

Abstract
Objective:
To examine changes in levels of plasma amyloid-β (Aβ) peptides, Aβ42 and Aβ40, in relation to onset of Alzheimer disease (AD) in adults with Down syndrome (DS).
Methods:
Plasma Aβ42 and Aβ40 were measured at initial examination and at follow-up in a community-based cohort of 225 adults with DS who did not have dementia at baseline and were assessed for cognitive/functional abilities and health status and followed at 14- to 20-month intervals. We used Cox proportional hazards modeling to estimate the cumulative incidence of AD by Aβ peptide change group (increasing, no change, or decreasing), adjusting for covariates.
Results:
Sixty-one (27.1%) of the participants developed AD. At follow-up, a decrease in Aβ42 levels, a decrease in the Aβ42/Aβ40 ratio, and an increase in Aβ40 levels were related to conversion to AD. Compared with the group with increasing levels of Aβ42, the likelihood of developing AD was 5 times higher for those whose plasma Aβ42 levels decreased over follow-up (hazard ratio [HR] = 4.9, 95% confidence interval [CI] 2.1–11.4). Decreasing Aβ42/Aβ40 was also strongly related to AD risk (HR = 4.9, 95% CI 1.8–13.2), while decreasing Aβ40 was associated with lower risk (HR = 0.4, 95% CI 0.2–0.9).
Conclusions:
Among adults with DS, decreasing levels of plasma Aβ42, a decline in the Aβ42/Aβ40 ratio, or increasing levels of Aβ40 may be sensitive indicators of conversion to AD, possibly reflecting compartmentalization of Aβ peptides in the brain.
Amyloid-β (Aβ) plays a critical role in the development of Alzheimer disease (AD). Aβ peptides Aβ40 and Aβ42 are the 2 major species generated by sequential proteolytic cleavage by β and δ secretases of the amyloid precursor protein (APP).1 Brain levels of Aβ42 increase early in the development of dementia,2 and studies of Aβ peptides in CSF have consistently shown that low levels of Aβ42 and high concentrations of tau protein are associated with AD and predict incipient AD.3–5 Reduced CSF Aβ42 is also associated with high brain amyloid load as imaged with Pittsburgh Compound B.5
Results from studies examining the relation of plasma concentrations of Aβ42 and Aβ40 to risk of AD have been less consistent, and the usefulness of plasma Aβ as a risk biomarker remains controversial.6–8 Several studies have reported that either elevated or decreasing plasma Aβ42 or Aβ42/Aβ40 ratio levels in older adults without dementia are associated with increased risk for mild cognitive impairment (MCI), cognitive decline, and AD,6,7,9–14 but others have not found this relationship.15,16 Still others have found that risk of AD is associated with low plasma levels of Aβ42, Aβ40, or the Aβ42/Aβ40 ratio.8,17–19 Although differences in Aβ assays, in study design and in populations examined are likely to influence findings, these inconsistencies may also be related to the timing of Aβ measures in relation to the development of AD, given the dynamic rather than static nature of the underlying neuropathology.20
Individuals with Down syndrome (DS) have increased levels of Aβ40 and Aβ42 peptides in plasma together with increased risk for AD neuropathology and clinical dementia.21–25 The nearly universal presence of AD neuropathology as well as the increased levels of Aβ peptides have been attributed, at least in part, to the triplication and overexpression of the gene for APP, located on chromosome 21,26 but large individual differences in plasma Aβ levels and the wide range of age at onset of AD within this population suggest a more complex underlying mechanism. In previous studies of this population, we found that high initial levels of plasma Aβ42, but not Aβ40, predicted onset of dementia over a 4-year follow-up period.11 The aim of the present study was to expand our previous work by examining the relation of change in levels of Aβ40, Aβ42, and the Aβ42/Aβ40 ratio to onset of dementia with longitudinal follow-up and repeated measures of Aβ peptides.
METHODS
Study population.
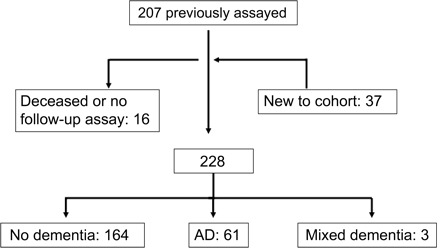
The sample included 225 members of a community-based sample of adults with confirmed DS, without dementia at their initial examination and with multiple measurements of Aβ peptides. Of these, 191 were among the 207 adults with DS in our previous examination of the relation of initial levels of Aβ peptides to dementia risk and 37 were new to the cohort11 (figure 1). All individuals were 40 years of age or older (range 40–78) and resided in New York, Connecticut, New Jersey, or eastern Pennsylvania. Participants were recruited with the help of state and voluntary service provider agencies and were eligible for inclusion in the present study if 1) a family member or correspondent provided informed consent, 2) he or she either provided consent or signed a form acknowledging their assent and willingness to participate, and 3) he or she was willing and able to provide blood samples and had been evaluated at least twice.
Figure 1. Description of sample.
Standard protocol approvals, registrations, and patient consents.
Recruitment, informed consent, and study procedures were approved by the Institutional Review Boards of the New York State Institute for Basic Research in Developmental Disabilities, Columbia University Medical Center, and The Johns Hopkins University School of Medicine.
Clinical assessment.
Assessments included evaluations of cognition, functional and vocational abilities, behavioral/psychiatric conditions, and health status. Assessments were repeated at 14- to 20-month intervals. Cognitive function was evaluated with a test battery designed for use with individuals varying widely in their premorbid levels of intellectual functioning, as previously described.27 Structured interviews were conducted with caregivers to collect information on adaptive behavior and medical history. Past and current medical records were reviewed for all participants. Participants showing cognitive and functional declines indicative of dementia were evaluated by the study neurologist.
Classification of dementia.
To determine the occurrence of dementia and dementia subtypes in participants, data from all available sources were reviewed during a consensus conference. Following recommendations of the AAMR-IASSID Working Group for the Establishment of Criteria for the Diagnosis of Dementia in Individuals with Developmental Disability,28 participants were classified into 2 groups: 1) dementia, if there was a history of progressive memory loss, disorientation, and functional decline over a period of at least 1 year and if there were no other medical or psychiatric conditions that might result in or mimic dementia present (e.g., untreated hypothyroidism, stroke) (n = 64); and 2) without dementia, if they were without cognitive or functional decline or if they exhibited less substantial cognitive and functional declines that did not meet criteria for dementia (n = 164). All participants classified as having dementia showed strong and consistent declines in cognition and function over the course of follow-up.
AD was the predominant form of dementia, accounting for 95% of the cases. Participants with evidence of dementia with stroke during the neurologic evaluations or from clinical histories were excluded (n = 3), leaving 61 participants with AD for analysis (figure 1). Age at meeting consensus conference criteria for dementia was used to estimate age at onset of AD, recognizing that it is difficult to document the onset of initial symptoms in this population with precision.
Plasma Aβ42 and Aβ40.
Participants were asked to provide a 10-mL venous nonfasting blood sample (K3 EDTA lavender-top tube) at each assessment cycle, although it was not always possible to obtain a blood sample at every cycle. Plasma levels of Aβ42 and Aβ40 were measured blind to dementia status using a combination of monoclonal antibody 6E10 (specific to an epitope present on 1–16 amino acid residues of Aβ) and rabbit antisera R165 (vs Aβ42) and R162 (vs Aβ40) in a double antibody sandwich ELISA as described previously.7,22 The detection limit for these assays was 5 pg/mL for Aβ40 and 10 pg/mL for Aβ42. Aβ40 and Aβ42 levels from each sample were measured twice using separate aliquots. Reliability between measurements was substantial for both peptides (r = 0.93 and r = 0.97 for Aβ40 and Aβ42, p < 0.001), and we used the mean of the 2 measurements in statistical analyses.
APOE genotypes.
APOE genotyping employed standard PCR–restriction fragment length polymorphism methods using HhaI (CfoI) digestion of an APOE genomic PCR product spanning the polymorphic (cys/arg) sites at codons 112 and 158. Acrylamide gel electrophoresis was used to assess and document the restriction fragment sizes.29 Participants were classified according to the presence or absence of an APOE ϵ4 allele.
Covariates.
Level of intellectual disability was classified into 2 groups based on IQ scores obtained before onset of dementia: mild/moderate (IQ 35–70) and severe/profound (IQ <35). Ethnicity was coded as white or other. We examined statin use, body mass index, and obesity as potential confounders but found that they were not related to Aβ levels nor to risk of AD and they were therefore not considered further in analyses.
Statistical analyses.
In preliminary analyses, we used χ2 tests for categorical variables and Student t tests and analyses of variance to compare demographic characteristics and Aβ peptide levels by dementia status. For inferential analyses, we examined the relation of change in Aβ peptides to risk of dementia. Among participants who developed AD over the follow-up period, change was calculated as the difference between the sample associated with the assessment interval at which AD was first diagnosed (incidence visit) and the baseline visit. For participants who remained without dementia throughout the follow-up period, change was calculated as the difference between Aβ peptide levels at the last follow-up and the initial measurement. In preliminary analyses, we examined change in Aβ as a continuous variable and found an inverse relation between change in Aβ42 and risk for AD (β = −0.032, p = 0.012), an inverse relation between change in the Aβ42/Aβ40 ratio (β = −1.6, p = 0.03), but no relation between change in Aβ40 and risk for AD (β = 0.001, p = 0.549). We then categorized change in Aβ by change groups to detect potential nonlinear threshold effects. We classified Aβ peptide change into 3 groups based on 0.5 standard deviations of change: 1) no change (no change ± 0.5 SD change); 2) increasing (>0.5 S.D change); and 3) decreasing (<0.5 SD change). We used Cox proportional hazards models to estimate the cumulative incidence of dementia and hazard ratio (HR) of dementia by change in Aβ peptide group, with increasing levels (>0.5 SD change) as the reference group. The time to event variable was time from initial Aβ peptide measurement to onset of dementia for incident cases and time from the initial Aβ peptide measurement to last assessment for those who remained without dementia. Because levels of Aβ42 and Aβ40 were correlated, we used models containing measures of both peptides in all analyses to determine if independent relationships with dementia status were present. All models were adjusted for sex, ethnicity, level of intellectual disability, and the presence of the APOE ϵ4 allele. (These analyses were repeated using change criteria of 0.25 SD and 0.75 SD. Findings were consistent in all 3 sets of analyses, and only the 0.5 SD results are presented here.)
RESULTS
Relation of Aβ peptides to demographic characteristics.
Table 1presents demographic characteristics and Aβ peptide levels by dementia status. The mean length of follow-up was 4.1 (±1.9) years. Over the course of follow-up, 61 participants (27.1%) developed dementia. The mean time from baseline to onset of dementia in cases was 2.8 (±1.6) years. Levels of Aβ40, but not Aβ42, increased modestly with age (r = 0.13, p = 0.07). Participants who subsequently developed dementia were older at baseline than those who remained without dementia throughout the follow-up period (53.7 vs 50.3 years, p = 0.001), but did not differ by sex, ethnicity, or level of intellectual disability. The frequency of the APOE ϵ4 allele was greater in those who developed dementia (26.2% vs 18.3%), but this difference failed to reach significance (table 1).
Table 1. Demographic characteristics.

a p < 0.001.
b p < 0.01.
c p < 0.05.
Relation of change in Aβ peptide levels to incidence of AD.
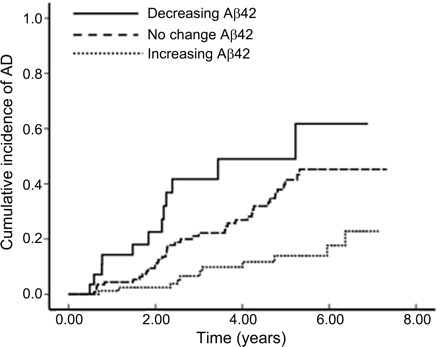
Increases in plasma Aβ40 peptides, decreases in Aβ42 peptides, and decreases in the Aβ42/Aβ40 ratio were significantly related to conversion to dementia over the follow-up period (table 2). Compared with those with increasing plasma Aβ40 levels, those with decreasing levels were 60% less likely to develop dementia over the follow-up period (HR = 0.4, 95% CI 0.2–0.9) (table 2). Compared with those with increasing levels of plasma Aβ42, those with no change in Aβ42 levels were 2.5 times as likely to develop AD (HR = 2.6, 95% CI 1.3–5.2), while those with decreasing levels of plasma Aβ42 were 5 times as likely to develop AD (HR = 4.9, 95% CI 2.1–11.4) (table 2) (figure 2). No change and decrease in the ratio of Aβ42/Aβ40 were also strongly related to the development of dementia. Compared with those with an increasing Aβ42/Aβ40 ratio over the follow-up period, those whose Aβ42/Aβ40 ratio did not change and those with a decreasing Aβ42/Aβ40 ratio were 4 to 5 times as likely to have developed AD (HR = 3.9, 95% CI 1.8–8.4 for those in the no change group; HR = 4.9.7, 95% CI 1.8–13.2 for those in the decreasing group) (table 2).
Table 2. Relation of change in Aβ peptide levels to incidence of Alzheimer disease.

a Cox proportional hazard model: hazard ratio (95% confidence interval), adjusted for sex, level of intellectual disability, ethnicity, and presence of the APOE ϵ4 allele.
b p < 0.05.
c p < 0.01.
Figure 2. Cumulative incidence of dementia by Aβ42 change group.
AD = Alzheimer disease.
DISCUSSION
In a previous study, we found that adults with DS with high initial levels of plasma Aβ42, but not Aβ40 or the ratio of Aβ42/Aβ40, had a 2.5-fold increased risk of incident dementia.11 The current study extends this analysis in the same study group to show that declining levels of Aβ42 and of the Aβ42/Aβ40 ratio are also associated with increased risk of incident dementia. Among adults with DS, the association of elevated Aβ42 with increased risk of dementia is consistent with studies showing elevated plasma levels of Aβ42 in other high-risk populations. Studies of families with AD mutations have shown elevated plasma Aβ42 levels in both symptomatic and nonsymptomatic individuals.30,31 Plasma Aβ42 and Aβ42/Aβ40 ratio levels were elevated in unaffected familial AD mutation carriers compared with unaffected individuals with familial AD without mutations. However, Aβ42 levels were lower in mutation carriers with incipient AD characterized as having a Clinical Dementia Rating = 0.5, supporting the hypothesis that Aβ42 decreases prior to overt disease.30 First-degree relatives of patients with late-onset AD without known mutations or genetic variants have also been found to have increased plasma Aβ42.32 Among elderly individuals without dementia, plasma levels of Aβ42 were also increased in women with MCI, who are at high risk of progression to AD,9 and high baseline levels and greater reductions in plasma levels of Aβ42 during follow-up have been associated with greater cognitive decline.10 In contrast, a prospective study examining plasma Aβ42 showed no difference in patients with MCI who progressed to AD.16 Other studies have found that low plasma Aβ42 or a low ratio of plasma Aβ42/Aβ40 was associated with more rapid cognitive decline or with the presence of frank disease.17,18,33
These inconsistencies may be related, at least in part, to the timing of Aβ measures in relation to disease onset and progression. If conversion to AD is associated with a decline in plasma Aβ42 or in the Aβ42/Aβ40 ratio, then plasma Aβ levels may already be low in those with MCI or incipient AD. Thus, given the long preclinical period for AD, it is important to control for stage of disease, and this can best be done in prospective, longitudinal studies with repeated peptide measurements in elders who are free of cognitive impairment at their initial assessment. In the current study, the development of dementia in adults with DS who were without dementia at baseline was strongly related to decreases in plasma Aβ42, decreases in the Aβ42/Aβ40 ratio, and increases in plasma Aβ40, suggesting that change may be a more sensitive biomarker of risk than level. Compared with individuals with increasing levels of Aβ peptides, incident dementia was 2.5 to 5 times more likely among those whose plasma levels of Aβ42 or the Aβ42/Aβ40 ratio did not change or declined by more than 0.5 standard deviations. These findings parallel those from a multiethnic cohort from Northern Manhattan.12 In that study, decline in plasma Aβ42 levels and in the Aβ42/Aβ40 ratio over a 4.5-year period was associated with a 3-fold increase in the likelihood of AD. In another study, higher initial plasma Aβ42 levels and greater reductions in Aβ42 levels were associated with more rapid cognitive decline in healthy elders without dementia.10 Similarly, an increase in the plasma Aβ40/Aβ42 ratio (comparable to a decreased Aβ42/Aβ40 ratio) measured at midlife predicted greater decline in a global measure of cognition and in cognitive status as measured by the Telephone Interview for Cognitive Status.14 These findings suggest that decline in plasma Aβ42 or the Aβ42/Aβ40 ratio might serve as a sensitive biomarker for incipient AD and may reflect aggregation of Aβ42 in senile plaques.7,12 Our results are also consistent with findings from longitudinal assessments of Aβ peptides in CSF, where decreases in Aβ42 are correlated with cognitive decline in episodic memory and cognitive speed.34 Prior studies, however, have not found significant correlations between levels of CSF and matched plasma samples of Aβ42 and Aβ40,35,36 suggesting that Aβ levels in CSF and plasma are not related in a simple way and that the source of Aβ synthesis in these 2 compartments is different.36 It has been suggested that some Aβ in brain parenchyma is eliminated through the vascular spaces, contributing to Aβ levels in blood,37 but other studies suggest that platelets are a major source of Aβ in plasma unrelated to CNS processes.38 Thus, there is as yet no consensus regarding how plasma Aβ is related to progressive CNS amyloid pathology. In most previous studies, the relationship between CSF and plasma Aβ levels was examined in cross-sectional samples and compared patients with MCI or established AD with controls without dementia.35,36 Future longitudinal studies comparing sequential measures of matched plasma and CSF Aβ levels may be an effective approach to address this issue.
In the current study, increasing levels of plasma Aβ40 were associated with increased risk of AD. In cross-sectional studies, increased levels of Aβ40, along with increased levels of Aβ42, have been found in plasma of adults with DS.21–24 We are not sure what role increasing Aβ40 may play in the pathogenesis of AD in DS. We speculate that increasing levels among adults with DS without dementia, who already have very high levels of Aβ40, may be a marker of rate of aging.
These new findings add to previous evidence indicating that change in levels of Aβ peptide in both blood and CSF are sensitive to progression of neuropathology in AD, both in the general population and among adults with DS. Our study is limited by a relatively small sample size with small numbers in high-risk groups. In addition, individual differences in Aβ peptide levels, as well as changes in these levels, are imperfectly related to risk of dementia, and the determinants of individual differences in both initial levels and the trajectory of change in Aβ need to be explored further before these assays can be used to inform diagnosis or predict individual risk. Differences in the formation of oligomeric forms of Aβ, in levels of autoantibodies to Aβ, or in other protective factors may modify the effect risk biomarkers. Thus, plasma Aβ levels are not useful for predicting future onset or to confirm diagnoses for specific individuals with DS at the present time.
Footnotes
- Aβ
- amyloid-β
- AD
- Alzheimer disease
- APP
- amyloid precursor protein
- CI
- confidence interval
- DS
- Down syndrome
- HR
- hazard ratio
- MCI
- mild cognitive impairment
AUTHOR CONTRIBUTIONS
Statistical analysis was conducted by Dr. Schupf, Dr. Zigman, and Dr. Tang.
DISCLOSURE
Dr. Schupf serves as a consultant to Janssen AI and receives research support from the NIH (R01AG014673 [PI], P01HD035897 [coinvestigator], P01AG07232 [coinvestigator], R01AG028786 [coinvestigator], U01 AG023749 [coinvestigator], P50 AG08702 [coinvestigator], R01AG007370 [coinvestigator], R21CA125461 [coinvestigator], R01AG036040 [coinvestigator], and AG034189 [coinvestigator]) and the Alzheimer's Association. Dr. Zigman serves as an Advisor to the DSM-V Neurodevelopment Disorders Work Group and receives research support from the NIH (P01HD35897 [coinvestigator] and R01AG014673 [coinvestigator]). Dr. Tang receives research support from the NIH (R01AG007370 [coinvestigator], R01AG016206 [coinvestigator], R01NS036630 [coinvestigator], P50AG008702 [coinvestigator], R01AG028506 [coinvestigator], and P01AG007232 [coinvestigator]). D. Pang reports no disclosures. Dr. Mayeux serves on scientific advisory boards for PsychoGenics Inc. and Quintiles and receives research support from the NIH (P01AG007232 [PI], P50AG008702 [PI], R37AG015473 [PI], 5R01AG014673 [coinvestigator], R01 AG028786 [coinvestigator], U01AG023749 [coinvestigator], U01AG023749-05S2 [coinvestigator], U24AG026395 [PI], and R01AG036040 [PI]). Dr. Mehta receives research support from the NIH (P01AG07232 [coinvestigator] and R01AG022374 [coinvestigator]). Dr. Silverman serves on the editorial board of the American Journal on Intellectual and Developmental Disabilities; serves as a consultant for Johnson & Johnson; and receives research support from the NIH (PO1HD035897 [PI], P30HD024061 [coinvestigator], UL1RR025005 [coinvestigator], and RO1HD065160 [coinvestigator]).
REFERENCES
- 1. Selkoe DJ. Normal and abnormal biology of the beta-amyloid precursor protein. Annu Rev Neurosci 1994;17:489–517 [DOI] [PubMed] [Google Scholar]
- 2. Naslund J, Haroutonian V, Mohs R, et al. Correlation between elevated levels of amyloid B-peptide in the brain and cognitive decline. JAMA 2000;283:1571–1577 [DOI] [PubMed] [Google Scholar]
- 3. Blennow K, Vanmechelen E. CSF markers for pathogenic processes in Alzheimer's disease: diagnostic implications and use in clinical neurochemistry. Brain Res Bull 2003;61:235–242 [DOI] [PubMed] [Google Scholar]
- 4. Hansson O, Zetterberg H, Buchhave P, et al. Prediction of Alzheimer's disease using the CSF Abeta42/Abeta40 ratio in patients with mild cognitive impairment. Dement Geriatr Cogn Disord 2007;23:316–320 [DOI] [PubMed] [Google Scholar]
- 5. Fagan AM, Roe CM, Xiong C, Mintun MA, Morris JC, Holtzman DM. Cerebrospinal fluid tau/beta-amyloid(42) ratio as a prediction of cognitive decline in nondemented older adults. Arch Neurol 2007;64:343–349 [DOI] [PubMed] [Google Scholar]
- 6. Mayeux R, Tang MX, Jacobs DM, et al. Plasma amyloid beta-peptide 1–42 and incipient Alzheimer's disease. Ann Neurol 1999;46:412–416 [DOI] [PubMed] [Google Scholar]
- 7. Mayeux R, Honig LS, Tang MX, et al. Plasma A[beta]40 and A[beta]42 and Alzheimer's disease: relation to age, mortality, and risk. Neurology 2003;61:1185–1190 [DOI] [PubMed] [Google Scholar]
- 8. van Oijen M, Hofman A, Soares HD, Koudstaal PJ, Breteler MM. Plasma Abeta(1–40) and Abeta(1–42) and the risk of dementia: a prospective case-cohort study. Lancet Neurol 2006;5:655–660 [DOI] [PubMed] [Google Scholar]
- 9. Assini A, Cammarata S, Vitali A, et al. Plasma levels of amyloid beta-protein 42 are increased in women with mild cognitive impairment. Neurology 2004;63:828–831 [DOI] [PubMed] [Google Scholar]
- 10. Pomara N, Willoughby LM, Sidtis JJ, Mehta PD. Selective reductions in plasma Abeta 1–42 in healthy elderly subjects during longitudinal follow-up: a preliminary report. Am J Geriatr Psychiatry 2005;13:914–917 [DOI] [PubMed] [Google Scholar]
- 11. Schupf N, Patel B, Pang D, et al. Elevated plasma beta-amyloid peptide Abeta(42) levels, incident dementia, and mortality in Down syndrome. Arch Neurol 2007;64:1007–1013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Schupf N, Tang MX, Fukuyama H, et al. Peripheral Abeta subspecies as risk biomarkers of Alzheimer's disease. Proc Natl Acad Sci USA 2008;105:14052–14057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sobow T, Flirski M, Kloszewska I, Liberski PP. Plasma levels of alpha beta peptides are altered in amnestic mild cognitive impairment but not in sporadic Alzheimer's disease. Acta Neurobiol Exp 2005;65:117–124 [DOI] [PubMed] [Google Scholar]
- 14. Okereke OI, Xia W, Selkoe DJ, Grodstein F. Ten-year change in plasma amyloid beta levels and late-life cognitive decline. Arch Neurol 2009;66:1247–1253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Fukumoto H, Tennis M, Locascio JJ, Hyman BT, Growdon JH, Irizarry MC. Age but not diagnosis is the main predictor of plasma amyloid beta-protein levels. Arch Neurol 2003;60:958–964 [DOI] [PubMed] [Google Scholar]
- 16. Hansson O, Zetterberg H, Vanmechelen E, et al. Evaluation of plasma Abeta(40) and Abeta(42) as predictors of conversion to Alzheimer's disease in patients with mild cognitive impairment. Neurobiol Aging 2010;31:357–367 [DOI] [PubMed] [Google Scholar]
- 17. Graff-Radford NR, Crook JE, Lucas J, et al. Association of low plasma Abeta42/Abeta40 ratios with increased imminent risk for mild cognitive impairment and Alzheimer disease. Arch Neurol 2007;64:354–362 [DOI] [PubMed] [Google Scholar]
- 18. Lambert JC, Schraen-Maschke S, Richard F, et al. Association of plasma amyloid beta with risk of dementia: the prospective Three-City Study. Neurology 2009;73:847–853 [DOI] [PubMed] [Google Scholar]
- 19. Sundelof J, Giedraitis V, Irizarry MC, et al. Plasma beta amyloid and the risk of Alzheimer disease and dementia in elderly men: a prospective, population-based cohort study. Arch Neurol 2008;65:256–263 [DOI] [PubMed] [Google Scholar]
- 20. Jack CR, Jr, Lowe VJ, Weigand SD, et al. Serial PIB and MRI in normal, mild cognitive impairment and Alzheimer's disease: implications for sequence of pathological events in Alzheimer's disease. Brain 2009;132:1355–1365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Matsubara E, Ghiso J, Frangione B, et al. Lipoprotein-free amyloidogenic peptides in plasma are elevated in patients with sporadic Alzheimer's disease and Down's syndrome. Ann Neurol 1999;45:537–541 [PubMed] [Google Scholar]
- 22. Mehta PD, Dalton AJ, Mehta SP, Kim KS, Sersen EA, Wisniewski HM. Increased plasma amyloid beta protein 1–42 levels in Down syndrome. Neurosci Lett 1998;241:13–16 [DOI] [PubMed] [Google Scholar]
- 23. Schupf N, Patel B, Silverman W, et al. Elevated plasma amyloid beta-peptide 1–42 and onset of dementia in adults with Down syndrome. Neurosci Lett 2001;301:199–203 [DOI] [PubMed] [Google Scholar]
- 24. Tokuda T, Fukushima T, Ikeda S, et al. Plasma levels of amyloid beta proteins Abeta1–40 and Abeta1–42(43) are elevated in Down's syndrome. Ann Neurol 1997;41:271–273 [DOI] [PubMed] [Google Scholar]
- 25. Zigman WB, Schupf N, Sersen E, Silverman W. Prevalence of dementia in adults with and without Down syndrome. Am J Ment Retard 1996;100:403–412 [PubMed] [Google Scholar]
- 26. Rumble B, Retallack R, Hilbich C, et al. Amyloid A4 protein and its precursor in Down's syndrome and Alzheimer's disease. N Engl J Med 1989;320:1446–1452 [DOI] [PubMed] [Google Scholar]
- 27. Silverman W, Schupf N, Zigman W, et al. Dementia in adults with mental retardation: assessment at a single point in time. Am J Ment Retard 2004;109:111–125 [DOI] [PubMed] [Google Scholar]
- 28. Aylward EH, Burt DB, Thorpe LU, Lai F, Dalton A. Diagnosis of dementia in individuals with intellectual disability. J Intellect Disabil Res 1997;41:152–164 [DOI] [PubMed] [Google Scholar]
- 29. Hixson JE, Vernier DT. Restriction isotyping of human apolipoprotein E by gene amplification and cleavage with HhaI. J Lipid Res 1990;31:545–548 [PubMed] [Google Scholar]
- 30. Ringman JM, Younkin SG, Pratico D, et al. Biochemical markers in persons with preclinical familial Alzheimer disease. Neurology 2008;71:85–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Scheuner D, Eckman C, Jensen M, et al. Secreted amyloid beta-protein similar to that in the senile plaques of Alzheimer's disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer's disease [see comments]. Nat Med 1996;2:864–870 [DOI] [PubMed] [Google Scholar]
- 32. Ertekin-Taner N, Younkin LH, Yager DM, et al. Plasma amyloid beta protein is elevated in late-onset Alzheimer disease families. Neurology 2008;70:596–606 [DOI] [PubMed] [Google Scholar]
- 33. Locascio JJ, Fukumoto H, Yap L, et al. Plasma amyloid beta-protein and C-reactive protein in relation to the rate of progression of Alzheimer disease. Arch Neurol 2008;65:776–785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Stomrud E, Hansson O, Zetterberg H, Blennow K, Minthon L, Londos E. Correlation of longitudinal cerebrospinal fluid biomarkers with cognitive decline in healthy older adults. Arch Neurol 2010;67:217–223 [DOI] [PubMed] [Google Scholar]
- 35. Mehta PD, Pirttila T, Patrick BA, Barshatzky M, Mehta SP. Amyloid beta protein 1–40 and 1–42 levels in matched cerebrospinal fluid and plasma from patients with Alzheimer disease. Neurosci Lett 2001;304:102–106 [DOI] [PubMed] [Google Scholar]
- 36. Le Bastard N, Aerts L, Leurs J, Blomme W, De Deyn P, Engelborghs S. No correlation between time-linked plasma and CSF Abeta levels. Neurochem Int 2009;55:820–825 [DOI] [PubMed] [Google Scholar]
- 37. Matsubara E, Hirai S, Amari M, et al. Alpha 1-antichymotrypsin as a possible biochemical marker for Alzheimer-type dementia. Ann Neurol 1990;28:561–567 [DOI] [PubMed] [Google Scholar]
- 38. Chen M, Inestrosa NC, Ross GS, Fernandez HL. Platelets are the primary source of amyloid beta-peptide in human blood. Biochem Biophys Res Commun 1995;213:96–103 [DOI] [PubMed] [Google Scholar]