

Summary
Using an established model of myocardial hypertrophy and fibrosis after angiotensin II (AngII) infusion, our aim was to characterize the early cellular element involved in the development of myocardial fibrosis in detail. Male Lewis rats were infused with saline or AngII (0.7 mg/kg per day) for up to seven days. Collagen deposition and cellular infiltration were identified by histology stains. Infiltrating cells were grown in vitro and examined by flow cytometry and immunostaining. Chemokine expression was measured using qRT-PCR. AngII infusion resulted in multifocal myocardial cellular infiltration (peak at three days) that preceded collagen deposition. Monocyte chemotactic protein (MCP)-1 transcripts peaked after one day of AngII exposure. Using a triple-labelling technique, the infiltrating cells were found to express markers of leucocyte (ED1+), mesenchymal [α-smooth muscle actin (SMA)+] and haematopeotic progenitor cells (CD133+) suggesting a fibroblast progenitor phenotype. In vitro, ED1+/SMA+/CD133+ cells were isolated and grown from AngII-exposed animals. Comparatively few cells were cultured from untreated control hearts, and they were found to be ED1−/SMA+/CD133−. We provide evidence that myocardial ECM deposition is preceded by infiltration into the myocardium by cells that express a combination of haematopoietic (ED1, CD133) and mesenchymal (SMA) cell markers, which is a characteristic of the phenotype of fibroblast precursor cells, termed fibrocytes. This suggests that fibrocytes rather than (as is often presumed) leucocytes may have effector functions in the initiation of myocardial fibrosis.
Keywords: fibrocytes, heart failure, hypertension, mesenchymal progenitor cells, myocardial fibrosis, renin–angiotensin system
Myocardial fibrosis is a pathophysiological process characterized by the deposition of extracellular matrix (ECM) proteins within myocardial tissue and is a common pathological feature of many cardiovascular diseases including hypertrophy and hypertension (Wynn 2008). Accumulation of ECM proteins within the myocardium enhances cardiac stiffness and eventually impairs cardiac function, leading to what is known as diastolic heart failure (Kim & Iwao 2000). Despite intense study, the mechanisms involved in the initiation and progression of myocardial fibrosis have yet to be fully characterized and are often suggested to be secondary to a primary inflammatory response (Pinzani & Rombouts 2004; Swartz 2009).
Recent work suggests that angiotensin II (AngII), a part of the renin–angiotensin system, may play a key role in the development of myocardial fibrosis (Kim & Iwao 2000). This is supported by clinical evidence that patients with cardiovascular diseases and associated myocardial fibrosis have increased serum levels of AngII (Kim & Iwao 2000; Brasier et al. 2002; Zhao et al. 2004). Inhibition of the renin–angiotensin system, and more specifically using AngII receptor blockers, has demonstrated a significant clinical benefit in the prevention of cardiac remodelling including myocardial fibrosis (Billet et al. 2008). Using rodent models, in which AngII is given exogenously, several groups have demonstrated the development of myocardial hypertrophy and fibrosis (Liu et al. 2003; Billet et al. 2008; Sopel et al. 2011). Taken together, this evidence strongly supports a role for AngII in the development of myocardial fibrosis; however, the direct mechanisms responsible and the effector cells involved have yet to be completely elucidated.
It has been described previously that exposure to AngII, as well as to other fibrotic stimuli, results in the rapid cellular infiltration that precedes the development of myocardial fibrosis (Mann 1999; Fujisawa et al. 2001). This temporal relationship suggests that the initial cellular component of the fibrotic response has an effector role in the stimulation of myocardial fibrosis. Until recently, much of the evidence has suggested that this cellular infiltration was mononuclear and consisted of cells from the monocyte/macrophage lineage, as demonstrated by positive immunocytochemical staining for specific molecules present on myeloid cells, ED1/CD11b (Huang et al. 2010;Liu et al. 2003). Such evidence supported the paradigm that AngII mediates the development of myocardial fibrosis by promoting the recruitment of leucocytes (largely macrophages), which could then initiate a fibrotic response (Kim & Iwao 2000).
However, the exact phenotype of the initial infiltrating cells has thus far been poorly defined. We believe that these cells are the first direct responder to increased circulating levels of AngII and as such could reasonably be assumed to mediate downstream effects. In this study, we provide evidence that the early cellular populations recruited to the myocardium, using a well-described model of AngII exposure known to result in myocardial hypertrophy and fibrosis, are not leucocytes. Rather they are circulating progenitor cells that express haematopoietic and mesenchymal markers. This information will offer a broadened understanding of the process of myocardial fibrosis and may be useful in planning future alternative avenues of intervention.
Materials and methods
Animals
All work was approved by Dalhousie University’s University Committee on Laboratory Animals following the guideline of the Canadian Council on Animal Care. Male Lewis rats (Charles River Laboratories, St. Constant, QC, USA) weighing 300–375 g were housed in the Carlton Animal Care Facility, Dalhousie University, and provided food and water ad libitum. Animals were anesthetized with a ketamine (100 mg/kg)/xylazine (5 mg/kg) cocktail, and a 1- to 2-cm mid-scapular skin incision was made using aseptic techniques. Osmotic minipumps (Durect Corporation, Cupertino, CA, USA) containing either saline (control group, n = 24) or AngII (0.7 mg/kg per day; Sigma-Aldrich Canada Ltd., Oakville, ON, USA, n = 34) dissolved in saline were placed subcutaneously. The incision was closed using 4-0 nylon sutures, and animals were allowed to recover. Rats were sacrificed 4h, 1, 2, 3 or 7 days following implantation of the minipumps. The hearts were harvested and weighed to calculate the cardiac mass index (heart/body weight ratio). The heart tissue was processed for further analysis as outlined below.
Haemodynamic measurements
Blood pressure was assessed using a non-invasive tail cuff system (Kent Scientific, Torrington, CT, USA). Animals were subjected to 5 days of measurements prior to the initiation of the 7-days experiment to allow for acclimatization and to prevent artificial elevation of blood pressure. Blood pressures were measured prior to and daily after implantation of mini-osmotic pumps for a minimum of five consecutive readings per animal.
Cell isolation and culture
Hearts from untreated, naïve rats (n = 4) or rats infused with AngII for 3 days (n = 6) were harvested under sterile conditions and used for cell isolation. Briefly, hearts were initially mechanically minced and then enzymatically digested in a collagenase solution (50 μg/ml Collagenase II; Cedar Lane, Burlington, ON, USA) in Roswell Park Memorial Institute 1640 (RPMI) media (Gibco; Life Technologies, Burlington, ON, USA) at 37 °C with agitation for 45 min. The cell isolates were washed twice with complete RPMI (10% heat-inactivated foetal calf serum, 2 mM l-glutamate, 100 μg/ml streptomycin and 100 U/ml penicillin) and then either poured through a 70-μm-pore filter to generate a single-cell suspension for flow cytometry or directly plated on T-75 flasks coated with 0.1% gelatin for culture. Of the whole homogenate samples, a small but equal proportion of each sample was plated onto a 0.1% gelatin-coated coverslip. Cellular isolates were incubated at 37°C with 5% CO2 for 3 days, after which all non-adherent cellular debris was removed and fresh medium was supplied. In all groups, digital images were taken using an inverted microscope for cell counting. Areas of cell growth were selected randomly (six/flask containing cells isolated from one heart) for cell counts. Cells grown in T-75 flasks were dissociated from the flask using 0.05% trypsin in serum free RPMI and were prepped for analysis by flow cytometry. Cells grown on coverslips were fixed in 4% paraformaldehyde and stored at 4 °C until stained.
Flow cytometry
Cells isolated out of the heart were analysed by flow cytometry either directly after isolation from the heart (naïve n = 2; AngII n = 2) or after expansion in culture (naïve n = 2; AngII n = 4). A single-cell suspension was isolated from whole heart homogenate directly after isolated. Cells were permeabilized for intracellular staining using Cytofix/Cytoperm kit (BD Bioscience, San Jose, CA, USA) as per manufacture’s protocol. Non-specific protein interactions were blocked by incubating the cells with goat IgG. Cells were incubated with polyclonal rabbit anti-CD133 (Abcam, Cambrdge, MA, USA), monoclonal mouse anti-CD68 (ED1; AbD Serotec, Oxford, UK) and polyclonal rabbit anti-Collagen 1a (Coll1a; Rockland, Gilbertsville, PA, USA). The cells were incubated with host-specific secondary antibodies conjugated with PerCP, Fitc and PE respectively. Positive, isotype, secondary-only and negative controls were completed alongside experimental staining for compensation purposes and to control for non-specific binding. Samples were fixed in a 0.4% formaldehyde solution and stored at 4 °C until analysis. Samples were analysed using an FACScalibur Flow Cytometer (BD Bioscience).
Expression of CD133 was assessed in isolated cells cultured for 3 days to determine whether their phenotype had changed during culture. Cells were permeabilized and blocked with goat IgG. Cells were then incubated with anti-CD133 followed by incubation with an FITC-conjugated secondary. Isotype, secondary-only and negative controls were completed alongside experimental staining. Samples were fixed and stored at 4 °C until analysis. Samples were analysed using an FACScalibur flow cytometer (BD Bioscience).
Histology
Hearts were fixed in 4% formaldehyde for 24 h and incubated in a series of increasing percentages of ethanol to dehydrate tissues, which was followed by incubation in a clearing agent, xylene. Hearts were embedded in paraffin wax and serially sectioned (5 μm). Basic myocardial histology and cellular infiltration were examined using heart cross sections stained with haematoxylin and eosin (H&E). A blinded observer analysed heart sections for cell and nuclear morphology at a 63× magnification. Light microscopy was performed using a Zeiss Axiovision 4.6 digital image analysis program (Carl Zeiss, Toronto, ON, USA). Pictures were captured using an AxioCam HRc camera (Carl Zeiss) and analysed in Adobe Photoshop 5.0.
Collagen deposition
Collagen staining was accomplished using Sirius red and quantified as described previously. Picrosirius red staining is a well-described histochemical stain for fibrillar type I and type III collagen, which are believed to be the main types of collagen seen with myocardial fibrosis (Haudek et al. 2006). Briefly, slides were examined under a microscope with the 10× objective, and areas of collagen deposition were captured with a digital camera. The image analysis software was used to quantify the amount of tissue positive for Sirius red. Images from areas containing the most collagen staining were taken from a cross section from individual hearts, with a minimum of four animals/experimental group, at a 10× magnification. Images were analysed for collagen content to compare areas of fibrosis between different experimental groups. Percentage collagen content was calculated and then averaged, and experimental groups were compared with control animals.
Immunohistochemistry
Immunohistochemistry was performed to assess the phenotypes of infiltrating cells. Briefly, slides were processed in a standard fashion as previously described (Sopel et al. 2011). Sections were incubated with a relevant primary antibody against CD68 (ED1, AbD Serotec), α-smooth muscle actin (SMA; 1A4, Sigma-Aldrich Canada Ltd.) or CD133 (Abcam) and were developed using the LSAB technique, as per manufacturer’s protocol (DakoCytomation, Burlington, ON, USA). Light microscopy was performed using a Zeiss Axiovision 4.6 digital image analysis program (Carl Zeiss). Pictures were captured using an AxioCam HRc camera (Carl Zeiss) and analysed in Adobe Photoshop 5.0. Among the images analysed, one image of maximal ED1+ myocardial infiltration from a minimum of four animals/experimental group captured at 25× was used for quantification. A blinded observer counted ED+ cells, the resulting tallies were averaged, and experimental groups were compared with control groups.
Immunofluorescence
Triple labelling using a combination of immunofluorescent probes and immunohistochemistry was performed to identify cells expressing multiple cell markers in paraffin-embedded heart samples. Briefly, slides were processed in a standard fashion as previously described (Sopel et al. 2011). Tissues were incubated with anti-CD133 (AbCam) and developed using the LSAB technique (DakoCytomation). The tissues were then incubated with primary antibodies of anti-CD68 (ED1, AbD Serotec) and anti-SMA (C04018, Biocare Medical, Concord, CA, USA) followed by host-specific secondary antibodies conjugated with Alexa488 and Cy3 respectively. Images were taken of AxioCam HRc camera (Carl Zeiss) using light microscopy to visualize the CD133 and fluorescent microscopy at the 488 and 535 wavelengths to assess fluorescent staining. The micrograph of the CD133 staining was analysed using Adobe Photoshop 5.0, the positive brown staining was selected, and a coloured mask representing the positive cells was generated. The two fluorescent images and the coloured mask were overlaid to assess co-localization.
Isolated cells that were paraformaldehyde-fixed on coverslips were stained using an altered protocol from those described above. Antigen retrieval was not required for formaldehyde-fixed samples. Non-specific protein binding was blocked by incubating the tissues with a 10% normal goat serum. Samples were then incubated with the primary antibodies anti-CD68 (ED1, AbD Serotec), anti-CD133 (Abcam) and anti-SMA (Biocare Medical), which was followed by incubation with host-specific secondary antibodies conjugated with Alexa488 Cy3 and Cy5 respectively. Lastly, tissues were incubated with Hoescht (Invitrogen, Burlington, ON, USA). Tissues were washed and wet-mounted. Fluorescent microscopy was preformed using an AxioCam HRc camera (Carl Zeiss). Pictures were taken at 488 and 535, and 650 and 350 wavelengths, and resulting images were overlaid. Images were analysed in Adobe Photoshop 5.0.
Relative quantitative RT-PCR (qRT-PCR)
Relative quantitative RT-PCR was used to analyse mRNA transcript levels within myocardial tissue as described previously (Hart-Matyas et al. 2010). Briefly, using the TRIzol reagent (Invitrogen), RNA was isolated from snap-frozen heart tissue, as per manufacturer’s protocol. Isolated total RNA (2 μg) was used to synthesize cDNA using oligo(dT)20 in SuperScript III First Strand Synthesis SuperMix kit (Invitrogen) as per the manufacturer’s protocols. Twenty-five nano gram of input cDNA with 0.5 μM of each of the forward and reverse primers for MCP-1 or reference gene and 1× iQ SYBR Green Supermix (Bio-Rad Laboratories, Mississauga, ON, USA) was subjected to qPCR using iQ Multicolour Real-Time PCR Detection System thermocycler (Bio-Rad). Standard curves, for efficiency, and no-template control samples were run along with the samples during thermocycling. A melting curve was performed after thermocycling was complete to ensure target specificity. Expression and normalization of the target gene, MCP-1, were calculated relative to the three housekeeping genes (Tub1a, LDHb and HPRT) using qBase version 1.3.5, as described previously (Hellemans et al. 2007). The primers used were designed against the mRNA sequence of specified genes using Primer3 (version 0.4.0, Table 1).
Table 1.
Primer sequences of the target genes
| Gene symbol | Forward primer | Reverse primer | Base pairs |
|---|---|---|---|
| MCP-1 | atctgtgctgaccccaataagg | gtggttgtggaaaagagagtgg | 196 |
| Tub1a | aaccatgcgtgagtgtatctcc | aactgtgggttccaggtctacg | 226 |
| HPRT | aaaggacctctcgaagtgttgg | ctttactggccacatcaacagg | 213 |
| LDHb | gtcatcaaccagaagctgaagg | agggaagaagcaaactgtgacc | 194 |
Forward and reverse primers (5′–3′ orientation) for the specific genes have a product length of the corresponding base pairs.
Statistical methods
Data are reported as mean ± SEM. Continuous variables were analysed with GraphPad Prism (La Jolla, CA) by comparing each experimental group with a control group using an unpaired two-tailed t-test. Upregulation of MCP-1 mRNA by qRT-PCR was compared to baseline gene expression and analysed by a one-sample t-test. Statistical significance was defined as a P ≤ 0.05.
Results
Haemodynamic measurements
The blood pressures of both AngII (n = 4)- and saline (n = 2)-infused animals were measured over the 7-day infusion period (Fig. 1). An elevation of blood pressure was evident as early as one day after starting of AngII infusion, compared with saline controls. Blood pressure levels remained elevated in AngII-infused animals over the seven days infusion, supporting AngII infusion as a model of hypertension.
Figure 1.
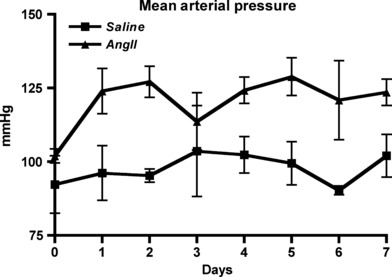
Mean arterial pressure measurements from animals infused with AngII (triangle) and saline (square) over a 7-days period.
Myocardial collagen deposition
AngII infusion is an established model of myocardial hypertrophy and fibrosis (Billet et al. 2008). Using this model, AngII exposure (7 days) resulted in significant myocardial hypertrophy (heart weight/body weight) when compared to saline control (P < 0.05; Table 2). Sirius red staining of the myocardium of AngII infused rats revealed multifocal areas of extensive collagen deposition throughout the ventricles of the heart (Figs 2 and S1). Saline-infused rats showed limited, background levels of collagen content within the interstitium. Quantification of collagen deposition by digital image analysis confirmed a significant increase in collagen content in areas of the hearts from 3-days and 7-days AngII-infused animals as compared to control animals (P < 0.05, Figs 2 and S1).
Table 2.
Hypertrophic assessment
| Saline | 3-days AngII | 7-days AngII | |
|---|---|---|---|
| Heart weight/body weight ratio (mg/g) | 2.7 ± 0.1 | 3.0 ± 0.1 | 3.2 ± 0.2* |
P<0.05 when compared to saline control.
Figure 2.
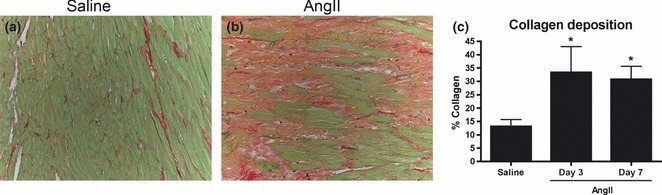
Sirius red stain for collagen. To evaluate collagen deposition in the experimental groups, heart sections were stained with Sirius red and fast green. Representative sections are shown from control animals (a) and 7-days AngII-exposed animals (b). Collagen in most afflicted areas of the heart sections was quantified using image analysis software (c), *P < 0.05. Images were captured at 10×.
Myocardial histology
Infusion of AngII is also known to be associated with cellular and structural changes as well. Hearts from saline control animals demonstrated normal myocardial morphology (Fig. 3). In contrast, heart tissue from AngII-exposed animals exhibited multifocal areas of cellular infiltration, cardiomyocyte loss and ECM extensive. These changes could be seen throughout the left and right ventricles and were evident in both perivascular and interstitial tissues. The most prominent feature of these changes was dense areas of cellular infiltration that could be seen as early as one day following the initiation of onset of AngII infusion and progressed over the 7 days (Fig. 3).
Figure 3.

Cellular infiltration. Hearts stained with H&E from control animals receiving saline (a), AngII for 1 days (b), AngII for 3 days (c) or AngII for 7 days (d). AngII-infused animals revealed areas of marked cellular accumulation, cardiomyocyte loss and replacement by extracellular matrix. Images were captured at 25×. qRT-PCR was used to assess myocardial expression of MCP-1 transcript levels relative to three housekeeping genes (e). Relative expression of MCP-1 mRNA was significantly increased in AngII-infused animals 1, 2 and 3 days, *P < 0.05.
To investigate factors in the environmental milieu that may be recruiting the infiltrating cells, we used relative qRT-PCR to assess the transcript levels of chemotactic factors. Specifically, we assessed the transcript levels of MCP-1, a chemotactic cytokine for monocytes/macrophages and some progenitor cell types. Myocardial expression of MCP-1 mRNA was quantified following 4 h, 1, 2 and 3 days of AngII infusion (Fig. 3). There was a significant increase in MCP-1 mRNA expression compared with the baseline expression of the three normalized housekeeping genes in the myocardium of animals exposed to AngII for 1, 2 and 3 days (P < 0.05). Furthermore, peak expression of MCP-1 mRNA occurred following one day of AngII infusion with an increase of approximately 20-fold above the baseline expression of the housekeeping genes.
Myocardial infiltrating cells
Next the infiltrating cell phenotype was characterised by assessing cell morphology and cell-specific marker expression. The infiltrating cells were mononuclear with no evident polymorphonuclear cells at any time point. Immunohistochemistry for cell-specific markers was used to characterize the infiltrating cell types within the myocardium. Approximately 50–75%, of the cellular infiltrate at 3 and 7 days was positive for a monocyte/macrophage marker, ED1 (Fig. 4). Quantification confirmed an increase in ED1+ cells at days 1, 3 and 7 in AngII-infused animals with peak infiltration seen at 3 days (P < 0.01, Fig. 4). Additionally, infiltrating cells at 3 and 7 days were largely positive for a myofibroblast marker, SMA (Fig. 4); however, owing to the diffuse nature of SMA staining, quantification of SMA+ cells was not performed. Similarly, we investigated the presence of a progenitor cell population within the myocardium of AngII-stimulated animals by staining for a haematopoietic progenitor cell marker, CD133. No CD133 positive staining cells were evident in saline control animals (Fig. 4). In AngII-infused animals, the infiltrating cell population at 3 and 7 days appeared largely positive for CD133, suggesting that infiltrating cells potentially express all three markers.
Figure 4.
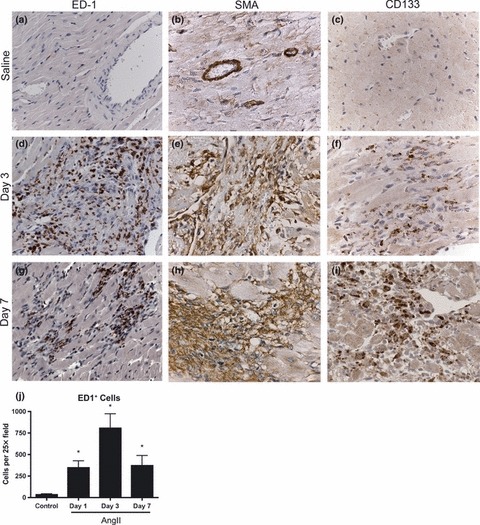
Immunohistochemistry for ED1 (a, d, g), smooth muscle actin (b, e, h) and CD133 (c, f, i) was used to identify the cellular infiltrate in heart sections. Representative sections from control animals receiving saline (a, b, c), AngII for 3 days (d, e, f) and AngII for 7 days (g, h, i) are shown. ED1+ cellular infiltration was quantified by counting three fields (HPF), n = 4 per group, *P < 0.01. Images were captured at 63×.
A correlation between the areas staining positive for ED1, SMA and CD133 suggested that many infiltrating cells might be co-expressing these markers. We used triple immunolabelling to characterize cell expression of ED1, SMA and CD133. We found that in the myocardium of AngII-infused animals, a significant population of ED1+ cells was also positive for SMA and CD133 (triple-positive) (Fig. 5). No double or triple labelling was apparent in the myocardium of saline control animals (data not shown). Expression of both haematopoetic (ED1, CD133) and mesenchymal (SMA) markers is not a generally accepted phenotype of mature macrophages although it is a recognised phenotype found in a specific mesenchymal progenitor cell type originating from a circulating CD14+ population (Hong et al. 2005; Frid et al. 2006; Billet et al. 2008).
Figure 5.
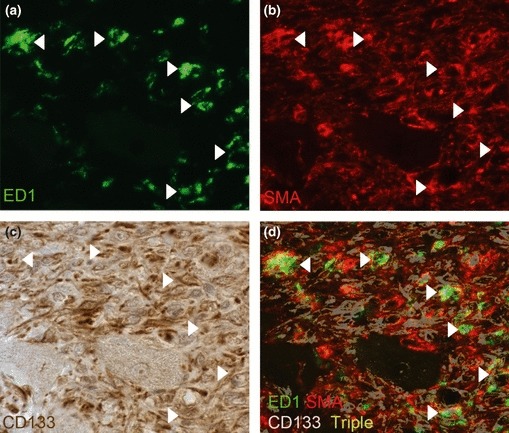
Triple labelling for ED1, smooth muscle actin (SMA) and CD133 in vivo. Immunofluorescent labelling for ED1 (green; a) and SMA (red; b) followed by immunohistochemistry for CD133 (brown; c). Triple labelling of cells is indicated in the composite image of the three stains where a representative coloured mask (purple) of CD133 positivity was generated and overlaid the dual-staining image of ED1 and SMA. Triple-positive cells are indicated by a white overlay colouring (arrows). Images were captured at 63×.
In vitro culture and characterization of infiltrating cells
Infiltrating cells were isolated from the myocardium of three day AngII-infused rats and evaluated by FACS analysis for the expression of ED1, SMA and CD133. Using this approach, cells isolated from AngII-exposed animals had a large population of cells that were ED1+/SMA+/CD133+ (greater than fourfold) when compared to saline control animals (Fig. 6).
Figure 6.
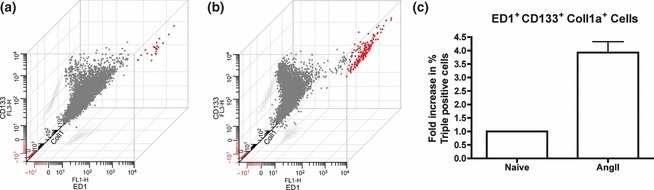
Isolation of infiltrating cells. A single-cell suspension of cells directly isolated out of naïve and AngII-exposed hearts was labelled for flow cytometry for ED1, Coll1a and CD133. Representative 3-axis dot plot of flow cytometry data from naïve (a) and AngII-(b) exposed animals is shown with ED-1 expression on the x-axis, CD133 expression on the y-axis and Coll1a expression on the z-axis. Triple-positive cells are displayed in red. Percentage positive triple-labelled cells normalized to naïve controls was determined (c).
The cultured cells were isolated in vitro and examined to confirm the phenotype of these cells. After three days of culturing heart homogenates, an adherent spindle-shaped cellular population was evident in cultures from the myocardium of AngII-treated animals when compared to the few cells obtained from the myocardiums of untreated animals (Fig. 7). When quantified, 136 ± 22 cells per high-powered view (hpv) could be identified in cultures from AngII-exposed animals compared to 2 ± 1 cells per hpv in cultures from untreated, naïve animals (P < 0.01, Fig. 7). Triple label immunofluorescence was used and confirmed that cultured cells from AngII-exposed animals were positive for ED1, SMA and CD133 (Fig. 7). In contrast, cells seen in cultures from naïve animals were only SMA+ (Fig. 7). CD133 expression was assessed by flow cytometry and suggested that 63 ± 5% of cells seen in cultures from AngII-exposed animals were positive (Fig. 7). Taken together, these data support our in vivo findings that infiltrating cells are largely of a mesenchymal progenitor cell population referred to as fibrocytes.
Figure 7.
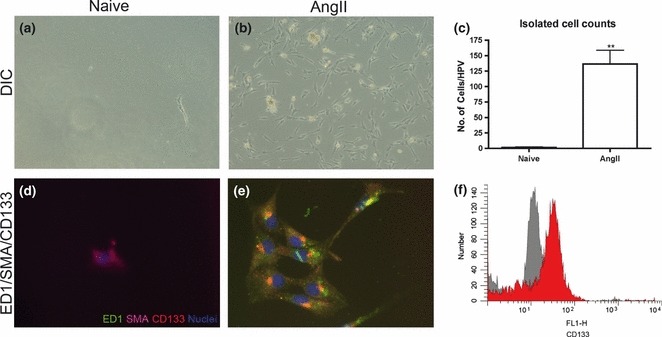
Infiltrating cells were cultured from whole-heart homogenates from naïve, untreated and 3-days AngII-infused mice. Photomicrographs were taken of adherent cell populations isolated from naive (a) and 3-days AngII-exposed (b) animals using differential interference contrast (DIC) microscopy (10×). Cell counting (six high-power fields of view (HPV) per plate) was used to quantify the number of adherent cells between the two groups (c). Triple-labelling immunofluorescence was used to demonstrate co-localization of ED1 (green), smooth muscle actin (red) and CD133 (purple) along with Hoeschst (blue) as a nuclear stain. Images were overlaid to show co-localization of markers in cells from untreated (d) and AngII-exposed (e) myocardiums (40×). A representative histogram of the CD133 expression (red) compared with the negative control (grey) determined by flow cytometry (F). *P < 0.05.
Discussion
Despite the fact that AngII infusion in rodents is a well-established model to study hypertension, hypertrophy and fibrosis, there is limited evidence regarding the exact cell types responsible for the fibrotic response. Previous work has suggested that an increase in circulating AngII initiates an inflammatory response within the myocardium that recruits leucocytes. These inflammatory cells are thought to be activated and produce pro-fibrotic factors (Kim & Iwao 2000). Support for this theory includes the identification of mononuclear cells within the infiltrating cell population, some of which stain positive for ED1 and CD11b, which are indicative of a monocyte/macrophage cell type (Huang et al. 2010; Liu et al. 2003). However, cell phenotypes are dynamic, and cells can alter their molecular expression to respond appropriately to stimuli in their environmental milieu. For example, circulating myeloid precursor cells express specific markers of monocytes/macrophages but are a phenotypically heterogeneous population and can differentiate into non-immune cell types when presented with the appropriate stimuli (Sunderkotter et al. 2004; Tacke & Randolph 2006). Previously, it has been assumed that, owing to ED1/CD11b-positive cellular staining, macrophages are the cell type predominantly recruited early during AngII-induced myocardial fibrosis (Liu et al. 2003). However, these cells have not been shown to be mature macrophages and therefore could be an early myeloid precursor cell type. In fact, we have shown that the number of ED1+ cells peaks at day after startings AngII infusion, while cellular infiltration is still largely evident after 7-days infusion. Furthermore, we have provided evidence that the majority of ED1+ cells are also positive for SMA, which is not an accepted phenotype of mature macrophages. A cell population that is ED1+SMA+ is indicative of a progenitor cell type and, in fact, we found that many of the infiltrating cells expressed the haematopoietic progenitor cell marker CD133. This evidence would suggest that a mesenchymal progenitor cell type is the primary responder to potent fibrotic stimuli and potentially initiates the fibrotic response within the myocardium.
We interpret these findings to suggest that infusion of exogenous AngII stimulates the recruitment of a fibroblast precursor cell type, called fibrocytes, within the myocardium. Fibrocytes are thought to be derived from an early monocyte precursor lineage and as such express myeloid markers supporting findings by others that have identified cells populations expressing markers of monocytes/macrophages (Frid et al. 2006). The important phenotypic distinction is that fibrocytes are characterized by the co-expression of haematopoietic markers (CD133/CD34), leucocyte markers (ED1/CD11b) and mesenchymal markers (SMA/Col-1a) (Hong et al. 2005; Bellini & Mattoli 2007). Expression levels of these markers vary depending on the level of differentiation from early progenitor cells to late myofibroblast cells, which could explain why we found less ED1+ cells at 7-days AngII infusion (Bellini & Mattoli 2007). This may also explain why only 63 ± 5% of isolated cells were positive for CD133. Work from our group has recently described the early migration of CD133+/SMA+ fibroblast progenitor cells in mice myocardium after AngII exposure, supporting our findings. However, the present manuscript is the first to describe the migration of fibrocytes in rat myocardium.
Fibrocytes are thought to be recruited out of the circulation by gradients of SDF-1α/CXCL12, CCL21 and, importantly, MCP-1/CCL2 (Abe et al. 2001; Phillips et al. 2004; Moore et al. 2005; Sakai et al. 2006). This is of significance because we found an increase in transcript levels of MCP-1 within the myocardium, as early as 4 h of AngII infusion, which could act as a possible mechanism for recruiting fibrocytes into the myocardium. Supporting our findings, recent work by Haudek et al. 2010 using MCP-1 KO animals was able to show that MCP-1 was responsible for the recruitment of CD34+/CD45+ fibroblast precursor cells to the myocardium in response to AngII (Haudek et al. 2010). Furthermore, inhibition of migration of these cells resulted in a reduction in the development of fibrosis, confirming that fibrocytes likely play an effector role in the development of fibrosis (Haudek et al. 2010). Although we did not identify the cellular origin of the MCP-1, others have found that local endothelial cells, vascular smooth muscle cells, fibroblasts and myocytes can produce MCP-1 in response to a variety of stimuli, possibly including the increase in circulating AngII and/or an increase in haemodynamic pressure evident in this model (Deshmane et al. 2009). While fibrocytes have been implicated as an effector cell in several models of tissue fibrosis, including after AngII exposure, this has not been well established in models of myocardial fibrosis (Chu et al. 2010; Haudek et al. 2006; Keeley et al. 2009).
We are proposing that fibrocytes, a progenitor cell type, are a key effector cell in the initiation of AngII-induced myocardial fibrosis. We are not suggesting that inflammatory cells play no role in this process. However, given that fibrocytes have the capacity to produce pro-inflammatory mediators as well as contribute to fibrosis by producing collagen and fibronectin, we suggest that fibrocytes may act as a bridging cell type from an early inflammatory response to a corresponding fibrotic response later (Chesney et al. 1998; Quan et al. 2004; Keeley et al. 2009). Our findings, in addition to recent work from others, are providing a body of evidence in which bone marrow-derived progenitor cells (fibrocytes) may represent a significant source of cells capable of migrating into tissues and differentiating into myofibroblasts (Haudek et al. 2006, 2008, 2010). Our findings also suggest that local activation of resident fibroblasts may not be the primary source of cells responsible for the development of fibrosis after AngII exposure and open the way for potential therapeutic modalities able to limit the recruitment of fibrocytes. In the present study, our findings demonstrate that the majority of the infiltrating cells expressed ED1+/SMA+ and CD133+, suggesting that the majority were bone marrow in origin. Clearly, future work that will characterize the regulation of fibrocyte recruitment may allow us to intervene pharmacologically and inhibit this recruitment to the myocardium and potentially prevent excessive myocardial fibrosis associated with many cardiovascular conditions.
Acknowledgments
This work was supported in part by a grant from the Nova Scotia Health Research Foundation (#42311). Furthermore, we would like to thank Mary Li and Tanya Myers for their skilful technical assistance.
Supplementary material
Figure S1. Whole Heart analysis of CollagenContent. Hearts from 7d AngII animals (n = 4) andsaline control animals (n = 2) were dissected intofive sections as illustrated (a) and processed for histologicalexamination.
References
- Abe R, Donnelly SC, Peng T, Bucala R, Metz CN. Peripheral blood fibrocytes: differentiation pathway and migration to wound sites. J. Immunol. 2001;166:7556–7562. doi: 10.4049/jimmunol.166.12.7556. [DOI] [PubMed] [Google Scholar]
- Bellini A, Mattoli S. The role of the fibrocyte, a bone marrow-derived mesenchymal progenitor, in reactive and reparative fibroses. Lab. Invest. 2007;87:858–870. doi: 10.1038/labinvest.3700654. [DOI] [PubMed] [Google Scholar]
- Billet S, Aguilar F, Baudry C, Clauser E. Role of angiotensin II AT1 receptor activation in cardiovascular diseases. Kidney Int. 2008;74:1379–1384. doi: 10.1038/ki.2008.358. [DOI] [PubMed] [Google Scholar]
- Brasier AR, Recinos A, III, Eledrisi MS. Vascular inflammation and the renin-angiotensin system. Arterioscler. Thromb. Vasc. Biol. 2002;22:1257–1266. doi: 10.1161/01.atv.0000021412.56621.a2. [DOI] [PubMed] [Google Scholar]
- Chesney J, Metz C, Stavitsky AB, Bacher M, Bucala R. Regulated production of type I collagen and inflammatory cytokines by peripheral blood fibrocytes. J. Immunol. 1998;160:419–425. [PubMed] [Google Scholar]
- Chu PY, Mariani J, Finch S, et al. Bone marrow-derived cells contribute to fibrosis in the chronically failing heart. Am. J. Pathol. 2010;176:1735–1742. doi: 10.2353/ajpath.2010.090574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshmane SL, Kremlev S, Amini S, Sawaya BE. Monocyte chemoattractant protein-1 (MCP-1): an overview. J. Interferon Cytokine Res. 2009;29:313–326. doi: 10.1089/jir.2008.0027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frid MG, Brunetti JA, Burke DL, et al. Hypoxia-induced pulmonary vascular remodeling requires recruitment of circulating mesenchymal precursors of a monocyte/macrophage lineage. Am. J. Pathol. 2006;168:659–669. doi: 10.2353/ajpath.2006.050599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujisawa G, Dilley R, Fullerton MJ, Funder JW. Experimental cardiac fibrosis: differential time course of responses to mineralocorticoid-salt administration. Endocrinology. 2001;142:3625–3631. doi: 10.1210/endo.142.8.8339. [DOI] [PubMed] [Google Scholar]
- Hart-Matyas M, Nejat S, Jordan JL, Hirsch GM, Lee TDG. IFN-[gamma] and Fas/FasL pathways cooperate to induce medial cell loss and neointimal lesion formation in allograft vasculopathy. Transpl. Immunol. 2010;22:157–164. doi: 10.1016/j.trim.2009.10.004. [DOI] [PubMed] [Google Scholar]
- Haudek SB, Xia Y, Huebener P, et al. Bone marrow-derived fibroblast precursors mediate ischemic cardiomyopathy in mice. Proc. Natl. Acad. Sci. U.S.A. 2006;103:18284–18289. doi: 10.1073/pnas.0608799103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haudek SB, Trial J, Xia Y, Gupta D, Pilling D, Entman ML. Fc receptor engagement mediates differentiation of cardiac fibroblast precursor cells. Proc. Natl Acad. Sci. U.S.A. 2008;105:10179–10184. doi: 10.1073/pnas.0804910105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haudek SB, Cheng J, Du J, et al. Monocytic fibroblast precursors mediate fibrosis in Angiotensin-II-induced cardiac hypertrophy. J. Mol. Cell. Cardiol. 2010;49:499–507. doi: 10.1016/j.yjmcc.2010.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellemans J, Mortier G, De Paepe A, Speleman F, Vandesompele J. qBase relative quantification framework and software for management and automated analysis of real-time quantitative PCR data. Genome Biol. 2007;8:R19. doi: 10.1186/gb-2007-8-2-r19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong KM, Burdick MD, Phillips RJ, Heber D, Strieter RM. Characterization of human fibrocytes as circulating adipocyte progenitors and the formation of human adipose tissue in SCID mice. FASEB J. 2005;19:2029–2031. doi: 10.1096/fj.05-4295fje. [DOI] [PubMed] [Google Scholar]
- Huang XR, Chung AC, Yang F, et al. Smad3 mediates cardiac inflammation and fibrosis in angiotensin II-induced hypertensive cardiac remodeling. Hypertension. 2010;55:1165–1171. doi: 10.1161/HYPERTENSIONAHA.109.147611. [DOI] [PubMed] [Google Scholar]
- Keeley EC, Mehrad B, Strieter RM. The role of circulating mesenchymal progenitor cells (fibrocytes) in the pathogenesis of fibrotic disorders. Thromb. Haemost. 2009;101:613–618. [PMC free article] [PubMed] [Google Scholar]
- Kim S, Iwao H. Molecular and cellular mechanisms of angiotensin ii-mediated cardiovascular and renal diseases. Pharmacol. Rev. 2000;52:11–34. [PubMed] [Google Scholar]
- Liu J, Yang F, Yang X-P, Jankowski M, Pagano PJ. NAD(P)H oxidase mediates angiotensin II-induced vascular macrophage infiltration and medial hypertrophy. Arterioscler. Thromb. Vasc. Biol. 2003;23:776–782. doi: 10.1161/01.ATV.0000066684.37829.16. [DOI] [PubMed] [Google Scholar]
- Mann DLMD. Mechanisms and models in heart failure: a combinatorial approach. Circulation. 1999;100:999–1008. doi: 10.1161/01.cir.100.9.999. [DOI] [PubMed] [Google Scholar]
- Moore BB, Kolodsick JE, Thannickal VJ, et al. CCR2-mediated recruitment of fibrocytes to the alveolar space after fibrotic injury. Am. J. Pathol. 2005;166:675–684. doi: 10.1016/S0002-9440(10)62289-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips RJ, Burdick MD, Hong K, et al. Circulating fibrocytes traffic to the lungs in response to CXCL12 and mediate fibrosis. J. Clin. Invest. 2004;114:438–446. doi: 10.1172/JCI20997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinzani M, Rombouts K. Liver fibrosis: from the bench to clinical targets. Digestive and Liver Disease. 2004;36:231–242. doi: 10.1016/j.dld.2004.01.003. [DOI] [PubMed] [Google Scholar]
- Quan TE, Cowper S, Wu SP, Bockenstedt LK, Bucala R. Circulating fibrocytes: collagen-secreting cells of the peripheral blood. Int. J. Biochem. Cell Biol. 2004;36:598–606. doi: 10.1016/j.biocel.2003.10.005. [DOI] [PubMed] [Google Scholar]
- Sakai N, Wada T, Yokoyama H, et al. Secondary lymphoid tissue chemokine (SLC/CCL21)/CCR7 signaling regulates fibrocytes in renal fibrosis. Proc. Natl. Acad. Sci. U.S.A. 2006;103:14098–14103. doi: 10.1073/pnas.0511200103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sopel MRN, Lee TDG, Legare JF. Myocardial fibrosis in response to angiotensin ii is preceded by the recruitment of mesenchymal progenitor cells. Lab. Invest. 2011;91:565–578. doi: 10.1038/labinvest.2010.190. [DOI] [PubMed] [Google Scholar]
- Sunderkotter C, Nikolic T, Dillon MJ, et al. Subpopulations of mouse blood monocytes differ in maturation stage and inflammatory response. J. Immunol. 2004;172:4410–4417. doi: 10.4049/jimmunol.172.7.4410. [DOI] [PubMed] [Google Scholar]
- Swartz RD. Idiopathic retroperitoneal fibrosis: a review of the pathogenesis and approaches to treatment. Am. J. Kidney Dis. 2009;54:546–553. doi: 10.1053/j.ajkd.2009.04.019. [DOI] [PubMed] [Google Scholar]
- Tacke F, Randolph GJ. Migratory fate and differentiation of blood monocyte subsets. Immunobiology. 2006;211:609–618. doi: 10.1016/j.imbio.2006.05.025. [DOI] [PubMed] [Google Scholar]
- Wynn TA. Cellular and molecular mechanisms of fibrosis. J. Pathol. 2008;214:199–210. doi: 10.1002/path.2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Q, Ishibashi M, Hiasa K, Tan C, Takeshita A, Egashira K. Essential role of vascular endothelial growth factor in angiotensin II-induced vascular inflammation and remodeling. Hypertension. 2004;44:264–270. doi: 10.1161/01.HYP.0000138688.78906.6b. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.