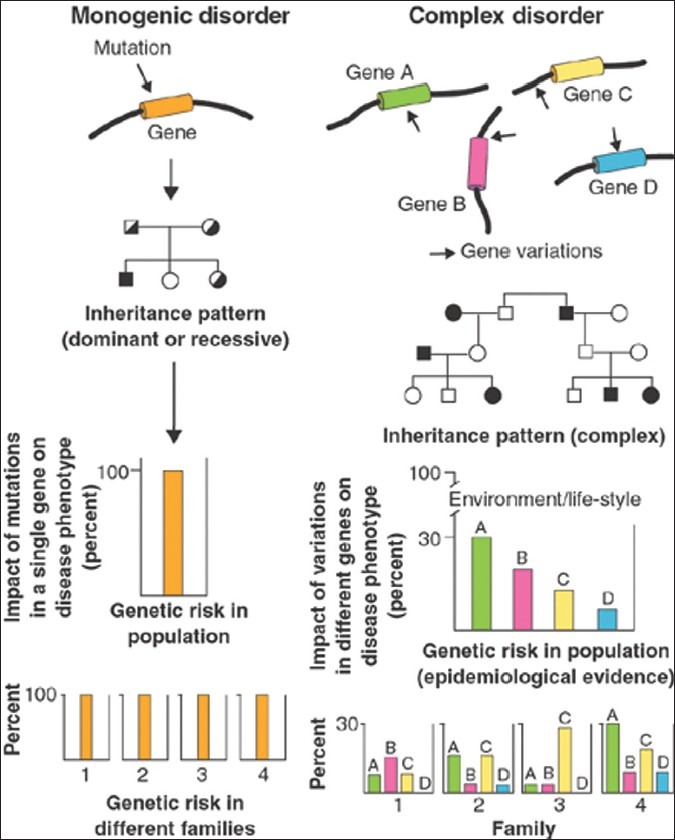
Picture adapted from Peltonen L and McKusick VA (2001)
Polymorphisms and mutations
A major difference in the genetic basis for simple Mendelian diseases and complex genetic diseases is the number of genes involved, and the contribution of each gene to the overall disease phenotype. The fact that the genetic alteration is predictably associated with a disease phenotype indicates that there is no redundancy or compensation in the particular biological system that can overcome the effect of the underlying genetic defect. Such a genetic alteration is termed ‘mutation’ as in the case of Mendelian diseases.
The genetic alterations that contribute to complex diseases are individually of much smaller effect and are generally called ‘generally polymorshims’ because they are prevalent in the population. When a specific allele occurs, in at least 1% of the population, it is said to be genetic polymorphism. In contrast to mutations that have been casually linked with Mendelian diseases, genetic polymorphisms are often not directly casually linked, but rather specific alleles are reported to be found more frequently in diseased individuals than in non-affected controls.[5]
Evidence for the role of genetic variants in periodontitis
Clinical and scientific data from a variety of sources suggest that the genetic variants are major determinants of syndromic and non-syndromic periodontitis. The evaluation of the quality of supporting studies requires an understanding of the formal genetic analytical methods that have been used.
Familial aggregation
Familial aggregation of a trait or disease can suggest genetic etiology. It results from shared genes, environmental exposures, and similar socio-economic influences. There is literature reporting familial aggregation of periodontal diseases, but due to different terminology, classification systems and lack of standardized methods of clinical examination, it is difficult to compare reports directly. Most familial reports are for early-onset periodontitis (EOP) now called as aggressive periodontitis (AgP).[6]
Twin studies
Monozygous (MZ) twins are genetically identical and dizygous (DZ) twins share on average 50% of their genes in common. Differences in disease experienced between MZ twins must be due to environmental factors, and between DZ twins they could arise from both environmental and genetic differences. The difference in concordance between MZ and DZ twins for a particular phenotype can be used to estimate the effects of the extra shared genes in MZ twins, if the environment for twin test is the same (reared together twins). Studying disease presentation in twins is useful for differentiating the variations due to environment from those due to genetic factors and for estimating the amount of heredity in a phenotype. Twins studies of periodontitis have been limited in scope and generally of small numbers. Most twin studies have studied chronic periodontitis.[7]
Segregation analysis
Genes segregate in families as predicted by Mendels laws. Segregation analysis is used to determine if trait transmission appears to fit a Mendelian or other mode of genetic transmission. Segregation analysis does not find a specific gene responsible for trait. Marazita and co-workers, 1994, performed the most definitive segregation analysis in North American families for aggressive periodontitis and found support for autosomal-dominant transmission with approximately 70% penetrance.[6]
Linkage analysis
Linkage analysis is a technique used to localize the gene for a trait to a specific chromosomal location. A specific trait can be followed as it segregates through families of interest and determine if the trait appears to segregate with a known genetic polymorphism. Linkage can prove the basis of disease. To date, linkage studies have been performed on two families with localized aggressive periodontitis. Boughman et al., identified an autosomal-dominant form of localized aggressive periodontitis in an extended family from Southern Maryland.[8] Hart et al., evaluated support for linkage in the region of chromosome 4 in a different population of families.[9]
Association studies
Genes contributing to common, complex diseases such as periodontitis have proved to be more difficult to isolate. A hypothetical example of the logic in play, in the study of gene polymorphism associations with the disease is: in periodontitis (condition A), bone is lost (abnormality B) and since bone is removed by osteoclasts, which are influenced by a specific gene (gene C), the association of a known polymorphism (polymorphism D) of gene C periodontal diseases studies.
Two types of association analysis are commonly used: population based and family based approaches.
![]()
Candidate genes for periodontal disease
The progression of periodontal disease is governed by the subject's host response, which is determined to some extent by previous experience (acquired immunity) but is predominantly influenced by the person's genetic make up. Thus, a range of genetic variations or deficiencies in the host response can increase the likelihood of periodontitis.
Several immune response traits have been associated with clinical forms of periodontitis and some of these factors the underlying genetic determinants are known. Although it is unlikely that polymorphisms in all these genetic determinants impart differential susceptibility to periodontal disease, it is reasonable to expect that multiple genes will be found to be important and that knowledge of these may permit determination of individual susceptibility. The key will be able to identify the genetic factors that are important enough to impart significant clinical risk.
In general, a gene may be considered as a candidate for a causative or modifying role in periodontitis if the physiological process determined by the genes has been associated with the presence or severity of disease.
Currently, little is known about which genes may be involved in periodontitis as disease modifying genes. The table summarizes candidate gene polymorphisms investigated in relation to periodontitis [Table 1].
Table 1.
Candidate genes and corresponding encoded proteins for which gene polymorphisms have been investigated as putative risk factors for periodontitis

Cytokine gene polymorphisms
Differences in the expression of cytokine are of great interest in periodontal research and this can be explained by genetic differences. Some individuals carry polymorphisms of host response genes that code for hyper secretion of certain cytokines in response to stimuli. Gene polymorphisms for various cytokines have been studied in relation to periodontal disease.
Interleukin-1
Polymorphisms in the IL-1 cluster have been the main locus of attention in recent studies because of the fundamental role of IL-1β in the pathogenesis of periodontal disease. Kornman et al., suggested that periodontitis associated genotype (PAG) had an approximately 7 times greater chance of having severe periodontitis than those who were PAG negative. PAG is a composite IL-1 genotype found by the combination of two rare alleles at separate single nucleotide polymorphisms (SNP) in this cluster at position -889 in the IL-1A promoter and at + 3954 (now referred to as +3953) of the IL-1B gene.[10]
Frequency of allele 2 of the IL-1B +3953 SNP was significantly increased in patients with advanced periodontitis.[11,12] Recently, a study tested polymorphisms derived from genes of the IL-1 cluster for association with generalized aggressive periodontitis (GAP) through both allelic association and by constructing a linkage disequilibrium map of the 2q13-14 disease candidate region. This study suggested that there is some evidence for an association between GAP and the IL-1β (+3953) polymorphisms.[13]
Currently, a genetic test is being marked for severe chronic periodontitis, called periodontal susceptibility test (PST). It tests for the presence of specific polymorphisms of the IL-1α and IL-1β genes. The genotype is determined from a finger stick blood or saliva samples.
Other cytokines
There is some preliminary evidence that genotype with respect to the IL-2 SNP might be associated with severity in aggressive periodontitis.[14] No significant difference was found in the frequency of IL-4 gene polymorphisms between control and periodontitis group in African–American-Brazilian population.[15] Analysis IL-6 gene polymorphisms in Czech patients with chronic periodontitis suggested that -572 G/C polymorphisms of IL-6 gene might be one of the protective factors associated with lower susceptibility to chronic periodontitis.[16] A study of the distribution of the genes related to IL-10 found no association between the genes for the cytokine and aggressive periodontitis compared with healthy controls.[17] Gonsales et al., investigated the influence of IL-10 promoter polymorphisms at position -824 and other at -597 on aggressive and chronic periodontitis and suggested that this polymorphism had no association with periodontal disease.[18] The frequency of genotypes at both positions was greater in moderate adult periodontitis. Six different IL-18 gene polymorphism (-656, -607, -137, +113, +127, and codon 35/3) were studied and none of the polymorphisms were associated with destructive periodontal disease.[19] Craandijk et al., found no significant association between a different series of four TNF-α gene polymorphisms and periodontitis patients.[20]
Receptor gene polymorphisms
FcγR gene polymorphisms
The Fc-gamma receptor is the receptor present on phagocytes, which binds IgG. Polymorphisms that influence the binding affinity between the FcγR and IgG of different subclasses are considered important in susceptibility to periodontal disease. There are three main classes of FcR: FcγRI, FcγR II, FcγR III. Sugita et al., reported that FcγR IIIb has a NA1-NA2 polymorphism. NA1 is a more efficient opsono-phagocytic agent than NA2. NA1 is found to a greater extent in Japanese patients who are resistant chronic periodontitis.[21] Meisel et al., analysed the association between FcγR IIIa (high affinity receptor) and FcγR IIIb (low affinity receptor) and chronic periodontitis in German population. They found that FcγR IIIa was associated with chronic periodontitis but this is in linkage disequilibrium with FcγR IIIb, which is also associated chronic periodontitis.[22]
Toll-like receptor gene polymorphisms
Toll-like receptors are signal molecules essential for the cellular response to bacterial cell wall components. Different functional effective polymorphisms of TLR-4 gene (Asp 299 Arg 677 Trp; Arg753 Gln) have been associated with impaired LPS signal transduction. Schroeder et al., suggested that genetic variants of TLR-4 might act as risk factor for the development of chronic periodontitis.[23]
CD14 gene polymorphisms
The R-allele in the promoter region of CD14 at position -260(-159) enhances the transcriptional activity of the gene.[24] Two studies with Caucasian subjects investigated the CD14-260 polymorphism in chronic periodontitis, but did not find any significant associations.[25,26] A higher frequency of the N –allele and the N/N genotype of CD14-1359 polymorphism was found in patients in severe periodontitis.
CARD15 gene polymorphisms
The 3020insC and 2104 C >T polymorphisms of the CARD15 (NOD2) gene leads to impaired activation of nuclear factor-kappa B, resulting in altered transcription of pro inflammatory cytokine genes and reduced expression of these cytokines.[27] No role for the CARD15 3020insC and 2104 C>T polymorphism was found for periodontitis in Caucasians.[28]
Polymorphism of RANK gene
RANKL and its receptor RANK have been implicated in increased rates of bone resorption in periodontal disease. Key mediators of osteoclast differentiation and activation, involve receptor activator of nuclear factor-κB (RANK), RANK ligand (RANKL), and osteoprotegrin (OPG). An association analysis with allelotypes showed that SNPs identified in the RANK/RANKL/OPG genes have no significant association with AgP in the Japanese population.[29]
Vitamin D receptor gene polymorphisms
Vitamin D receptor gene polymorphism has regulatory effects on bone mineral density and bone turnover. Hennig et al., suggested that genotype of Taql VDR gene might be a risk indicator for susceptibility to EOP.[30]
N-formyl peptide receptor polymorphisms
The high affinity FMLP receptor (FPR1) of phagocytic cells interacts with bacterial FMLP and mediates chemotaxis, degranulation, and superoxide production. These cellular functions are disrupted in PMN's from aggressive periodontitis patients. Zhang et al., studied the prevalence of polymorphisms at nt329T-C (codon 110 phenylalanine-serine), and at nt378C-G (codon 126 cysteine-tryptophan) in the 583 bp interval of the FMLP receptor gene in localised, generalised, and aggressive periodontitis.[31] Six SNP's were identified including two located in the FPR1 second extra cellular loop that were significantly associated with the AgP phenotype in African –American patients. These data do not support the hypothesis that the FPR1 SNP's C329T>C and C378C>G play an etiologic role in AgP, but do suggest that SNP's in the second extracellular loop may be etiologically important.
Antigen-antibody gene polymorphisms
Class II human leukocyte antigens
HLA are involved in genetically predetermined humoral immune response via recognition of foreign antigens. MHC class II molecules (HLA-DP,-DQ,-DR) are expressed on cells that immuno survey host cells including B and T cells, macrophages and accessory cells for the presence of foreign peptides. Studies have suggested that patients with the HLA-DRB1 1501-DQB1 0602 genotype may have an accelerated T cell response to Porphyromonas. gingivalis and an increased susceptibility to EOP in Japanese patients.[32]
Immunoglobulin g2 variations
It is of interest that levels of IgG2 vary among populations regardless of disease status. A number of different biological mechanical may be involved in IgG2 variations, suggesting possible genetic and environmental heterogeneity. The IgG molecules carry genetically determined variations in the gamma heavy chains, termed Gm allotypes and present in also referred to as G2m (n).
Choi et al., assessed allotypes for the IgG subclasses and levels of IgG2 antibody to Actinobacillus actinomycetemcomitans and P. gingivalis in localized juvenile periodontitis (LJP) and rapidly progressing periodontitis (RPP) patients. The RPP patients who were positive for G2m (23) allotype had elevated antibody to P. gingivalis.[33]
Polymorphisms in genes encoding enzymes
Cathepsin C gene polymorphism
Cathepsin C is a lysosomal protease present in neutrophils and macrophages as well as epithelial cells. Hart and co-workers, 2000, identified and localized a gene on chromosome 11, which is responsible for a severe form of pre-pubertal periodontitis in a family of Jordanian descent.[34] Other mutations in the CTS C gene have been linked to the Papillon-Lefevre syndrome, a disease which is also associated with a severe form of pre-pubertal periodontitis.[35,36] Therefore, certain functional CTS C gene mutation needs to be considered causative for pre-pubertal periodontitis (PPP).
Matrix metallo proteinase gene polymorphisms
MMP-1 is an important mediator of connective tissue destruction in periodontal disease. Cao Z et al., evaluated the association between the MMP-1 promoter gene polymorphism and chronic periodontitis susceptibility and/or severity in a Chinese population. This study suggests that a single nucleotide polymorphism in the MMP-1 promoter region of -1607 bp may be associated with severe chronic periodontitis in a Chinese population.[37]
Polymorphisms in cyclooxygenase-2 gene
Prostaglandin E2 (PGE2) is considered to be an important mediator of tissue destruction in periodontitis. Cyclooxygenase (COX) is known to catalyze the production of prostaglandins. COX-2, which is induced in an inflammatory response, is responsible for prostaglandin synthesis at sites of inflammation. Candidate genes in the chromosome 9q32-33 region include the gene for cyclooxygenase-2. A single nucleotide polymorphism of COX-2–765 has been shown to alter the expression of the COX-2 gene. Ho et al., suggested that the –765G to C polymorphism of the COX-2 gene is associated with a decreased risk for periodontitis in Taiwanese, especially in AgP.[38]
Polymorphisms in genes encoding for myeloperoxidase (MPO) and N-acetyl transferase (NAT-2)
MPO and NAT2 are enzymes participating in the metabolism of xenobiotics including arylamines from tobacco smoke. MPO is also implicated in defence against bacterial challenge and inflammatory tissue destruction. Miesel et al., reported that polymorphic variants of both NAT2 and MPO affect the extent of the periodontal state. Subjects designated as rapid acetylators (bearing at lease one wild-type allele NAT2) or bearing the highly expressed MPO variant (homozygous -463 G/G) are at increased risk of periodontitis when smoking.[39] Kocher et al., reported the association between bone loss in periodontal disease and polymorphism of NAT2. Mean bone loss was reported to be higher in the group with slow acetylator phenotype.[40]
Polymorphisms in genes encoding vasoactive enzymes
The presence of lymphotoxin-α (TNF-β), angiotensin-converting enzyme (ACE) and endothelin-1 (ET-1) gene polymorphs were evaluated in Caucasian patients with adult periodontititis. Significant differences were reported with regard to three-locus combination of genotypes between diseased and healthy subjects.[41]
Discussion
Sir William Osler noted, “If it were not for the great variability among individuals, medicine might as well be a science not an art”. This statement embodies the influence of genetic variability on treatment outcomes both in medicine and dentistry. The technological advances stemming from the human genome project have changed the face of biological investigations and have placed genomics at the forefront of biomedical sciences. The individual risk for disease as well as response to treatment will be highly influenced by genetic prediction, resulting in changes in the delivery of healthcare and treatment based genomic approach to targeting molecular pathways disrupted in disease.
Periodontal disease results in destruction of the supporting structures and most of the destructive process, involved are host derived. The processes leading to destruction and regeneration of the destroyed tissues are of great interest to both researchers and clinicians. The information contained within the human genome can potentially lead to a better understanding of the control mechanisms modulating the production of inflammatory mediators as well as provides potential therapeutic targets. With the availability of human genome sequence and the knowledge of the complement of the genes, it should be possible to identify the metabolic pathways involved in periodontal destruction and regeneration. Most forms of periodontitis represent a life long account of interactions between the genome, behaviour and environment. Predicting these metabolic pathways, as well as their interactions with the modifiers represents a daunting task.
A significant paradigm has occurred in the perception of periodontal disease over the last 20 years. Increasingly, the emphasis for risk is being placed on host genetic and other non-microbial environmental factors as modifiers of bacterially induced diseases. A multitude of host factors are involved in responses to microbial challenge and in subsequent immune responses. Genetic polymorphisms exist in many if not most of the inflammatory and immune mediators. The allele or polymorphism in question may not be always active and may need environmental or other gene activity to be bought into play. Researchers must also consider the biological plausibility of the effect of gene polymorphism in question having a role in the periodontal disease process.
The requirements in providing a disease and polymorphism association are:
The polymorphism must influence the gene product.
Biases in the study population should be recognised and controlled.
Confounders such as smoking and socio-economic class must be controlled.
The affected gene product should be a part of the disease etiopathology.
An important problem related to research in the etiopathogenesis of periodontitis is that, whatever the cause of the disease is, the symptoms are the same: deepening of periodontal pocket, loss of attachment, and alveolar bone. It is likely that overlapping clinical phenotypes exist between different forms of periodontitis. It is important that globally accepted definitions of the case (chronic and aggressive forms) of periodontitis are used in future studies, to allow valid comparisons to be made between gene polymorphisms data from different parts of the world. Other complications arise related to the variable presentation overtime of periodontal disease.
The distribution and frequency of gene polymorphism may vary considerably between different cohorts, particularly those of differing racial origins. Consequently, gene association studies may require investigations of large homogenous cohorts and data obtained from one study may not be readily generalised to other patients’ cohorts. In addition, the use of ‘gene’ approach risks overlooking other genes involved in the regulation of inflammation and consequently missing genetic loci that could be strongly associated with the disease risk. The development of newer technologies such as genome wide “SNP chips” has created the possibility of carrying out more systematic studies to identify gene loci associated with a risk of disease.
The development of periodontitis in an individual depends on the collective presence of a number of environmental risk factors in conjunction with a number of genetic factors at a given time point in life. The more genetic factors an individual has inherited, the greater the genetic predisposition and higher the chance of early development of periodontitis. A multitude of polymorphisms in genes most of which code for various aspects of host immune response, have been explored. However, even among studies with subjects of same ethnic background no consistent results have been obtained. Often, researchers have found some significant associations by defining small groups of individuals or after stratification. The carriage of specific combinations of alleles within a given locus, and among various genes has only been sparsely investigated.
The genetic studies in relation to periodontitis are hampered by population heterogeneity and differences in patient selection and diagnostic criteria. However, it is possible that inconsistent results may reflect the underlying complexity and heterogeneity of genetic influence in periodontitis. Another problem encountered was that many studies had investigated putative genetic risk factors without considering other established risk factors for periodontitis as co-varieties (e.g., smoking, age, gender). Further the vast majority of studies have not encountered the infectious component (gene–environment interaction) it is recommended strongly that whenever possible the bacterial microorganisms, are appropriate surrogate measures of bacterial infections should be included as co-variates in the analysis.
Studies applying the candidate gene approach could be guided by results from genome wide searches or by results from gene expression signatures or family linkage analysis. Furthermore, these type of studies need to be large scale, in consortium-based approaches, because single studies are underpowered.
The sequencing of the human genome along with recent achievements in genomics provides an unparalleled opportunity to advance our understanding of the role of genetic factors in human health and disease, to allow more precise definition of non-genetic factors involved, and to apply this insight rapidly to the prevention, diagnosis and treatment of the diseases.
The current practical utility of genetic knowledge in periodontitis is limited. However, performing clinical periodontal assessments of siblings of AgP probands is one of the most useful activities that can be performed to ensure early diagnosis of this disease. By careful clinical diagnostic procedures, susceptible patients may be detected early and therapy instigated, which may prevent the more significant disease aspects from occurring. In the pursuit of better genetic diagnostic tests for chronic and AgP, research must be planned using plausible biological arguments. The bias and misinterpretation of genetic association with disease should be avoided.
Conclusions
Seamless collaborations among clinicians, epidemiologists, geneticists, mathematicians, and computer experts will be needed to solve the genetic underpinnings of complex disease that affects the lives of millions. If we can design and build an unprecedented and noble structure, resting on firm bedrock of the “Human Genome Project”, then the true promise of genomics research for benefitting human kind can be realised.
Despite major advances in awareness of genetic risk factors for periodontal disease, we are still away from determining the genetic basis of both aggressive and chronic periodontitis. To date, major gene mutations, which result in the periodontitis phenotype in otherwise systemically healthy individuals, have not been identified and no specific genetic risk factor for the disease has been identified. The studies fail to quantity the magnitude of contribution of a particular disease associated allele to disease risk. This failure to quarantine the sensitivity and specificity of test precludes determination of the clinical utility of the association.
An interesting approach, which is emerging, is the development of the susceptibility profile concept for specific diseases. For example, the risk of Alzheimer's disease is being considered to be substantially influenced by a total of ten genetic polymorphisms of inflammation-related molecules. It is feasible that a similar situation may pertain in periodontitis, where several relatively common high-risk polymorphisms could be inherited by an individual, giving them a cumulative high susceptibility profile. For such a high-risk profile to be proved in periodontal disease, much testing across different populations would be needed. Such useful genetic information would be invaluable in strategies aimed at preventing the development of periodontal disease and its therapeutic intervention.
Footnotes
Source of Support: Nil
Conflict of Interest: None declared.
References
- 1.Kinane DF, Hart TC. Genes and gene polymorphisms associated with periodontal disease. Crit Rev Oral Biol Med. 2003;14:430–49. doi: 10.1177/154411130301400605. [DOI] [PubMed] [Google Scholar]
- 2.Hart TC, Zhang Y, Gorry MC, Hart PS, Cooper M, Marazita ML, et al. A mutation in the SOS1 gene causes hereditary gingival fibromatosis type 1. Am J Hum Genet. 2002;70:943–54. doi: 10.1086/339689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tabor HK, Risch NJ, Myers RM. Opinion: Candidate gene approaches for studying complex genetic triats: Practical considerations. Nat Rev Genet. 2002;3:391–7. doi: 10.1038/nrg796. [DOI] [PubMed] [Google Scholar]
- 4.Peltonen L, McKusick VA. Genomics and medicine. Dissecting human disease in the postgenomic era. Science. 2001;291:1224–9. doi: 10.1126/science.291.5507.1224. [DOI] [PubMed] [Google Scholar]
- 5.Schork NJ, Fallin D, Lanchbury JS. Single nucleotide polymorphisms and the future of genetic epidemiology. Clin Genet. 2000;58:250–64. doi: 10.1034/j.1399-0004.2000.580402.x. [DOI] [PubMed] [Google Scholar]
- 6.Marizata ML, Burmeister JA, Gunsolley JC, Koertage TE, Lake K, Schenkein HA. Evidence for autosomal dominant inheritance and race specific heterogeneity in early–onset periodontitis. J Periodontol. 1994;65:623–30. doi: 10.1902/jop.1994.65.6.623. [DOI] [PubMed] [Google Scholar]
- 7.Michalowicz BS, Aeppli D, Virag JG. Periodontal findings in adult twins. J Periodontol. 1991;62:293–9. doi: 10.1902/jop.1991.62.5.293. [DOI] [PubMed] [Google Scholar]
- 8.Boughman JA, Halloran SL, Roulston D, Schwartz S, Suzuki JB, Weitkamp LF, et al. An autosomal dominant form of juvenile periodontitis: Its localization to chromosome 4 and linkage to dentinogenesis imperfecta and gc. J Craniofac Genet Dev Biol. 1986;6:341–50. [PubMed] [Google Scholar]
- 9.Hart TC, Marizata ML, McCanna KM, Schenkein HA, Diehl SR. Reevaluation of chromosome 4q candidate region for early onset periodontitis. Human Genet. 1993;91:416–22. doi: 10.1007/BF00217764. [DOI] [PubMed] [Google Scholar]
- 10.Kornman KS, Crane A, Wang HY, di Giovine FS, Newmann MG, Pirk FW, et al. The interleukin-1 genotype as a severity factor in adult periodontal disease. J Clin Periodontol. 1997;24:72–7. doi: 10.1111/j.1600-051x.1997.tb01187.x. [DOI] [PubMed] [Google Scholar]
- 11.Galbraith GM, Hendley TM, Sanders JJ, Palesch Y, Pandey JP. Polymorphic cytokine genotypes as markers of disease severity in adult periodontitis. J Clin Periodontol. 1999;26:705–9. doi: 10.1034/j.1600-051x.1999.t01-1-261101.x. [DOI] [PubMed] [Google Scholar]
- 12.Rogers MA, Figliomenni L, Baluchova K, Tan AE, Davis G, Henry PJ, et al. Do interleukin-1 polymorphisms predict the development of periodontitis or the success of dental implants. J Periodontal Res. 2002;37:37–41. doi: 10.1034/j.1600-0765.2002.00651.x. [DOI] [PubMed] [Google Scholar]
- 13.Scapoli C, Trombelli L, Mamolini E, Collins A. Linkage disequilibrium analysis of case-control data: An application to generalized aggressive periodontitis. Genes Immun. 2005;6:44–52. doi: 10.1038/sj.gene.6364152. [DOI] [PubMed] [Google Scholar]
- 14.Scarrel-Caminaga RM, Trevilatto PC, Souza AP, Brito RB, Line SR. Investigation of an interleukin-2 polymorphism in patients with different levels of chronic periodontitis. J Clin Periodontol. 2002;29:587–91. doi: 10.1034/j.1600-051x.2002.290701.x. [DOI] [PubMed] [Google Scholar]
- 15.Carla CP, Jose RG, Arthur BN, Mário TJ, Márcio FM, Joerg M, et al. Interleukin-4 gene polymorphism and its relation to periodontal disease in a Brazilian population of African heritage. J Dent. 2004;32:241–6. doi: 10.1016/j.jdent.2003.11.004. [DOI] [PubMed] [Google Scholar]
- 16.Holla LI, Fassmann A, Stejskalova A, Znojil V, Vanek J, Vacha J. Analysis of the interleukin-6 gene promoter polymorphisms in czech patients with chronic periodontitis. J Periodontol. 2004;75:30–6. doi: 10.1902/jop.2004.75.1.30. [DOI] [PubMed] [Google Scholar]
- 17.Kinane DF, Hodge P, Eskdale J, Ellis R, Gallagher G. Analysis of genetic polymorphisms at the interleukin-10 and tumor necrosis factor loci in early onset periodontits. J Periodontal Res. 1999;34:379–86. doi: 10.1111/j.1600-0765.1999.tb02270.x. [DOI] [PubMed] [Google Scholar]
- 18.Gonzales JR, Michel J, Diete A. Analysis of genetic polymorphisms at the interleukin-10 loci in aggressive and chronic periodontitis. J Clin Periodontol. 2002;29:816–22. doi: 10.1034/j.1600-051x.2002.290905.x. [DOI] [PubMed] [Google Scholar]
- 19.Folwaczny M, Glas J, Török HP, Tonenchi L, Paschos E, Bauer B, et al. Polymorphisms of the interleukin-18 gene in periodontitis patients. J Clin Periodontol. 2005;32:530–4. doi: 10.1111/j.1600-051X.2005.00711.x. [DOI] [PubMed] [Google Scholar]
- 20.Craandijk J, Van Krugten MV, Verweij CL, Vandervelden U, Loos BG. Tumor necrosis factor alpha gene polymorphisms in relation to periodontits. J Clin Periodontol. 2002;29:28–34. doi: 10.1034/j.1600-051x.2002.290105.x. [DOI] [PubMed] [Google Scholar]
- 21.Sugita N, Kobayashi T, Ando Y, Yoshihara A, Yamamoto K, Vande-Winkel JG, et al. Increased frequency of increased gamma RIIIb-NA1 allele in periodontitis resistant subjects in an elderly Japanese population. J Dent Res. 2001;30:914–8. doi: 10.1177/00220345010800031301. [DOI] [PubMed] [Google Scholar]
- 22.Meisel P, Carlssson LE, Sawaf H, Fanghaenel J, Greinacher A, Kocher T. Polymorphisms of Fc Gamma receptors RIIa, RIIIa and RIIIb in patients with adult periodontal disease. Genes Immun. 2001;2:258–62. doi: 10.1038/sj.gene.6363777. [DOI] [PubMed] [Google Scholar]
- 23.Schröder NW, Meister D, Wolff V, Christan C, Kaner D, Haban V, et al. Chronic periodontal disease is associated with single-nucleotide polymorphisms of the human TLR-4 gene. Genes Immun. 2005;6:448–51. doi: 10.1038/sj.gene.6364221. [DOI] [PubMed] [Google Scholar]
- 24.Hubacek JA, Rothe G, Pit’ha J, Skodova Z, Stanek V, Poledne R, et al. C (-260) – T Polymorphism in the promoter of the CD14 receptor gene as a risk factor for myocardial infarction. Circulation. 1999;99:3218–20. doi: 10.1161/01.cir.99.25.3218. [DOI] [PubMed] [Google Scholar]
- 25.Holla LI, Buckova D, Fassmann A, Halabala T, Vasku A, Vacha J. Promoter polymorphisms in the CD14 receptor gene and their potential association with the severity of chronic periodontits. J Med Genet. 2002;39:844–8. doi: 10.1136/jmg.39.11.844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Folwaczny M, Glas J, Torok HP, Fricke K, Folwaczny C. The CD14-159C-to-T promoter polymorphism in periodontal disease. J Clin Periodontol. 2004;31:991–5. doi: 10.1111/j.1600-051X.2004.00600.x. [DOI] [PubMed] [Google Scholar]
- 27.Girardin SE, Boneca IG, Viala J, Chamaillard M, Labigne A, Thomas G, et al. NOD2 is a general sensor of peptidoglycan muramyl dipeptide (MDP) detection. J Biol Chem. 2003;278:8869–72. doi: 10.1074/jbc.C200651200. [DOI] [PubMed] [Google Scholar]
- 28.Folwaczny M, Glas J, Torok HP, Mauermann D, Folwaczny C. The 3020insC mutation of the NOD2/CARD15 gene in patients with periodontal disease. Eur J Oral Sci. 2004;112:316–9. doi: 10.1111/j.1600-0722.2004.00137.x. [DOI] [PubMed] [Google Scholar]
- 29.Soedarsono N, Rabello D, Kamei H, Fuma D, Ishihara Y, Suzuki M, et al. Evaluation of RANK/RANKL/OPG gene polymorphisms in aggressive periodontitis. J Periodontal Res. 2006;41:397–404. doi: 10.1111/j.1600-0765.2006.00874.x. [DOI] [PubMed] [Google Scholar]
- 30.Hennig BJ, Parkhill JM, Chapple IL, Heasman PA, Taylor JJ. Association of a vitamin D receptor gene polymorphism with localized early onset periodontal disease. J Periodontol. 1999;70:1032–8. doi: 10.1902/jop.1999.70.9.1032. [DOI] [PubMed] [Google Scholar]
- 31.Zhang Y, Hart PS, Moritti AJ, Bouwsma OJ, Fisher EM, Dudlicek L, et al. Biochemical and mutational analysis of the cathepsin C gene (CTSC) in three North American families with papillon-lefevere syndrome. Hum Mutat. 2002;20:75. doi: 10.1002/humu.9040. [DOI] [PubMed] [Google Scholar]
- 32.Takashiba S, Ohyama H, Oyaizu K, Kogoe-kato N, Murayama Y. HLA genetics for diagnosis of susceptibility to early onset periodontits. J Periodontal Res. 1999;34:374–8. doi: 10.1111/j.1600-0765.1999.tb02269.x. [DOI] [PubMed] [Google Scholar]
- 33.Choi JI, Ha MH, Kim JH, Kim SJ. Immunoglobulin allotypes and immunoglobulin G subclass responses to actinobacillus actinomycetemcomitans and porphyromonas gingivalis in early onset periodontits. Infect Immun. 1996;64:4226–30. doi: 10.1128/iai.64.10.4226-4230.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hart TC, Hart PS, Michalec MD, Zhang Y, Firatli E, Van Dyke TE, et al. Haim-Munk syndrome and Papillon-lefeverre syndrome are allelic mutations in cathepsin C. J Med Genet. 2000;37:81–2. doi: 10.1136/jmg.37.2.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hart TC, Hart PS, Bowden DW, Michalec MD, Callison SA, Walker SJ, et al. Mutations of the cathepsin CG are responsible for Papillon-lefevere syndrome. J Med Genet. 1999;36:881–7. [PMC free article] [PubMed] [Google Scholar]
- 36.Toomes C, James J, Wood AJ, Wu CL, McCornick D, Lench N, et al. Loss-of-function mutations in the cathepsin C gene result in periodontal disease and palmo-plantar keratosis. Nat Genet. 1999;23:378–80. doi: 10.1038/70525. [DOI] [PubMed] [Google Scholar]
- 37.Cao Z, Li C, Zhu G. MMP-1 promoter gene polymorphism and susceptibility to chronic periodontitis in a Chinese population. Tissue Antigens. 2006;68:38–43. doi: 10.1111/j.1399-0039.2006.00615.x. [DOI] [PubMed] [Google Scholar]
- 38.Ho YP, Lin YC, Yang YH, Ho KY, Wu YM, Tsai CC. Cyclooxygenase-2 Gene–765 single nucleotide polymorphism as a protective factor against periodontitis in Taiwanese. J Clin Periodontol. 2008;35:1–8. doi: 10.1111/j.1600-051X.2007.01167.x. [DOI] [PubMed] [Google Scholar]
- 39.Miesel P, Siegeund A, Grimm R, Herrmann FH, John U, Schwahn C, et al. The interleukin-1 polymorphism, smoking and the risk of periodontal disease in the population based SHIP study. J Dent Res. 2003;82:189–93. doi: 10.1177/154405910308200308. [DOI] [PubMed] [Google Scholar]
- 40.Kocher T, Sawaf H, Fanghanel J, Timm R, Meisel P. Association between bone loss and periodontal disease and polymorphism of N-acetyl transferase (NAT2) J Clin Periodontol. 2002;29:21–7. doi: 10.1034/j.1600-051x.2002.290104.x. [DOI] [PubMed] [Google Scholar]
- 41.Holla LI, Fassman A, Vasku A, Znojil V, Vanek J, Vacha J. Interactions of lymphotoxin α (TNF-β) anqiotensin-converting enzyme (ACE), and endothelin-1 (ET-1) gene polymorphisms in adult periodontitis. J Periodontol. 2001;72:85–9. doi: 10.1902/jop.2001.72.1.85. [DOI] [PubMed] [Google Scholar]