

Summary
Background
The mechanisms underlying exacerbation of asthma induced by respiratory syncytial virus (RSV) infection have been extensively studied in human and animal models. However, most of these studies focused on acute inflammation and little is known of its long-term consequences on remodelling of the airway tissue.
Objective
The aim of the study was to use a murine model of prolonged allergen-induced airway inflammation to investigate the effect of RSV infection on allergic airway inflammation and tissue remodelling.
Methods
We subjected mice to RSV infection before or during the chronic phase of airway challenges with OVA and compared parameters of airway inflammation and remodelling at the end-point of the prolonged allergen-induced airway inflammation protocol.
Results
RSV infection did not affect the severity of airway inflammation in any of the groups studied. However, RSV infection provoked airway remodelling in non-sensitized, allergen-challenged mice that did not otherwise develop any of the features of allergic airways disease. Increased collagen synthesis in the lung and thickening of the bronchial basal membrane was observed in non-sensitized allergen-challenged mice only after prior RSV infection. In addition, fibroblast growth factor (FGF)-2 but not TGF-β1 was increased in this group following RSV infection.
Conclusion
Our data show for the first time that RSV infection can prime the lung of mice that are not previously systemically sensitized, to develop airway remodelling in response to allergen upon sole exposure via the airways. Moreover, our results implicate RSV-induced FGF-2 in the remodelling process in vivo.
Keywords: airway allergic inflammation, airway remodelling, asthma, RSV
Introduction
Recurrent wheezing in children has been associated with respiratory infections early in life, and one of the main viral agents thought to be responsible for this phenomenon is respiratory syncytial virus (RSV) [1, 2]. A considerable effort has been put into defining the mechanisms underlying exacerbations and/or predisposition to asthma induced by RSV infection. In parallel to clinical studies in children, animal models of allergic airway inflammation within their limitations have proven useful in unravelling the molecular basis of the contribution of RSV to the onset or exacerbation of asthma [3-5]. Exacerbation of acute allergic airway inflammation by prior exposure to RSV is well documented in mice [6-8]. RSV infection enhances airway responsiveness to bronchoprovocation, increasing cellular influx to the lung, eosinophilic infiltration, mucus production, and T helper type 2 (Th2) type cytokine production in lung tissue and draining lymph nodes. Extensive studies involving the use of knock-out animals, blocking antibodies directed against cytokines and chemokines of interest, or T cell transfer delineate roles for the cytokines IL-4, IL-13, and to a greater extent IL-5, the chemokines regulated upon activation, normal T cell expressed and secreted (RANTES) and macrophage inflammatory protein-1α (MIP-1α), and in particular CD8+ T cells [9-14]. Controversy regarding the deleterious effect of RSV infection on the development of asthma later in life stems from reports of a protective effect of RSV infection before the onset of allergic airway inflammation illustrated by decreased AHR in infected animals [15]. However, such a protective effect has been more convincingly described for other respiratory viruses such as influenza virus [16]. It is noteworthy that the protective effect mediated by RSV is only observed during the acute phase of allergic inflammation, when RSV influences the Th1/Th2 compartments towards Th1, but still leads to increased AHR in the chronic phase of asthma facilitating the development of a Th2 response [15]. In studies where animals are subjected to RSV infection subsequent to the establishment of an asthmatic phenotype, exacerbation of allergic airway inflammation is observed and results in increased AHR, mucus secretion, cytokine production and lung cellular infiltrates [17].
However, none of these studies address the question of the long-term effect of RSV infection in conjunction with asthma in terms of remodelling of the airway tissue. Features characteristic of human asthma include structural changes of the airway, mainly illustrated by goblet cell hyperplasia and increased deposition of extracellular matrix protein such as collagen type I and IV, fibronectin, elastin and tenascin in the subepithelial region of bronchioles, together with proliferation of smooth muscle cells, fibroblasts and myofibroblasts [18]. It is believed that remodelling leads to thickening of the airway wall resulting in accelerated loss of lung capacity seen in severe asthmatics [19, 20].
Here, we investigated effects of RSV infection on remodelling in asthma, using an established murine model of chronic allergen-induced airway inflammation, in which mice exhibit all the main features of airway remodelling upon prolonged allergen challenge [21].
Materials and methods
Animals and viruses
Female BALB/c mice aged 6–8 weeks were purchased from Harlan. RSV strain A2 (Dr G. Wertz, University of Alabama, USA) was propagated and titrated for infectivity by plaque assay in HEp-2 monolayers.
Induction of allergic airway disease and respiratory syncytial virus infections
Airway inflammation and remodelling were induced in mice sensitized with OVA in alum as previously described [21]. Briefly, OVA mice were sensitized using 0.01 mg/mouse OVA (Sigma-Aldrich, Gillingham, Dorset) in 0.2 mL of alum (Au-Gel-S; Serva Electrophoresis, Heidelberg, Germany) i.p. on days 0 and 10, while control mice received the same volume of phosphate-buffered saline (PBS) in alum. OVA and alum mice were then challenged daily for 20 min with 5% OVA in aerosolized from days 20 to 26. Prolonged inflammation and remodelling were induced by subsequent similar OVA exposure three times a week. Mice were infected with 106 PFU of RSV A2 in 50 μL volume intranasally on day – 12 or on day 27 of the allergen-induced inflammation protocols or independently. Mice were killed at day 55 for examination of inflammation and remodelling parameters. Some mice from each group were also culled at day 35 to generate lung homogenates for ELISA analysis. An overview of the protocols is shown in Fig. 1.
Fig. 1.

Study design. Schematic overview of the protocols used to study the effect of respiratory syncytial virus (RSV) infection on remodelling in a murine model of asthma, as detailed in ‘Materials and methods’. Airway inflammation and remodelling were induced in female BALB/c mice i.p. sensitized with OVA in alum (⇑), and subjected to serial aerosolized OVA challenges (↑). Prolonged allergic inflammation (chronic phase) was induced by subsequent exposure to aerosolized OVA three times a week until mice were killed at day 55 (Allergy Only protocol, OVA/OVA and Alum/OVA control groups). Mice were infected intranasally with RSV and left to recover for 12 days before the onset of allergen-induced airway inflammation (RSV/Allergy protocol, RSV-OVA/OVA and RSV-Alum/OVA control groups) or at the peak of the allergic inflammatory response at day 27 (Allergy/RSV protocol OVA/RSV-OVA and Alum/RSV-OVA control groups). RSV infection only control mice were infected at day – 12 (RSV day – 12) or day 27 (RSV day 27) and killed at day 55.
Assessment of pulmonary inflammation
Cell recovery. Bronchoalveolar lavage (BAL) was performed as described [21]. Briefly, the airways of the mice were lavaged three times with 0.4 mL of PBS via a tracheal cannula. BAL fluid was centrifuged (700 g, 5 min, 4 °C); cells were counted, and then pelleted onto glass slides by cytocentrifugation (5 × 104 cells/slide). Differential cell counts were performed on Giemsa (Shandon, Runcorn, UK)-stained cytospins and related to total cell counts.
Histology
Paraffin-embedded lung sections (4 μm) were stained with haematoxylin and eosin and periodic acid-Schiff according to standard protocols. For pulmonary eosinophilia assessment, lung sections were stained with Congo Red and eosinophils were enumerated on sections at × 40 and numbers expressed as a percentage of total cell infiltrates.
Collagen synthesis
Soluble collagen was measured in lung tissue homogenates using a colorimetric protein measurement kit (Sircol collagen assay, Biocolor, Belfast, UK).
Quantification of extracellular matrix deposition
Image analysis was performed on lung sections prepared as described and stained with Sirus Red following standard protocol, using Scion Image Analysis software package (Scion Corporation, Frederick, MD, USA) adapted from the literature [22]. Digital photographs of four bronchioles per tissue section were taken at × 40 under polarizing light and these images were converted into monochrome. Ten measurements of 20 μm lines from each of the four bronchioles were drawn at a right angle from the basement membrane into the submucosa and the mean density along the 20 μm was calculated and expressed as pixels per square micrometre.
Immunohistochemistry
Paraffin-embedded lung tissue sections (4 μm) were cut from alum and OVA mice and dewaxed in Histoclear for 3 × 5 min and rehydrated. Endogenous peroxidase activity was blocked by incubation with 1.5% H2O2 in methanol for 30 min. Thereafter, endogenous avidin and biotin were blocked using a blocking kit (Vector Laboratories, Peterborough, UK). All incubations were carried out in humidified chambers and slides were washed between steps for 15 min each in PBS containing 0.1% saponin (Sigma). Non-specific binding was blocked using 10% donkey serum in PBS for 20 min. Sections were subsequently incubated with polyclonal goat antibody against fibroblast growth factor (FGF)-2 or normal goat IgG as a negative control (both Santa Cruz Biotechnology Inc., Autogenbioclear Ltd, Wiltshire, UK) for 90 min. Sections were then incubated with biotinylated donkey anti-goat secondary antibody (Jackson Immunoresearch Laboratories Inc., Baltimore, MD, USA) for 30 min, followed by streptavidin complex (DAKO, Ely, UK) for another 30 min. Slides were developed for approximately 5 min in diaminobenzidine (DAB; Vector Laboratories, Peterborough, UK) and counterstained in Gills haematoxylin. Sections were subsequently dehydrated, mounted and studied by light microscopy. FGF-2 intensity and localization of staining were analysed semi-quantitatively by grading immunostained sections using the following scoring system: 0 = no staining; 1 = weak staining restricted to epithelial cell nuclei; 2 = moderate staining localized to the cytoplasm of bronchial epithelial cells, alveolar epithelium, vascular epithelium; 3 = as in 2, but stronger intensity and additional staining throughout cellular infiltrates; 4 = as in 3, but heavy staining. Grading was performed blind by two independent investigators. For blocking studies, FGF-2 antibody was pre-incubated with a 10-fold excess of FGF-2 blocking peptide (Santa Cruz Biotechnology) at 37 °C for 45 min.
Enzyme-linked immunosorbent assay
Cytokines postulated to be involved in tissue fibrosis were analysed in BAL supernatant and lung tissue homogenate supernatants. ELISA kits (R&D, Abingdon, UK) for TGF-β and FGF-2 were used according to the manufacturer’s instructions. Although both of the kits were specific for the human cytokines, they also cross-react with the mouse orthologues.
Statistical analysis
Results were expressed as mean ± SEM with a group size of four to 15 from three different experiments. Data was analysed by ANOVA (Kruskal-Wallis) or Mann-Whitney U-test where appropriate and statistical significance was accepted when P < 0.05.
Results
Airway inflammation
Allergic pulmonary inflammation is characterized by leucocyte influx to the airways and by peribronchiolar and perivascular infiltrates in the lung. BAL was collected and cell content was analysed by differential cell counts of Wright-Giesma-stained cytospins. On day 55, total leucocyte influx was increased in all sensitized and challenged mice compared with their respective alum/OVA controls, and these infiltrates mainly consisted of macrophages. Eosinophils were found in BAL of all OVA/OVA, but not in corresponding alum/OVA control groups (Fig. 2a). In parallel, lung eosinophilia was assessed on Congo Red-stained lung tissue sections (Fig. 2b). All OVA-sensitized and challenged mice showed lung eosinophilia [21]. Although significant increases were observed in all OVA/OVA groups compared with corresponding alum/OVA controls, no difference in eosinophil recruitment was observed between RSV-infected and non-infected groups.
Fig. 2.
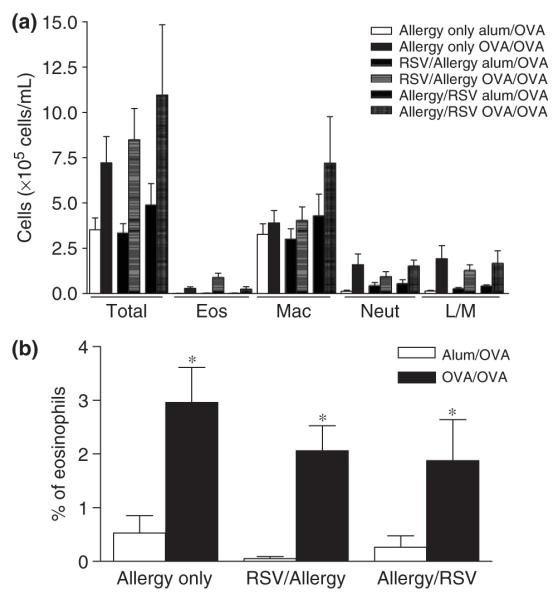
Effect of primary respiratory syncytial virus (RSV) infection on airway inflammation. Mice were killed 24 h after the final OVA challenge, BAL (a) were collected and lung sections prepared and stained with Congo Red (b), as described in ‘Materials and methods’. Values are expressed as mean ± SEM; n = 6–15 (a) and 4 (b) in three different experiments. *Significantly different from Allergy only Alum/OVA group.
Further histological examination of lung tissue for peribronchial cellular infiltration, goblet cell hyperplasia and mucus secretion revealed no difference between RSV-infected and non-infected groups. We also examined levels of IFN-γ, IL-4, IL-5 and IL-13 in both BAL and lung tissue at day 35 and day 55, but found that levels were not significantly changed by RSV infection (data not shown). Hence, RSV infection did not affect parameters of inflammation in a detectable manner.
Airway remodelling
Extracellular matrix deposition is a feature of airway remodelling following persistent allergen challenge [21]. We infected mice with RSV either before sensitization and allergen challenge or during challenges and examined levels of lung collagen biochemically or matrix deposition around the airways microscopically by image analysis. OVA/OVA mice showed increased levels of soluble collagen in lung tissue compared with alum/OVA controls [21]. In both RSV-infected groups, non-sensitized mice exhibited increased levels of soluble collagen, reaching those seen in corresponding infected sensitized and challenged mice. Moreover, soluble collagen was significantly increased in RSV-alum/OVA group compared with Allergy only alum/OVA controls (Fig. 3). These results indicated that RSV infection occurring before encounter with inhaled allergen leads to increased levels of soluble collagen in the lungs of alum control mice that would otherwise not develop features of airway remodelling.
Fig. 3.
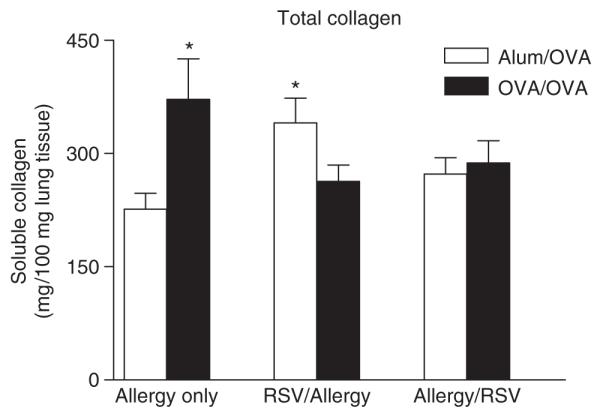
Effect of primary respiratory syncytial virus (RSV) infection on total collagen production. Soluble collagen was measured by Sircol assay in lung tissue homogenates prepared as described in ‘Materials and methods’. Results are expressed as mean ± SEM; n = 8–16 in three independent experiments. *Significantly different from Allergy only Alum/OVA group.
Matrix deposition was investigated on lung sections of mice in each group stained with Sirius Red, and collagen deposition along the basal membrane of airways was quantitatively measured by image analysis as described in ‘Materials and methods’. Under regular light, collagen appears red on Sirus Red-stained sections, as shown in Fig. 4a. Lung sections of OVA/OVA mice showed peribronchial and perivascular infiltrates, goblet cell hyperplasia and collagen deposition in the subepithelial area as well as in the perivascular regions, as we have demonstrated previously [21]. The same characteristic features were observed for RSV-OVA/OVA and OVA/RSV-OVA groups. As expected, cellular infiltrates and collagen deposition remained very low in alum/OVA control mice [21]. In contrast, dramatic increases in subepithelial and perivascular collagen deposition occurred in alum/OVA groups infected with RSV. Quantitative analysis confirmed that levels of subepithelial collagen deposition in the lungs of RSV-infected alum/OVA mice were significantly increased compared with non-infected alum/OVA controls (Fig. 4b). Cumulatively, these results demonstrate that infection by RSV, regardless of timing, provokes airway remodelling in non-sensitized, allergen-challenged mice that otherwise do not develop structural airway changes in the absence of systemic sensitization.
Fig. 4.

Effect of primary respiratory syncytial virus (RSV) infection on subepithelial collagen deposition. (a) Representative photomicrographs of paraffin-embedded (4 μm) Sirus Red-stained day 55 lung sections from alum and OVA groups of mice given Allergy only, RSV/Allergy, Allergy/RSV and RSV only protocols, taken under non-polarizing light conditions. (b) For quantification of subepithelial collagen deposition, random measurements from the basement membrane into the submucosa (10 measurements of 20 μm in length) were taken and the mean density was calculated from four bronchioles per mouse. Data is expressed as mean ± SEM; n = 4 mice in three independent experiments. *Significantly different from Allergy only Alum/OVA group.
In previous work, we reported that TGF-β1 plays a crucial role in airway remodelling [23]. We therefore investigated whether TGF-β1 could mediate the remodelling changes induced by RSV in alum/OVA control mice. Quantitative analysis of active TGF-β1 production in lung tissue by ELISA revealed no significant difference between RSV-infected and non-infected groups of alum/OVA mice (data not shown). However, numerous other mediators are implicated in airway remodelling, particularly pro-fibrotic factors [18]. Of these, fibroblast growth factor-2 was a potential candidate, as its secretion by epithelial cells in vitro is found to be increased by RSV infection [24]. Therefore, we determined the extent of expression of FGF-2 in the lung by measuring levels of FGF-2 in lung homogenate and BAL by ELISA as well as by immunostaining in lung sections. Although we did not detect any difference in the absolute levels of FGF-2 in the lung by ELISA, we found clear differences in FGF-2 staining patterns between lungs from alum/OVA and OVA/OVA mice (Fig. 5a and data not shown). In non-infected alum/OVA mice, staining was weak and FGF-2 expression was limited to bronchial epithelial and vascular endothelial cell nuclei (Figs 5a-i). After OVA sensitization and challenges, FGF-2 staining localized to the cytoplasm of bronchial epithelial cells, to the vascular endothelium and alveolar areas (Figs 5a-ii). Analysis of RSV-infected OVA/OVA mice revealed that although RSV infection elicited a similar pattern of FGF-2 staining, the intensity of staining was stronger and additional FGF-2 deposition was observed throughout the cellular infiltrates (Figs 5a-iv and vii). Observation at higher magnification identified macrophages as a likely source of FGF-2 among the infiltrating cells (inset in Figs 5a-iii). Most importantly, lung sections of alum/OVA mice in both RSV-infected groups also stained strongly positive for FGF-2 (Figs 5a-iii and vi), revealing a pattern similar to that found in OVA/OVA mice. FGF-2 production in the lung of RSV-alum/OVA and alum/RSV-OVA could not solely be attributed to RSV infection, as RSV only infected mice did not exhibit such high levels of FGF-2 (Figs 5a-v and viii).These results were confirmed by semi-quantitative analysis of histology slides using an arbitrary scoring system based on the intensity and localization of FGF-2-specific staining for multiple mice in each group (Fig. 5b).
Fig. 5.

FGF-2 production in the lung. (a) Representative photomicrographs of paraffin-embedded (4 μm) lung sections from alum and OVA groups of mice given Allergy only, respiratory syncytial virus (RSV)/Allergy, Allergy/RSV and RSV only protocols, immunostained with anti-FGF-2 antibody. Positive staining is brown. Black arrow shows macrophage at a higher magnification. (b) Semi-quantitative analysis of FGF-2 secretion using a scoring system as described in ‘Materials and methods’. Data is expressed as mean ± SEM; n = 5 mice in three independent experiments.
Discussion
Associations between respiratory tract infections, RSV in particular, and asthma have been recognized for many years [25]. However, whilst the effects of RSV infection on the predisposition or the exacerbation of acute asthma have been well studied, relatively little is known about the effect of respiratory tract infection on airway remodelling. Here, we used a murine model of chronic allergen exposure to determine the consequences of primary RSV infection on allergen-induced structural changes in airway tissues. Mice underwent a well-described protocol resulting in chronic allergen-induced airway inflammation and were subjected to RSV infection either before the onset of allergen sensitization, or during allergen challenges. Although prior RSV infection had no effect on mice immunized and challenged with OVA, infection of unsensitized mice provoked airway remodelling after long-term aerosol challenge.
Our data showed no apparent differences in lung inflammation between RSV-infected and non-infected groups. Leucocyte recruitment to the lung as well as cytokine expression in the lung was similar between allergen-challenged mice irrespective of prior exposure to RSV. These results are not in accordance with previous studies of RSV-enhanced allergic airway inflammation, which attributed enhanced pathology to increased inflammation [6]. However, the reason for this difference might reflect the acute protocols used previously vs. our chronic allergic challenge protocol, as well as the timing of infection in our study.
The primary aim of this study was to investigate the effect of RSV infection on airway remodelling after prolonged allergen challenge. OVA/OVA mice at day 55 of allergen challenge showed signs of airway remodelling, as we have described previously [21]. Similar pathological features were observed in RSV-infected OVA/OVA mice with no enhancement in severity of airway remodelling being observed, probably due to the fact that this model of chronic allergen challenge already induced robust changes in tissue pathology. These results are not in accordance with recent work by Becnel et al. [26], who reported that RSV infection enhanced severity of airway remodelling in a murine model of allergen-induced airway inflammation. However, several key differences in their protocol might account for these discrepancies. RSV was given on day 1 of systemic allergen sensitization phase, whereas mice in our study were allowed to recover from RSV infection before undergoing sensitization, in an attempt to replicate the human situation where RSV infection in childhood precedes encounter with allergen. In addition, mice given RSV at the peak of allergen-induced allergic inflammation were killed during the peak of RSV-induced inflammation [26], whereas mice in our study further underwent prolonged allergen challenge. Moreover, the Becnel study did not include mice that had not been sensitized to OVA systemically, whereas we found that RSV infection did provoke remodelling in this group of mice. Alum-RSV/OVA and RSV-alum/OVA groups developed significantly enhanced collagen around the airways in contrast to uninfected alum/OVA controls.
AHR is one of the characteristic features of asthma. In mouse models of OVA-induced airway inflammation, AHR to bronchoconstrictors is only observed in animals that have been systemically sensitized to OVA before airway allergen exposure. Mice subjected to repeated OVA aerosol challenges only fail to develop an asthmatic phenotype accompanied with AHR [21, 27]. In the current study, we found that prior RSV infection had no effect on the development of AHR (data not shown). RSV infection before sensitization and challenge did not change the responses to methacholine. The induction of airway remodelling in alum/RSV-OVA mice was not accompanied by the induction of AHR. The association between airway remodelling and AHR is ambiguous with many in vivo mouse studies showing that changes in lung function can be uncoupled from airway structural cells. For example, mice genetically deficient in eosinophils are protected from development of airway remodelling but not from developing AHR [28]. Similarly studies deleting mast cells, or blocking TGF-β affect remodelling but not airway function [29, 30].
In previous work, we reported that TGF-β1 plays a crucial role in airway remodelling. In fact, therapeutic blockade of TGF-β1 in our model results in reduction of airway remodelling [30]. However, TGF-β1 did not seem to be involved in the fibrosis and remodelling observed in alum/RSV-OVA and RSV/alum-OVA mice, as active TGF-β1 levels assessed by ELISA in lung tissue were not elevated at any time-point studied. Other pro-fibrotic factors play a role in airway remodelling, as suggested by the fact that lung fibrosis does not always correlate with a high level of lung TGF-β1 [21]. Other candidates for involvement in airway remodelling include FGF-2, which is secreted by lung epithelial cells at an increased rate in asthmatic subjects compared with healthy individuals [31, 32]. In a model of allergen-induced airway inflammation and remodelling in infant rhesus monkeys, FGF-2 is implicated in remodelling of the basement membrane zone in animals exposed to allergen [33]. We selected FGF-2 as a likely mediator for the RSV/OVA-induced remodelling occurring in non-sensitized mice based on a report by Dosanjh et al. [24] who found that RSV infection increases FGF-2 secretion by human epithelial cells in vitro. In our model, FGF-2 expression was increased in OVA-sensitized and challenged mice compared with controls. RSV infection induced a further increase of FGF-2 secretion in OVA/OVA mice, but most importantly induced de novo secretion of FGF-2 in alum/OVA mice and controls only infected with RSV. Structural changes persisted long after resolution of RSV infection. We therefore postulate that RSV infection up-regulates FGF-2 in the epithelium of non-sensitized alum mice and primes them for development of remodelling after subsequent airway challenge. Interestingly, we found that there was no difference in levels of soluble FGF-2 by ELISA. It is conceivable that the differences we are detecting in pathology are due to increases of FGF-2 production within local microenvironments in the lung, even though the overall absolute amounts in the lung tissue are unchanged. Differences within these tightly packed cellular microenvironments might reflect changes in pathology, even though absolute values are unchanged by ELISA. Alternatively, it may not be possible to detect levels of FGF-2 protein, as it is known that FGF-2 is stored within the basement membrane zone of the airway epithelium through binding to perlecan, a heparin sulphate proteoglycan that is integral to the basement membrane zone [33]. In this latter study, FGF-2 was also found to be associated with remodelling of the lung in monkeys following allergen challenge.
Taken together our results suggest that RSV infection primes the lung epithelium to release pro-fibrotic factors upon allergen exposure irrespective of systemic sensitization. This suggests that in presence of infection, OVA is presented by mature APCs in the lung leading to local sensitization, and consequently to airway remodelling after prolonged allergen challenge. It has been proposed that activation of the endotoxin/TLR4 pathway simultaneous to exposure to OVA can compensate for an absence of systemic sensitization, and result in OVA-induced airway inflammation [34]. Monick et al. showed that one effect of RSV infection is the up-regulation of TLR4 and subsequent sensitization of epithelial cells to endotoxin [35]. Others have shown that activation of the lung epithelium via infection with adenoviral vectors expressing inflammatory cytokines also elicit an inflammatory response to inhaled OVA [36]. We postulate that in our model, infection by RSV of alum/OVA control mice primes the lung epithelium and renders these mice susceptible to develop allergen-induced airway remodelling upon inhaled allergen challenge only, in absence of systemic allergen sensitization.
In summary, here we show that RSV infection, whether occurring before or concomitantly to allergen aerosol exposure, provokes airway remodelling in animals that would otherwise not develop structural airway changes upon sole exposure to allergen via the airway. We postulate that RSV infection primes the lung epithelium to become susceptible to subsequent aerosol allergen challenge and therefore account for the inception of remodelling in absence of systemic allergen sensitization. Most clinical studies have been designed to ascertain the causal link between RSV infection in early childhood and the development and/or exacerbation of asthma later in life, and elucidate mechanisms by which RSV primes individuals to respond to otherwise innocuous allergens. In light of the work presented here, it would also be worthwhile looking in patients for evidence of an association between RSV infection and lung structural changes.
Acknowledgements
The authors would like to thank Lorraine Lawrence for technical assistance with histology. This work was supported by the Wellcome Trust (ref. no. 057704).
References
- 1.Openshaw PJ, Yamaguchi Y, Tregoning JS. Childhood infections, the developing immune system, and the origins of asthma. J Allergy Clin Immunol. 2004;114:1275–7. doi: 10.1016/j.jaci.2004.08.024. [DOI] [PubMed] [Google Scholar]
- 2.Openshaw PJ, Hewitt C. Protective and harmful effects of viral infections in childhood on wheezing disorders and asthma. Am J Respir Crit Care Med. 2000;162:S40–3. doi: 10.1164/ajrccm.162.supplement_1.maic-11. [DOI] [PubMed] [Google Scholar]
- 3.Lloyd CM, Gutierrez-Ramos JC. Animal models to study chemokine receptor function: in vivo mouse models of allergic airway inflammation. Methods Mol Biol. 2004;239:199–210. doi: 10.1385/1-59259-435-2:199. [DOI] [PubMed] [Google Scholar]
- 4.Openshaw PJ, Dean GS, Culley FJ. Links between respiratory syncytial virus bronchiolitis and childhood asthma: clinical and research approaches. Pediatr Infect Dis J. 2003;22:S58–64. doi: 10.1097/01.inf.0000053887.26571.eb. [DOI] [PubMed] [Google Scholar]
- 5.Openshaw PJ. Potential mechanisms causing delayed effects of respiratory syncytial virus infection. Am J Respir Crit Care Med. 2001;163:S10–3. doi: 10.1164/ajrccm.163.supplement_1.2011111. [DOI] [PubMed] [Google Scholar]
- 6.Schwarze J, Gelfand EW. Respiratory viral infections as promoters of allergic sensitization and asthma in animal models. Eur Respir J. 2002;19:341–9. doi: 10.1183/09031936.02.00254302. [DOI] [PubMed] [Google Scholar]
- 7.Alwan WH, Kozlowska WJ, Openshaw PJ. Distinct types of lung disease caused by functional subsets of antiviral T cells. J Exp Med. 1994;179:81–9. doi: 10.1084/jem.179.1.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.You D, Becnel D, Wang K, Ripple M, Daly M, Cormier SA. Exposure of neonates to respiratory syncytial virus is critical in determining subsequent airway response in adults. Respir Res. 2006;7:107. doi: 10.1186/1465-9921-7-107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schwarze J, Cieslewicz G, Joetham A, et al. Critical roles for interleukin-4 and interleukin-5 during respiratory syncytial virus infection in the development of airway hyperresponsiveness after airway sensitization. Am J Respir Crit Care Med. 2000;162:380–6. doi: 10.1164/ajrccm.162.2.9903057. [DOI] [PubMed] [Google Scholar]
- 10.Lukacs NW, Tekkanat KK, Berlin A, et al. Respiratory syncytial virus predisposes mice to augmented allergic airway responses via IL-13-mediated mechanisms. J Immunol. 2001;167:1060–5. doi: 10.4049/jimmunol.167.2.1060. [DOI] [PubMed] [Google Scholar]
- 11.Tang YW, Graham BS. Anti-IL-4 treatment at immunization modulates cytokine expression, reduces illness, and increases cytotoxic T lymphocyte activity in mice challenged with respiratory syncytial virus. J Clin Invest. 1994;94:1953–8. doi: 10.1172/JCI117546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.John AE, Berlin AA, Lukacs NW. Respiratory syncytial virus-induced CCL5/RANTES contributes to exacerbation of allergic airway inflammation. Eur J Immunol. 2003;33:1677–85. doi: 10.1002/eji.200323930. [DOI] [PubMed] [Google Scholar]
- 13.John AE, Gerard CJ, Schaller M, et al. Respiratory syncytial virus-induced exaggeration of allergic airway disease is dependent upon CCR1-associated immune responses. Eur J Immunol. 2005;35:108–16. doi: 10.1002/eji.200425439. [DOI] [PubMed] [Google Scholar]
- 14.Schwarze J, Cieslewicz G, Joetham A, Ikemura T, Hamelmann E, Gelfand EW. CD8 T cells are essential in the development of respiratory syncytial virus-induced lung eosinophilia and airway hyperresponsiveness. J Immunol. 1999;162:4207–11. [PubMed] [Google Scholar]
- 15.Peebles RS, Jr, Hashimoto K, Collins RD, et al. Immune interaction between respiratory syncytial virus infection and allergensensitization critically depends on timing of challenges. J Infect Dis. 2001;184:1374–9. doi: 10.1086/324429. [DOI] [PubMed] [Google Scholar]
- 16.Walzl G, Tafuro S, Moss P, Openshaw PJ, Hussell T. Influenza virus lung infection protects from respiratory syncytial virus-induced immunopathology. J Exp Med. 2000;192:1317–26. doi: 10.1084/jem.192.9.1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Makela MJ, Tripp R, Dakhama A, et al. Prior airway exposure to allergen increases virus-induced airway hyperresponsiveness. J Allergy Clin Immunol. 2003;112:861–9. doi: 10.1016/s0091-6749(03)02020-7. [DOI] [PubMed] [Google Scholar]
- 18.Chiappara G, Gagliardo R, Siena A, et al. Airway remodelling in the pathogenesis of asthma. Curr Opin Allergy Clin Immunol. 2001;1:85–93. doi: 10.1097/01.all.0000010990.97765.a1. [DOI] [PubMed] [Google Scholar]
- 19.Bumbacea D, Campbell D, Nguyen L, et al. Parameters associated with persistent airflow obstruction in chronic severe asthma. Eur Respir J. 2004;24:122–8. doi: 10.1183/09031936.04.00077803. [DOI] [PubMed] [Google Scholar]
- 20.Rasmussen F, Taylor DR, Flannery EM, et al. Risk factors for airway remodeling in asthma manifested by a low postbronchodilator FEV1/vital capacity ratio: a longitudinal population study from childhood to adulthood. Am J Respir Crit Care Med. 2002;165:1480–8. doi: 10.1164/rccm.2108009. [DOI] [PubMed] [Google Scholar]
- 21.McMillan SJ, Lloyd CM. Prolonged allergen challenge in mice leads to persistent airway remodelling. Clin Exp Allergy. 2004;34:497–507. doi: 10.1111/j.1365-2222.2004.01895.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Flood-Page P, Menzies-Gow A, Phipps S, et al. Anti-IL-5 treatment reduces deposition of ECM proteins in the bronchial subepithelial basement membrane of mild atopic asthmatics. J Clin Invest. 2003;112:1029–36. doi: 10.1172/JCI17974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McMillan SJ, Xanthou G, Lloyd CM. Manipulation of allergen-induced airway remodeling by treatment with anti-TGF-beta antibody: effect on the Smad signaling pathway. J Immunol. 2005;174:5774–80. doi: 10.4049/jimmunol.174.9.5774. [DOI] [PubMed] [Google Scholar]
- 24.Dosanjh A, Rednam S, Martin M. Respiratory syncytial virus augments production of fibroblast growth factor basic in vitro: implications for a possible mechanism of prolonged wheezing after infection. Pediatr Allergy Immunol. 2003;14:437–40. doi: 10.1046/j.0905-6157.2003.00090.x. [DOI] [PubMed] [Google Scholar]
- 25.Peebles RS, Jr, Hashimoto K, Graham BS. The complex relationship between respiratory syncytial virus and allergy in lung disease. Viral Immunol. 2003;16:25–34. doi: 10.1089/088282403763635429. [DOI] [PubMed] [Google Scholar]
- 26.Becnel D, You D, Erskin J, Dimina DM, Cormier SA. A role for airway remodeling during respiratory syncytial virus infection. Respir Res. 2005;6:122. doi: 10.1186/1465-9921-6-122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lloyd CM, Gonzalo JA, Coyle AJ, Gutierrez-Ramos JC. Mouse models of allergic airway disease. Adv Immunol. 2001;77:263–95. doi: 10.1016/s0065-2776(01)77019-8. [DOI] [PubMed] [Google Scholar]
- 28.Humbles AA, Lloyd CM, McMillan SJ, et al. A critical role for eosinophils in allergic airways remodeling. Science. 2004;305:1776–9. doi: 10.1126/science.1100283. [DOI] [PubMed] [Google Scholar]
- 29.Masuda T, Tanaka H, Komai M, et al. Mast cells play a partial role in allergen-induced subepithelial fibrosis in a murine model of allergic asthma. Clin Exp Allergy. 2003;33:546–9. doi: 10.1046/j.1365-2222.2003.01588.x. [DOI] [PubMed] [Google Scholar]
- 30.McMillan SJ, Xanthou G, Lloyd CM. Manipulation of allergen-induced airway remodeling by treatment with anti-TGF-beta antibody: effect on the Smad signaling pathway. J Immunol. 2005;174:5774–80. doi: 10.4049/jimmunol.174.9.5774. [DOI] [PubMed] [Google Scholar]
- 31.Redington AE, Roche WR, Madden J, et al. Basic fibroblast growth factor in asthma: measurement in bronchoalveolar lavage fluid basally and following allergen challenge. J Allergy Clin Immunol. 2001;107:384–7. doi: 10.1067/mai.2001.112268. [DOI] [PubMed] [Google Scholar]
- 32.Shute JK, Solic N, Shimizu J, McConnell W, Redington AE, Howarth PH. Epithelial expression and release of FGF-2 from heparan sulphate binding sites in bronchial tissue in asthma. Thorax. 2004;59:557–62. doi: 10.1136/thx.2002.002626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Evans MJ, Van Winkle LS, Fanucchi MV, et al. Fibroblast growth factor-2 in remodeling of the developing basement membrane zone in the trachea of infant rhesus monkeys sensitized and challenged with allergen. Lab Invest. 2002;82:1747–54. doi: 10.1097/01.lab.0000043911.94235.f3. [DOI] [PubMed] [Google Scholar]
- 34.Eisenbarth SC, Piggott DA, Huleatt JW, Visintin I, Herrick CA, Bottomly K. Lipopolysaccharide-enhanced, toll-like receptor 4-dependent T helper cell type 2 responses to inhaled antigen. J Exp Med. 2002;196:1645–51. doi: 10.1084/jem.20021340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Monick MM, Yarovinsky TO, Powers LS, et al. Respiratory syncytial virus up-regulates TLR4 and sensitizes airway epithelial cells to endotoxin. J Biol Chem. 2003;278:53035–44. doi: 10.1074/jbc.M308093200. [DOI] [PubMed] [Google Scholar]
- 36.Stampfli MR, Wiley RE, Neigh GS, et al. GM-CSF transgene expression in the airway allows aerosolized ovalbumin to induce allergic sensitization in mice. J Clin Invest. 1998;102:1704–14. doi: 10.1172/JCI4160. [DOI] [PMC free article] [PubMed] [Google Scholar]