

Abstract
The crosstalk between fibroblasts and keratinocytes is a vital component of the wound healing process, and involves the activity of a number of growth factors and cytokines. In this work, we develop a mathematical model of this crosstalk in order to elucidate the effects of these interactions on the regeneration of collagen in a wound that heals by second intention. We consider the role of four components that strongly affect this process: transforming growth factor-β, platelet-derived growth factor, interleukin-1 and keratinocyte growth factor. The impact of this network of interactions on the degradation of an initial fibrin clot, as well as its subsequent replacement by a matrix that is mainly composed of collagen, is described through an eight-component system of nonlinear partial differential equations. Numerical results, obtained in a two-dimensional domain, highlight key aspects of this multifarious process, such as re-epithelialization. The model is shown to reproduce many of the important features of normal wound healing. In addition, we use the model to simulate the treatment of two pathological cases: chronic hypoxia, which can lead to chronic wounds; and prolonged inflammation, which has been shown to lead to hypertrophic scarring. We find that our model predictions are qualitatively in agreement with previously reported observations and provide an alternative pathway for gaining insight into this complex biological process.
Keywords: mathematical biology, mathematical modelling, wound healing, fibroblast–keratinocyte crosstalk, prolonged inflammation, chronic hypoxia
1. Introduction
Wound healing is a complex, finely balanced process and, in the case of successful healing, is generally thought to comprise four interconnected and overlapping stages: haemostasis (clot formation), inflammation, proliferation and remodelling [1]. The formation of an initial fibrin clot, as well as its degradation and subsequent replacement with a stronger, mature matrix that is mainly comprised of collagen, occurs as a result of a series of interactions, involving the activity of a number of cells, growth factors (GFs) and cytokines [2,3]. Two key cells involved in this process are fibroblasts and keratinocytes [4], whose interaction, via a paracrine loop [5], is essential to the outcome of successful dermal remodelling [6] and re-epithelialization [7]. This dermal–epidermal crosstalk is currently an active area of biological research (see Werner et al. [8] for a comprehensive review). It is known that keratinocytes and fibroblasts cooperatively induce gel contraction in vitro [9]. More recently, it was observed that keratinocytes can regulate the modulation of fibroblasts into a phenotype that is more suited to the remodelling, rather than the deposition, of the extracellular matrix (ECM) [6,10], thus precipitating the subsequent stage of wound healing. Disruptions to this process can lead to serious pathologies such as hypertrophic scars [11], keloid scars [12] and non-healing wounds [13]. It is therefore of crucial importance to throw light on such networks. As it is difficult to conduct in vivo investigations into these processes in a non-invasive manner, realistic mathematical models that are based on known cell behaviours provide a useful framework for studying the wound healing process and its dysregulation.
One of the earliest mathematical models of wound healing was developed by Sherratt & Murray [14], who used two coupled reaction–diffusion equations to describe re-epithelialization, as well as the dependence of the rate of wound closure on the rate of cell proliferation and the migratory activity of the cells. Tranquillo & Murray [15] developed a model for the closure of a dermal wound that included the production of collagen by the fibroblasts and the effects of cell traction on the rate of wound closure. This model has provided the basis for most subsequent mechanochemical models of dermal wound healing, including those by Olsen et al. [16], Tranqui & Tracqui [17] and Vermolen & Javierre [18]. Cruywagen & Murray [19] developed a continuum mechanical model of the interactions between the dermal–epidermal layers of a developing embryo, assuming that ‘signal morphogens’ are emitted in one layer and have their paracrine effect in the other. Several models have also been developed to study the actions of GFs during the wound healing process, including the effect of keratinocyte growth factor (KGF) signalling on dermal–epidermal interactions [20] and the chemotactic and mitotic effect of platelet-derived growth factor (PDGF) on fibroblasts in dermal wound healing [21], while Murphy et al. [22–24] recently investigated the effects of transforming growth factor-β (TGF-β) and tension on dermal wound closure. There have also been several models that describe the laying down of collagen fibres in a wound, including those by Dale et al. [25] and Dallon et al. [26], who investigated the effect of TGF-β on this process, and more recently by Cumming et al. [27], who used a hybrid multiscale discrete-continuum model. Despite these numerous advances, to our knowledge, there are no mathematical models of wound healing that consider the fibroblast–keratinocyte crosstalk that we argue is central to the wound healing process.
In this work, we simulate the behaviour of a wound that is not surgically closed and which therefore heals by second intention (figure 1). In §2, we develop a mathematical model that describes a set of interactions between fibroblasts and keratinocytes that are vital to the proliferative phase of healing, as well as several critical GFs and cytokines that mediate this crosstalk. As illustrated in figure 1, during this stage, enzymes produced by fibroblast cells degrade the initial fibrin clot and synthesize a collagen-based ECM in its place [29], while keratinocyte cells migrate across the wound surface leading to re-epithelialization [7]. In §4, we show that our model captures essential features of the normal healing process, and can be used to describe abnormal wound healing pathologies. It is widely accepted that persistent, chronic hypoxia inhibits healing [30,31] and is one of the most common causes of chronic wounds [32] in which healing fails to produce anatomical and functional integrity [33]. In addition to chronic hypoxia, we consider the case of an excessively long inflammatory response, following Murphy et al. [24]. We find that when treatment is initiated within an appropriate time frame, our model predicts that a normal healing outcome is achievable in each pathological case.
Figure 1.
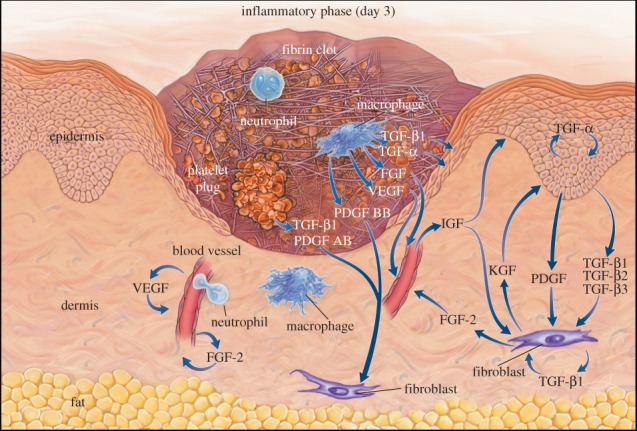
Taken with permission from Singer & Clark [28] (copyright ©1999 Massachusetts Medical Society). Schematic of a wound near the end of the inflammatory stage and the beginning of the proliferative stage. We observe that a ‘tongue’ of epidermal cells extends into the wound area owing to the migration and proliferation of keratinocytes. The progression of this tongue depends on the availability of a suitable substrate. Note that our model includes interleukin-1, which is not illustrated in the figure. In the model that we present in this work, the x-axis runs parallel to the surface of the skin, while the y-axis is perpendicular to the surface.
2. Mathematical model
We model a wound of partial thickness in which both the dermal and epidermal layers are damaged. We present our model in terms of spatial variables (x, y) and use this model to simulate wound healing in a two-dimensional domain. In particular, we focus on the complex network of interactions that comprise the crosstalk between keratinocyte and fibroblast cells, whose densities we represent by k(x, y, t) and f(x, y, t), respectively. Following the approach of Cumming et al. [27], we assume that a fibrin clot containing platelets is formed during the initial haemostasis phase and, on being degraded by enzymes produced by fibroblasts, is replaced with a matrix mainly composed of collagen that allows keratinocyte migration. In order to describe this process, the matrix in our model is represented by two species, c(x, y, t) and ρ(x, y, t), corresponding to the fibrin clot and ECM densities, respectively. Wound healing involves the activity of a large number of GFs and cytokines (see [34] for a recent review). We restrict our attention to the activity of those species that strongly impact the proliferation and migration of fibroblasts and keratinocytes, as well as the degradation and renewal of the ECM. Specifically, we consider the concentrations of interleukin-1, i(x, y, t); KGF, G(x, y, t); PDGF, P(x, y, t); and TGF-β, β(x, y, t).
In figure 2, we present a schematic of what we believe to be the most important interactions between these eight species during the dermal wound healing process. In this work, we develop a two-dimensional model of nonlinear reaction–diffusion partial differential equations to describe these interactions. We now discuss the biological phenomena that underpin the form of the equations.
Figure 2.
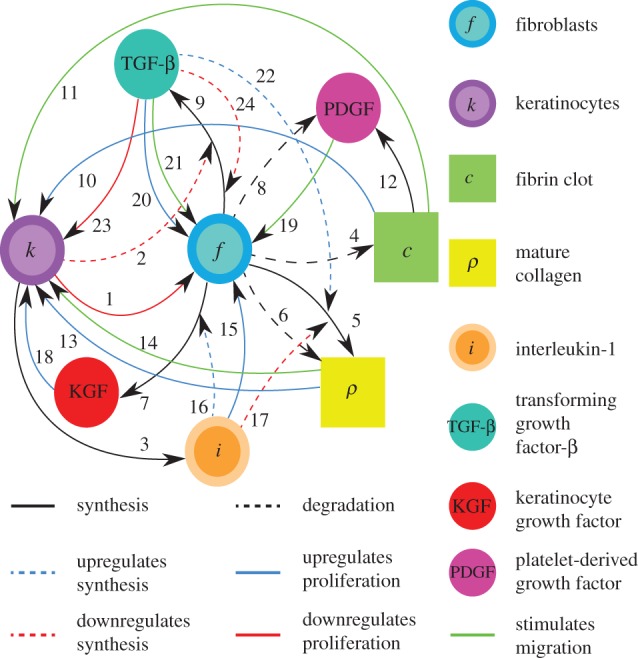
Schematic of the interactions between the components of the model. The references for each arrow are listed in the electronic supplementary material, table S1. Black solid line, synthesis; black dashed line, degradation; blue dashed line, upregulates synthesis; blue solid line, upregulates proliferation; red dashed line, downregulates synthesis; red solid line, downregulates proliferation; green solid line, stimulates migration.
(a). Keratinocytes
Re-epithelialization is driven by the proliferation and migration of keratinocytes. It has been observed that this migration is dependent on the presence of an appropriate proximate substrate, such as the newly formed collagen-based matrix [35,36], and is stimulated by both the presence of mature collagen [37] and fibronectin (which is present in the fibrin clot) [38]. We note that in several models of angiogenesis (e.g. [39]) it is assumed that endothelial cells require an ECM in order to migrate into the wound bed. In our model, we assume that keratinocyte migration is dependent on the densities of the fibrin clot and mature collagen. Owing to the rapid degradation of fibrin, this migration effectively occurs along the fibrin–collagen interface. This is consistent with the observation that the keratinocytes form an invasive ‘tongue’ prior to re-epithelialization, as shown in figure 1. The proliferation of keratinocytes is upregulated by the presence of fibrin [40] as well as mature collagen [41], and we model this using a logistic form with an intrinsic proliferation rate constant λ1. This proliferation is upregulated by the presence of KGF [42,43] and is downregulated by the presence of TGF-β [44], which we model using the coefficients λ2 and λ3, respectively. Assuming that this species has a maximum density 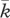 and random motility coefficient Dk, the fibrin clot has initial density
and random motility coefficient Dk, the fibrin clot has initial density 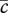 , and the density of mature collagen in the healed state is
, and the density of mature collagen in the healed state is 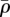 , we thus have:
, we thus have:
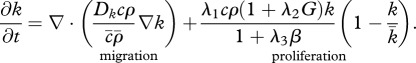 |
2.1 |
(b). Fibroblasts
The proliferation and migration of fibroblasts are critically important to successful dermal wound healing, as they replace lost collagen [29]. It has been observed that there is an increased migration of fibroblasts owing to a chemotactic response to both PDGF [21] and TGF-β [45], and we model this using terms with chemotactic coefficients χfP and χfβ, respectively. We model the proliferation of fibroblasts using a logistic form with an intrinsic proliferation rate constant λ4. Keratinocytes are observed to have a complex effect on the behaviour of fibroblasts: while they secrete interleukin-1, which upregulates fibroblast proliferation [46], they also physically control the growth of fibroblasts by forming colonies [47]. We model these processes using the coefficients λ5 and λ7, respectively. Fibroblast proliferation is further upregulated by the presence of TGF-β [48], which we model using the coefficient λ6. Assuming that this species has a maximum density 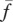 and random motility coefficient Df, we obtain:
and random motility coefficient Df, we obtain:
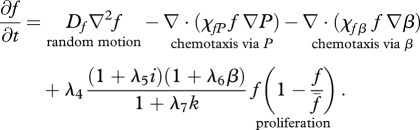 |
2.2 |
(c). Initial provisional matrix (fibrin clot)
The initial provisional matrix is degraded by enzymes produced by a variety of cells, particularly fibroblasts [49,50]. As an approximation to this more complicated process, we model the degradation of the provisional matrix as being (linearly) dependent on the fibroblast cell density at a rate characterized by the constant λ8. We thus have:
| 2.3 |
(d). Extracellular matrix
Fibroblasts produce collagen, the main component of the new ECM [51]. We model this dependence in line with Flegg et al. [52] and Tracqui et al. [53], where a kinetic term proportional to the fibroblast population is used. This production is dependent on the concentration of oxygen [54]; however, to avoid further complexity, we assume the dependence of collagen production on oxygen is incorporated into the coefficient λ9. Interleukin-1 is known to downregulate the production of collagen by fibroblasts [55] and TGF-β is known to upregulate this process [56]. We model these processes using the coefficients λ10 and λ11, respectively. As fibroblasts are known to degrade newly synthesized collagen [57], following Dallon and co-workers [26,58] we assume that this process occurs at a rate, characterized by the constant λ12, proportional to the collagen density. We thus have:
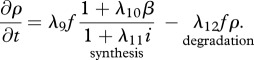 |
2.4 |
(e). Interleukin-1
An important cytokine in the wound healing process is interleukin-1 [59], whose production by keratinocytes [60] we model using a linear term with rate constant λ13. Cytokines and GFs tend to have a half-life of the order of a few minutes [61], and so this decay is assumed to decay linearly with rate constant λ14. Assuming this species diffuses at a rate characterized by the constant Di, we thus have:
| 2.5 |
(f). Keratinocyte growth factor
KGF is known to play an important role in epidermal wound healing [62]. Its production by fibroblasts, which is upregulated by the presence of interleukin-1 [42,43], is modelled using a nonlinear term with coefficients λ15 and λ16. The decay of this species is assumed to be linear with rate constant λ17. Assuming this species diffuses at a rate characterized by the constant DG, we thus have:
| 2.6 |
(g). Platelet-derived growth factor
PDGF is produced by degranulating platelets present in the initial fibrin clot [63]. The rate of production of PDGF decreases as the fibrin clot is degraded, and we model this production using a term with rate constant λ18. This GF decays naturally at a rate characterized by the constant λ19, and is further removed by fibroblasts through endocytosis [21] at a rate characterized by the constant λ20. Assuming this species diffuses at a rate characterized by DP, we thus have:
| 2.7 |
(h). Transforming growth factor-β
The protein TGF-β exists in a number of isoforms, each of which has different biochemical activities [64]. In our model, we consider the activity of the TGF-β1, as it is the most prevalent isoform in wounds [65] and its activity in the formation of scars is known to be significant [66]. TGF-β is secreted in a latent form and is then converted to an active form by enzymes [25] or by myofibroblasts [67]. For this investigation, we assume that this conversion occurs rapidly, and we represent both active and latent forms of TGF-β by a single species, β. Fibroblasts are one of the main sources of TGF-β in wounds [48], and their production of this GF is inhibited if TGF-β is already present [25]. We model this production using a saturation form, in line with the work of Murphy et al. [23], with the constant λ21 characterizing the rate of production by fibroblasts and the constant λ23 characterizing the half-maximal rate of production. It has been observed that this production by fibroblasts is downregulated by the presence of keratinocytes [68], and we model this using a downregulatory coefficient λ22. Modelling the decay of TGF-β using a linear term with rate constant λ24 and assuming that it diffuses at a rate characterized by the constant Dβ, we thus have:
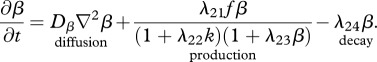 |
2.8 |
(i). Geometry, initial and boundary conditions
We assume that (x, y) represents a plane perpendicular to the surface of the skin, which includes both epidermal and dermal regions. Note that in figure 1, the x-axis runs parallel to the surface of the skin and the y-axis is perpendicular to the surface. As illustrated in the schematic in the electronic supplementary material, figure S1, we model a wound of depth Ly and length 2Lx, with symmetry around x = 0, and where y = 0 represents the lower edge of the wound. We assume that x = Lx represents the interface where the wound meets healthy tissue and that 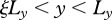 represents the epidermal region. For our chosen values of Lx, Ly and
represents the epidermal region. For our chosen values of Lx, Ly and 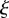 (table 1), this assumption corresponds to the physical extent of the human epidermis. Our computational domain also contains some of the surrounding unwounded tissue and extends to the region
(table 1), this assumption corresponds to the physical extent of the human epidermis. Our computational domain also contains some of the surrounding unwounded tissue and extends to the region 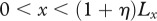 ,
, 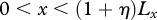 .
.
Table 1.
Parameter values for the model. Here, tw represents parameters estimated in this work. The dimensionless values of the diffusion and chemotactic coefficients (marked with asterisks) are those along the x-axis. The corresponding values along the y-axis are obtained by multiplying these terms by (Lx/Ly)2.
| dimensional | dimensionless | source | |
|---|---|---|---|
| Lx | 1.0 cm | 1 | tw |
| Ly | 0.3 cm | 1 | tw |
| ξ | 0.8 | tw | |
| η | 0.2 | tw | |
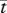 |
1 day | 1 | tw |
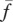 |
10−1 g cm−3 | 1 | [69], tw |
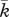 |
10−3 g cm−3 | 1 | tw |
 |
10−3 g cm−3 | 1 | [70], tw |
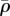 |
10−3 g cm−3 | 1 | [70], tw |
 |
1.39 × 10−7 g cm−3 | 1 | [71], tw |
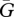 |
5.38 × 10−8 g cm−3 | 1 | [20], tw |
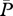 |
4.78 × 10−8 g cm−3 | 1 | [21], tw |
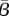 |
2.75 × 10−7 g cm−3 | 1 | [72] |
| Dk | 4.33 × 10−9 cm2 s−1 | *2.59 × 10−4 | [20] |
| Df | 1.7 × 10−10 cm2 s−1 | *1.02 × 10−5 | [73] |
| Di | 6.93 × 10−6 cm2 s−1 | *0.42 | [71], tw |
| DG | 9.5 × 10−6 cm2 s−1 | *0.57 | [20] |
| DP | 2.78 × 10−8 cm2 s−1 | *1.67 × 10−3 | [21] |
| Dβ | 2.94 × 10−7 cm2 s−1 | *1.76 × 10−2 | [22] |
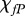 |
5.53 × 10−17 g−1 cm s−1 | *0.69 × 10−4 | tw |
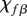 |
3.18 × 10−16 g−1 cm s−1 | *0.69 × 10−4 | tw |
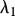 |
14 | tw | |
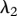 |
3.2 | [74] | |
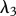 |
1 | [75] | |
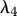 |
3.5 | tw | |
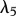 |
1.5 | [76], tw | |
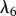 |
7.27 × 106 cm3 g−1 | 2 | [23,77] |
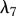 |
10 | tw | |
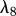 |
0.6 | [58] | |
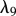 |
0.64 | [58] | |
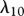 |
7.27 × 106 cm3 g−1 | 2 | [23,78] |
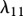 |
1 | [55] | |
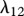 |
0.44 | [58] | |
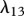 |
16.6 | tw | |
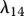 |
1.92 × 10−4 s−1 | 16.6 | [79], tw |
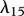 |
1.2 | tw | |
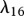 |
5 | [74], tw | |
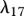 |
8.33 × 10−5 s−1 | 7.2 | [74] |
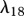 |
2.4 | tw | |
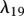 |
2.78× 10−5 s−1 | 2.4 | [21] |
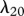 |
0.555 cm3 g−1 s−1 | 48 | [21], tw |
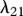 |
0.5 cm3 g−1 s−1 | 0.05 | [23, 80] |
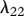 |
0.6 | [68], tw | |
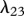 |
1.3 × 106 cm3 g−1 | 0.3575 | [81] |
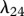 |
0.354 d−1 | 0.354 | [72] |
We assume that keratinocytes are initially present in the epidermal region outside the wounded area,  ,
, 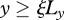 , while fibroblasts and the ECM are present in the region y < 0, as well as the dermal region outside the wounded area,
, while fibroblasts and the ECM are present in the region y < 0, as well as the dermal region outside the wounded area,  ,
, 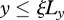 . In our model, interleukin-1 and KGF are produced by keratinocytes and fibroblasts, respectively, and so we assume that they are initially absent in the wounded area and present outside. As PDGF and TGF-β are produced by macrophages and platelets present in the fibrin clot [63], we assume that they are initially present in the wounded area and absent outside. For the GFs we use no-flux boundary conditions along each boundary, while for the keratinocytes and fibroblasts we use no-flux boundary conditions along x = 0 and y = Ly, and Dirichlet boundary conditions along
. In our model, interleukin-1 and KGF are produced by keratinocytes and fibroblasts, respectively, and so we assume that they are initially absent in the wounded area and present outside. As PDGF and TGF-β are produced by macrophages and platelets present in the fibrin clot [63], we assume that they are initially present in the wounded area and absent outside. For the GFs we use no-flux boundary conditions along each boundary, while for the keratinocytes and fibroblasts we use no-flux boundary conditions along x = 0 and y = Ly, and Dirichlet boundary conditions along 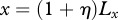 and
and 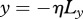 . These initial and boundary conditions are simulated using steep tanh functions and are detailed in the electronic supplementary material. We now present a set of numerical results obtained using this model that describe different wound healing outcomes. We include three movies in the electronic supplementary material that show the evolving densities of keratinocytes over the entire domain for each outcome, illustrating the development of the invasive ‘tongue’ and the subsequent migration of these cells across the wound.
. These initial and boundary conditions are simulated using steep tanh functions and are detailed in the electronic supplementary material. We now present a set of numerical results obtained using this model that describe different wound healing outcomes. We include three movies in the electronic supplementary material that show the evolving densities of keratinocytes over the entire domain for each outcome, illustrating the development of the invasive ‘tongue’ and the subsequent migration of these cells across the wound.
3. Material and methods
The model equations were non-dimensionalized (this is detailed in the electronic supplementary material) and numerical results were obtained by integrating the resulting system using the NAG subroutine D03RAF [82] for different choices of the dimensionless parameters over a two-dimensional domain, with 121 × 121 grid points. A schematic of our non-dimensionalized model is shown in figure S2 of the electronic supplementary material. The dimensional and dimensionless values for the parameters of our model are listed in table 1. While our model is intended to be a caricature of the network of interactions that comprise the fibroblast–keratinocyte crosstalk, we have as far as possible used parameter values that correspond to reported experimental data. We now discuss how these parameters were determined.
(a). Scaling, diffusion and random motility coefficients
The carrying capacity of fibroblast cells is roughly 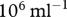 [69], while the volume of each cell, which weighs the same as water, is around
[69], while the volume of each cell, which weighs the same as water, is around 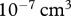 [16]. We thus take
[16]. We thus take 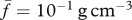 . Noting that the molecular weight of KGF is around 27 kDa [83] and taking the value for the KGF concentration from Wearing & Sherratt [20], we have
. Noting that the molecular weight of KGF is around 27 kDa [83] and taking the value for the KGF concentration from Wearing & Sherratt [20], we have 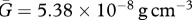 . Assuming that the density of PDGF is the same as that of KGF and noting that the molecular weight of PDGF is around 24 kDa [21], we take
. Assuming that the density of PDGF is the same as that of KGF and noting that the molecular weight of PDGF is around 24 kDa [21], we take 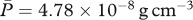 . The molecular weight of interleukin-1 was found to be in the range 12–70 kDa [71], and for our simulations we consider the higher value 70 kDa. Assuming that the density of interleukin-1 is also the same as that of KGF, we take
. The molecular weight of interleukin-1 was found to be in the range 12–70 kDa [71], and for our simulations we consider the higher value 70 kDa. Assuming that the density of interleukin-1 is also the same as that of KGF, we take 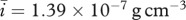 . We assume that the densities of the ECM and the fibrin clot are comparable and take both
. We assume that the densities of the ECM and the fibrin clot are comparable and take both 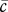 and
and 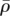 to be
to be 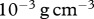 , which lies within the range of values previously used in simulations by Namy et al. [70]. We also assume that the density of the epidermal layer is similar to that of the dermal layer, and take
, which lies within the range of values previously used in simulations by Namy et al. [70]. We also assume that the density of the epidermal layer is similar to that of the dermal layer, and take 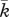 to be
to be 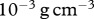 . Using our assumed value for the molecular weight of interleukin-1, we estimate the random motility coefficient by considering the molecular weight, m, and random motility coefficient, D, for another chemical using
. Using our assumed value for the molecular weight of interleukin-1, we estimate the random motility coefficient by considering the molecular weight, m, and random motility coefficient, D, for another chemical using 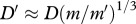 . Following Wearing & Sherratt [20], we consider the species N-formyl-methionyl-leucyl-phenylalanine, for which
. Following Wearing & Sherratt [20], we consider the species N-formyl-methionyl-leucyl-phenylalanine, for which 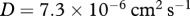 and
and 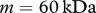 . We therefore obtain the estimate
. We therefore obtain the estimate 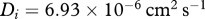 . Finally, we assume that the dimensional values of the chemotactic coefficients
. Finally, we assume that the dimensional values of the chemotactic coefficients 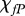 and
and 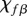 are
are 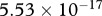 g cm−1 s−1 and
g cm−1 s−1 and 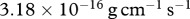 , respectively.
, respectively.
(b). Reaction kinetic coefficients
For our simulations, we assume that the intrinsic growth rate constants λ1 and λ4 have the dimensionless values λ1 = 14 and λ4 = 3.5, such that the wound closes within three weeks. It is known that KGF stimulates a 4.2-fold rise in keratinocyte proliferation [74]; the proliferation of keratinocytes will halve under the influence of TGF-β [75]; interleukin-1 stimulates a 2.5-fold rise in fibroblast proliferation [76], downregulates the production of collagen by fibroblasts by 50 per cent [55] and increases the production of KGF by fibroblasts by a factor of 6 [74]; and the presence of keratinocytes decreases the production of TGF-β by a factor of 1.6 [68]. We thus set the dimensionless values λ2 = 3.2, λ3 = 1, λ5 = 1.5, λ11 = 1, λ16 = 5 and λ22 = 0.6. We set the dimensionless value λ7 = 10 to ensure that keratinocytes sufficiently restrict the proliferation of fibroblasts. The half-life of interleukin-1 mRNA was found to be in the range 60–90 min [79]. For our simulations, we use the upper limit and take the dimensional value 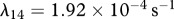 . The dimensionless values of
. The dimensionless values of 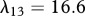 and
and 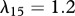 are chosen such that we obtain the steady state
are chosen such that we obtain the steady state 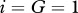 inside the wound, while the dimensionless value of λ18 = 2.4 is chosen such that there is a balance in the production and decay of PDGF in the absence of fibroblasts. For the degradation of PDGF by enzymes produced by fibroblasts, we use the upper limit of the range of values estimated by Haugh [21] and take the dimensional value
inside the wound, while the dimensionless value of λ18 = 2.4 is chosen such that there is a balance in the production and decay of PDGF in the absence of fibroblasts. For the degradation of PDGF by enzymes produced by fibroblasts, we use the upper limit of the range of values estimated by Haugh [21] and take the dimensional value 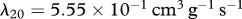 . Finally, in the model by Murphy et al. [23], the rate of TGF-β synthesis by fibroblasts is estimated to be
. Finally, in the model by Murphy et al. [23], the rate of TGF-β synthesis by fibroblasts is estimated to be 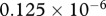 ng (cell d−1). This value was deduced from experiments by Wang et al. [80], where the maximum dilution of TGF-β was
ng (cell d−1). This value was deduced from experiments by Wang et al. [80], where the maximum dilution of TGF-β was 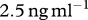 . Thus, we take the rate constant of TGF-β production for a given density,
. Thus, we take the rate constant of TGF-β production for a given density, 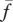 , of fibroblasts to be
, of fibroblasts to be 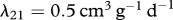 . The values of the parameters λ6, λ8, λ9, λ10, λ12, λ17, λ19, λ23 and λ24 were taken directly from the corresponding references listed in table 1.
. The values of the parameters λ6, λ8, λ9, λ10, λ12, λ17, λ19, λ23 and λ24 were taken directly from the corresponding references listed in table 1.
4. Results
(a). Normal wound healing
We simulate the normal healing case using the parameter values listed in table 1. Figure 3a shows how keratinocytes migrate into the wound bed at the interface between the intact ECM and the fibrin clot, reproducing the invasive ‘tongue’ seen in figure 1. These cells subsequently migrate across the wound site, leading to re-epithelialization, as seen in figure 3b,c. Although not shown here, we also find that fibroblasts migrate over the entire domain, completely degrading the fibrin clot and synthesizing mature collagen in the process. The large initial concentration of TGF-β in the wound is found to result in the production of an excessive amount of collagen by fibroblasts; however, the rapid drop in TGF-β concentration, coupled with increasing levels of interleukin-1, quickly downregulates this production, and the ECM density approaches its pre-wounded value. We now consider two pathologies that can arise owing to a disruption to the wound healing process.
Figure 3.
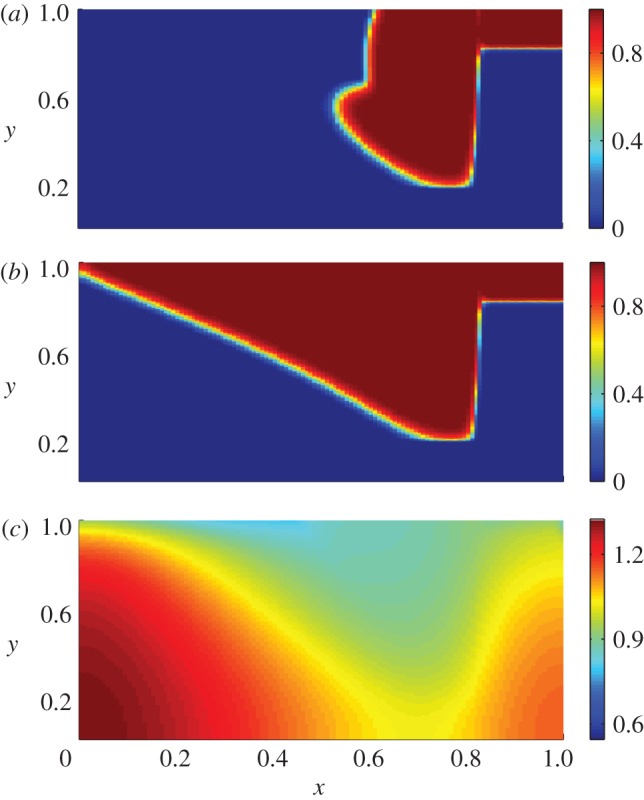
Numerical results for the non-dimensionalized system in the case of normal wound healing for the choice of parameters listed in table 1. (a) The spatial distribution of keratinocytes is shown at t = 8.33 days post-injury. We observe that keratinocytes have migrated into the wound space, reproducing the invasive ‘tongue’ observed in figure 1. Further, the distributions of (b) keratinocytes and (c) ECM for the same parameter values at t = 18.5 days post-injury are shown. For this later time, keratinocytes are mainly confined to the upper (epidermal) region of the wound, resulting in an increased concentration of interleukin-1 and, consequently, a reduced amount of ECM in this region. These results are consistent with the expected behaviour of normal wounds, in which collagen is deposited in the dermal regions by fibroblasts, and the previously discussed experimental observation that keratinocytes prevent the excess production of collagen [6,10].
(b). Prolonged inflammation
First, we consider the case of an excessively long inflammatory response. Murphy et al. [24] noted that poorly regulated inflammatory mediators can lead to pathological scarring and simulated this case by decreasing the decay rate of TGF-β. Such an assumption is justified by the observation that prolonged inflammation results in reduced levels of neutrophils, which in turn negatively affects the ability of chemoattractants to dissipate [84]. Thus, we follow the approach used by Murphy et al. [24] and model this case by decreasing the value of the parameter that characterizes the decay of TGF-β, λ24, by a factor of 10. We find that although keratinocytes quickly migrate across the domain to re-epithelialize the wound (figure 4a), the ECM levels in this case are significantly increased.
Figure 4.
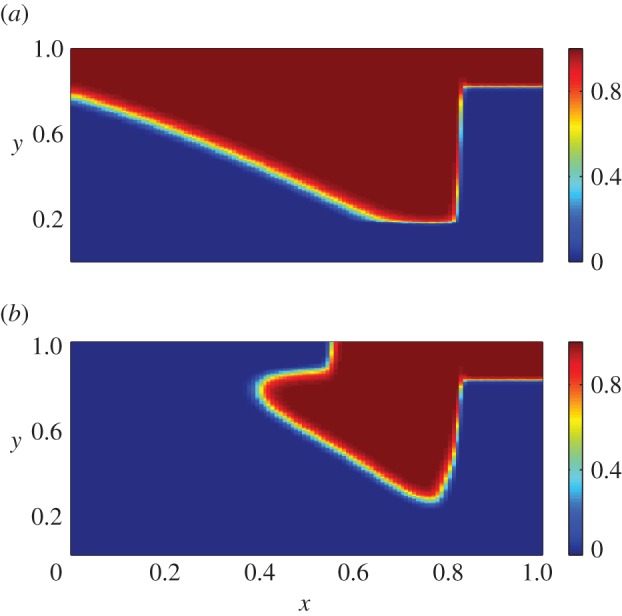
Spatial distributions of keratinocytes for the non-dimensionalized system at t = 18.5 days post-injury in the cases of (a) prolonged inflammation and (b) chronic hypoxia. For the case of prolonged inflammation, we use λ24 = 0.0354 (corresponding to a decrease in the decay rate of β), while for the case of chronic hypoxia, we use the values λ4 = 1.75, λ9 = 0.432 and λ21 = 0.016 (corresponding to a decrease in the proliferation rate of f and the production rates of 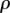 and β, respectively). We find that the wound takes far longer to re-epithelialize in the case of chronic hypoxia than in the case of prolonged inflammation.
and β, respectively). We find that the wound takes far longer to re-epithelialize in the case of chronic hypoxia than in the case of prolonged inflammation.
The average ECM density (relative to normal) is calculated by averaging the net ECM density in the wound bed at each timestep over the total number of grid points in this region. A smooth function is then obtained using spline interpolation, and this is then divided by the corresponding function obtained for the normal case. Figure 5 shows that the ECM levels in the prolonged inflammation case, shown as a thick red curve, are consistently much higher than normal. This arises owing to the prolonged presence of TGF-β in the wound (prolonged inflammation), which stimulates fibroblasts to produce an excessive amount of collagen and often leads to hypertrophic scarring.
Figure 5.
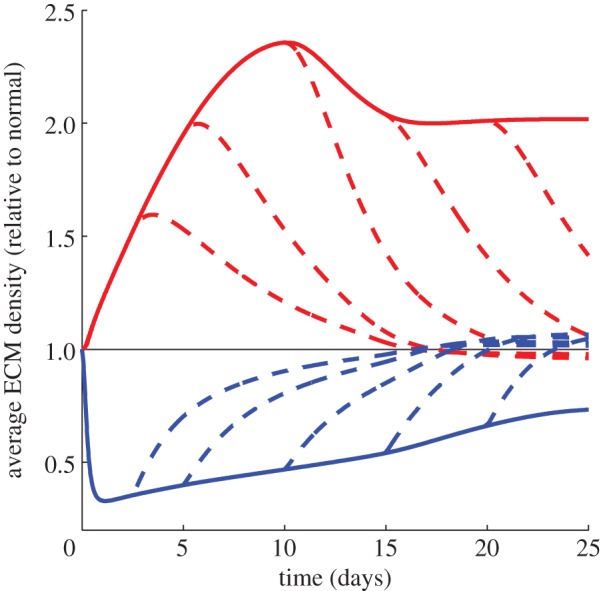
Average density of ECM (relative to normal) for different pathologies up to t = 25 days. The normal case is represented by a thin black line, while the case of prolonged inflammation and chronic hypoxia are represented by thick red and blue lines respectively. The thick dashed lines represent the situations where a treatment is applied in each case at t = 2.5, 5, 10, 15 and 20 days. In the case of prolonged inflammation, we observe that the ECM levels in the wound tend to a much higher value than for the normal wound, corresponding to hypertrophic scarring. On the other hand, in the case of chronic hypoxia, we observe that the ECM levels are around 0.5 times the normal value, and this significantly inhibits the rate of healing (figure 6). When treatment is applied in each case, the ECM levels eventually approach a value close to that obtained in the normal healing process.
(c). Chronic hypoxia
We consider the case of chronic wound hypoxia, where a significantly reduced concentration of oxygen inhibits the healing process [85]. Prolonged hypoxia is one of the most common causes of chronic wounds [32], where healing fails to produce anatomical and functional integrity [33]. Siddiqui et al. [86] noted that fibroblast cells placed in chronically low oxygen levels over 72 h exhibited a 50 per cent decrease in population doublings, a 1.48-fold reduction in collagen production and a 3.1-fold decrease in TGF-β production relative to a standard oxygen system. These observations confirm the findings that chronic hypoxia reduces collagen production and that fibroblast proliferation is greatly inhibited in this case [87]. We investigate this situation by changing the parameters λ4, λ9 and λ21 by these experimentally determined amounts. As seen in figure 4b, keratinocytes take much longer to migrate across the wound in this case.
The rate of wound closure is estimated by considering the extent of the non-epithelialized region in each case (see the electronic supplementary material for further details). Figure 6 shows the wound takes much longer to heal than in the normal case, which is in agreement with the conclusions of Oberringer et al. [31]. Additionally, as seen in figure 5, our model predicts that collagen is produced at a slower rate than in the normal case.
Figure 6.
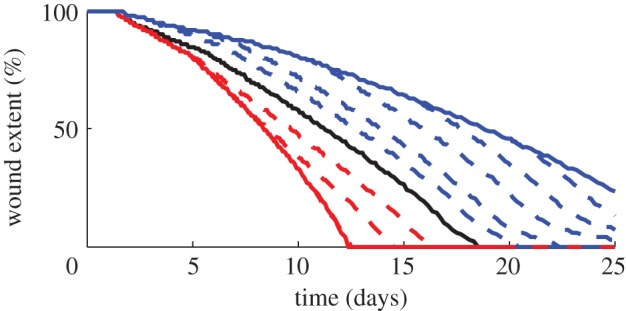
Percentage of wound extent for different pathologies up to t = 25 days. The normal case is represented by a thick black line, while the case of prolonged inflammation and chronic hypoxia are represented by thick red and blue lines, respectively. The thick dashed lines represent the situations where a treatment is applied in each case at t = 2.5, 5, 10, 15 and 20 days. We find that wound re-epithelializes at a much faster rate in the case of prolonged inflammation, although it leads to excessive collagen production (figure 5), while a chronic hypoxic wound takes a much longer time to re-epithelialize. When a treatment is applied in the case of hypoxia, the wound re-epithelializes at a rate closer to that of a normal wound. However, in the case of prolonged inflammation, the wound closes by t = 12.5 days, and so any treatment effected after this time does not impact the re-epithelialization process.
(d). Implementation of treatment
Finally, we simulate the application of a treatment for the two pathologies under consideration. Prolonged inflammation can be treated by blocking the TGF-β pathway, for instance, by suppressing the secretion of TGF-β by fibroblasts (see Murphy et al. [24] for a recent discussion). We therefore model the treatment of prolonged inflammation by returning the rate of TGF-β decay to the corresponding value for the normal healing case. We note that early treatment can significantly impact the wound healing outcome. From figure 5, we see that a treatment applied at early times can cause the ECM density to quickly approach the corresponding value for the normal case. Furthermore, figure 6 shows that a treatment applied at days 2.5 and 5 can cause the wound to re-epithelialize at a rate closer to the normal rate, while treatments applied after 12.5 days will not affect this process. These findings are in agreement with the results of Murphy et al. [24], who noted that a healing strategy implemented after around two weeks is less effective in preventing hypertrophic scarring than an early treatment.
Tissue oxygen levels can be increased by the use of hyperbaric oxygen therapy, an approach that has been used to treat chronic hypoxia [13]. We simulate this treatment at various times by returning the proliferation rate of fibroblasts and the rates of production of collagen and TGF-β to the corresponding values for the normal healing case. As seen in figure 5, we find that the application of a treatment causes the ECM density to approach the corresponding value for the normal case. Furthermore, figure 6 shows that on applying treatment, the wound re-epithelializes at a rate closer to that of a normal wound.
5. Discussion
Wound healing has been the subject of many mathematical investigations over the last two decades. These models have tended to concentrate on either epidermal or dermal wound healing; however, it is known that the crosstalk between epidermal cells (keratinocytes) and the primary cells of the dermis (fibroblasts) is critical to successful wound healing. The model we have developed here is more general than earlier models, as it explicitly takes these interactions into account. For the sake of simplicity, we assume that the fibrin clot is present initially, and so we do not model its formation, which occurs rapidly after wounding. As human wounds (healing by second intention) generally tend to heal by infilling rather than contraction, we do not include mechanical effects in our modelling. We note, however, that mechanical phenomena may play a role in severe hypertrophic scarring.
Our predictions, based on the numerical solutions of a coupled system of governing reaction–advection–diffusion partial differential equations, capture many key aspects of the wound healing process. In addition to reproducing a healing timeline (including the removal of a fibrin clot and re-epithelialization) that is qualitatively similar to the observed wound healing process, our model can simulate healing pathologies. To this end, we provide two illustrative examples of sub-optimal healing that lead to excessive collagen production and to a delay in healing, respectively.
We simulate the case of an excessively long inflammatory response by assuming that the decay rate of TGF-β is small. Our model predicts that this situation can give rise to hypertrophic scarring via the excessive production of collagen by fibroblasts, and that this outcome can be most effectively prevented by implementing a healing strategy effected within two weeks.
Sufficient local oxygen levels are required in order to facilitate normal fibroblast proliferation and collagen production [88], and TGF-β production. We simulate hypoxic conditions by changing the values of the parameters that correspond to these processes from their level in the normal healing situation by experimentally determined amounts. The significant delay in healing that we observe in the case of chronic hypoxia is also in agreement with the literature [13,31]. Our model predicts that hypoxic wounds can be successfully treated with the application of supplemental oxygen, which is consistent with recent reviews [13].
This model could be extended in ways that may better allow us to capture the healing process. For instance, the sharp profiles of keratinocytes (see figure 3) arise owing to the initial rectangular geometry, and more realistic behaviour could be obtained by using initial conditions that more closely represent the geometry of a dermal wound. Additionally, in order to prevent potential scenarios in which the wound cannot re-epithelialize, the dynamic generation of the fibrin clot could be incorporated into the model. Future models of this process could also consider the competition for space that occurs between fibroblasts, keratinocytes and mature collagen, which we ignore in this model to avoid increasing its complexity. While every attempt has been made to obtain parameter values that are in agreement with previous experimental results, several parameters in our model have been estimated in order to obtain appropriate steady states, and to qualitatively simulate observed healing behaviour. A detailed parameter sensitivity analysis, while outside the scope of the current work, will help to further validate this model. Although we have not attempted to address the full complexity of the fibroblast–keratinocyte crosstalk in this work, we find that our model captures the significant consequences of this critical biological process.
Acknowledgments
The authors would like to acknowledge funding from the following sources for R.C.S. to visit Queensland University of Technology (QUT): the Institute of Health and Biomedical Innovation (under the Visiting Researcher Scheme), the School of Mathematical Sciences at QUT and Western Kentucky University. Computational resources and services used in this work were provided by the HPC and Research Support Unit, QUT. This research was supported under the Australian Research Council's Discovery Projects funding scheme (project number DP0878011).
References
- 1.Enoch S., Leaper D. J. 2005. Basic science of wound healing. Surgery 23, 37–42 10.1383/surg.23.2.37.60352 (doi:10.1383/surg.23.2.37.60352) [DOI] [Google Scholar]
- 2.Werner S., Grose R. 2003. Regulation of wound healing by growth factors and cytokines. Physiol. Rev. 83, 835–870 10.1152/physrev.00031.2002 (doi:10.1152/physrev.00031.2002) [DOI] [PubMed] [Google Scholar]
- 3.Babu M., Wells A. 2001. Dermal–epidermal communication in wound healing. Wounds 13, 183–189 [Google Scholar]
- 4.Ågren M. S., Eaglstein W. H., Ferguson M. W. J., Harding K. G., Moore K., Saarialho-Kere U. K., Schultz G. S. 2000. Causes and effects of the chronic inflammation in venous leg ulcers. Acta Derm.Venereol. Supp. 210, 3–17 [PubMed] [Google Scholar]
- 5.Nowinski D., Lysheden A.-S., Gardner H., Rubin K., Gerdin B., Ivarsson M. 2004. Analysis of gene expression in fibroblasts in response to keratinocyte-derived factors in vitro: potential implications for the wound healing process. J. Invest. Dermatol. 122, 216–221 10.1046/j.0022-202X.2003.22112.x (doi:10.1046/j.0022-202X.2003.22112.x) [DOI] [PubMed] [Google Scholar]
- 6.Ghaffari A., Kilani R. T., Ghahary A. 2009. Keratinocyte-conditioned media regulate collagen expression in dermal fibroblasts. J. Invest. Dermatol. 129, 340–347 10.1038/jid.2008.253 (doi:10.1038/jid.2008.253) [DOI] [PubMed] [Google Scholar]
- 7.El Ghalbzouri A., Gibbs S., Lamme E., Van Blitterswijk C., Ponec M. 2002. Effect of fibroblasts on epidermal regeneration. Br. J. Dermatol. 147, 230–243 10.1046/j.1365-2133.2002.04871.x (doi:10.1046/j.1365-2133.2002.04871.x) [DOI] [PubMed] [Google Scholar]
- 8.Werner S., Krieg T., Smola H. 2007. Keratinocyte–fibroblast interactions in wound healing. J. Invest. Dermatol. 127, 998–1008 10.1152/physrev.00032.2002 (doi:10.1152/physrev.00032.2002) [DOI] [PubMed] [Google Scholar]
- 9.Souren J. E. M., Ponec M., Van Wijk R. 1989. Contraction of collagen by human fibroblasts and keratinocytes. In Vitro Cell. Dev. B 25, 1039–1045 10.1007/BF02624138 (doi:10.1007/BF02624138) [DOI] [PubMed] [Google Scholar]
- 10.Harrison C., Gossiel F., Bullock A., Sun T., Blumsohn A., Mac Neil S. 2006. Investigation of keratinocyte regulation of collagen I synthesis by dermal fibroblasts in a simple in vitro model. Br. J. Dermatol. 154, 401–410 10.1111/j.1365-2133.2005.07022.x (doi:10.1111/j.1365-2133.2005.07022.x) [DOI] [PubMed] [Google Scholar]
- 11.Ghahary A., Ghaffari A. 2007. Role of keratinocyte–fibroblast cross-talk in development of hypertrophic scar. Wound Repair Regen. 15, S46–S53 10.1111/j.1524-475X.2007.00225.x (doi:10.1111/j.1524-475X.2007.00225.x) [DOI] [PubMed] [Google Scholar]
- 12.Funayama E., Chodon T., Oyama A., Sugihara T. 2003. Keratinocytes promote proliferation and inhibit apoptosis of the underlying fibroblasts: an important role in the pathogenesis of keloid. J. Invest. Dermatol. 121, 1326–1331 10.1111/j.1523-1747.2003.12572.x (doi:10.1111/j.1523-1747.2003.12572.x) [DOI] [PubMed] [Google Scholar]
- 13.Thackham J. A., McElwain D. L. S., Long R. J. 2008. The use of hyperbaric oxygen therapy to treat chronic wounds: a review. Wound Repair Regen. 16, 321–330 10.1111/j.1524-475X.2008.00372.x (doi:10.1111/j.1524-475X.2008.00372.x) [DOI] [PubMed] [Google Scholar]
- 14.Sherratt J. A., Murray J. D. 1990. Models of epidermal wound healing. Proc. R. Soc. Lond. B 241, 29–36 10.1098/rspb.1990.0061 (doi:10.1098/rspb.1990.0061) [DOI] [PubMed] [Google Scholar]
- 15.Tranquillo R. T., Murray J. D. 1992. Continuum model of fibroblast-driven wound contraction: inflammation-mediation. J. Theor. Biol. 158, 135–172 10.1016/S0022-5193(05)80715-5 (doi:10.1016/S0022-5193(05)80715-5) [DOI] [PubMed] [Google Scholar]
- 16.Olsen L., Sherratt J. A., Maini P. K. 1995. A mechanochemical model for adult dermal wound contraction and the permanence of the contracted tissue displacement profile. J. Theor. Biol. 177, 113–128 10.1006/jtbi.1995.0230 (doi:10.1006/jtbi.1995.0230) [DOI] [PubMed] [Google Scholar]
- 17.Tranqui L., Tracqui P. 2000. Mechanical signaling and angiogenesis. The integration of cell-extracellular matrix couplings. CR Acad. Sci. III-Vie 323, 31–47 10.1016/S0764-4469(00)00110-4 (doi:10.1016/S0764-4469(00)00110-4) [DOI] [PubMed] [Google Scholar]
- 18.Vermolen F. J., Javierre E. 2010. Computer simulations from a finite-element model for wound contraction and closure. J. Tissue Viability 19, 43–53 10.1016/j.jtv.2009.11.003 (doi:10.1016/j.jtv.2009.11.003) [DOI] [PubMed] [Google Scholar]
- 19.Cruywagen G., Murray J. D. 1992. On a tissue interaction model for skin pattern formation. J. Nonlinear Sci. 2, 217–240 10.1007/bf02429856 (doi:10.1007/bf02429856) [DOI] [PubMed] [Google Scholar]
- 20.Wearing H. J., Sherratt J. A. 2000. Keratinocyte growth factor signalling: a mathematical model of dermal–epidermal interaction in epidermal wound healing. Math. Biosci. 165, 41–62 10.1016/S0025-5564(00)00008-0 (doi:10.1016/S0025-5564(00)00008-0) [DOI] [PubMed] [Google Scholar]
- 21.Haugh J. M. 2006. Deterministic model of dermal wound invasion incorporating receptor-mediated signal transduction and spatial gradient sensing. Biophys. J. 90, 2297–2308 10.1529/biophysj.105.077610 (doi:10.1529/biophysj.105.077610) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Murphy K. E., Hall C. L., McCue S. W., McElwain D. L. S. 2011. A two-compartment mechanochemical model of the roles of transforming growth factor β and tissue tension in dermal wound healing. J. Theor. Biol. 272, 145–159 10.1016/j.jtbi.2010.12.011 (doi:10.1016/j.jtbi.2010.12.011) [DOI] [PubMed] [Google Scholar]
- 23.Murphy K. E., Hall C. L., Maini P. K., McCue S. W., McElwain D. L. S. 2012. A fibrocontractive mechanochemical model of dermal wound closure incorporating realistic growth factor kinetics. Bull. Math. Biol. 74, 1143–1170 10.1007/s11538-011-9712-y (doi:10.1007/s11538-011-9712-y) [DOI] [PubMed] [Google Scholar]
- 24.Murphy K. E., McCue S. W., McElwain D. L. S. 2012. Clinical strategies for contractures from a predictive mathematical model of dermal repair. Wound Repair Regen. 20, 194–202 10.1111/j.1524-475X.2012.00775.x (doi:10.1111/j.1524-475X.2012.00775.x) [DOI] [PubMed] [Google Scholar]
- 25.Dale P. D., Sherratt J. A., Maini P. K. 1996. A mathematical model for collagen fibre formation during foetal and adult dermal wound healing. Proc. R. Soc. Lond. B 263, 653–660 10.1098/rspb.1996.0098 (doi:10.1098/rspb.1996.0098) [DOI] [PubMed] [Google Scholar]
- 26.Dallon J. C., Sherratt J. A., Maini P. K. 2001. Modeling the effects of transforming growth factor-β on extracellular matrix alignment in dermal wound repair. Wound Repair Regen. 9, 278–286 10.1046/j.1524-475X.2001.00278.x (doi:10.1046/j.1524-475X.2001.00278.x) [DOI] [PubMed] [Google Scholar]
- 27.Cumming B. D., McElwain D. L. S., Upton Z. 2009. A mathematical model of wound healing and subsequent scarring. J. R. Soc. Interface 7, 19–34 10.1098/rsif.2008.0536 (doi:10.1098/rsif.2008.0536) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Singer A. J., Clark R. A. F. 1999. Cutaneous wound healing. New Engl. J. Med. 341, 738–746 10.1056/NEJM199909023411006 (doi:10.1056/NEJM199909023411006) [DOI] [PubMed] [Google Scholar]
- 29.Clark R. A. F., Ghosh K., Tonnesen M. G. 2007. Tissue engineering for cutaneous wounds. J. Invest. Dermatol. 127, 1018–1029 10.1038/sj.jid.5700715 (doi:10.1038/sj.jid.5700715) [DOI] [PubMed] [Google Scholar]
- 30.Hunt T. K., Gimbel M. L. 2002. Hyperbaric oxygen and wound healing. In Hyperbaric surgery: perioperative care (eds Bakker D. J., Cramer F. S.), pp. 439–459 Flagstaff, AZ: Best Publishing Company [Google Scholar]
- 31.Oberringer M., Meins C., Bubel M., Pohlemann T. 2007. A new in vitro wound model based on the co-culture of human dermal microvascular endothelial cells and human dermal fibroblasts. Biol. Cell. 99, 197–207 10.1042/BC20060116 (doi:10.1042/BC20060116) [DOI] [PubMed] [Google Scholar]
- 32.Mathieu D. 2002. Hyperbaric oxygen therapy in the management of non-healing wounds. In Hyperbaric surgery: perioperative care (eds Bakker D. J, Cramer F. S.), pp. 317–339 Flagstaff, AZ: Best Publishing Company [Google Scholar]
- 33.Lazarus G. S., Cooper D. M., Knighton D. R., Margolis D. J., Percoraro R. E., Rodeheaver G., Robson M. C. 1994. Definitions and guidelines for assessment of wounds and evaluation of healing. Wound Repair Regen. 2, 165–170 10.1046/j.1524-475X.1994.20305.x (doi:10.1046/j.1524-475X.1994.20305.x) [DOI] [PubMed] [Google Scholar]
- 34.Barrientos S., Stojadinovic O., Golinko M. S., Brem H., Tomic-Canic M. 2008. Growth factors and cytokines in wound healing. Wound Repair Regen. 16, 585–601 10.1111/j.1524-475X.2008.00410.x (doi:10.1111/j.1524-475X.2008.00410.x) [DOI] [PubMed] [Google Scholar]
- 35.O'Toole E. A. 2001. Extracellular matrix and keratinocyte migration. Clin. Exp. Dermatol. 26, 525–530 10.1046/j.1365-2230.2001.00891.x (doi:10.1046/j.1365-2230.2001.00891.x) [DOI] [PubMed] [Google Scholar]
- 36.Leigh I. M. 2003. Biology of keratinocytes. In Dermatology (eds Bolognia J. L., Jorizzo J. L., Rapini R. P.), pp. 763–774 New York, NY: Mosby Elsevier [Google Scholar]
- 37.Pilcher B. K., Dumin J. A., Sudbeck B. D., Krane S. M., Welgus H. G., Parks W. C. 1997. The activity of collagenase-1 is required for keratinocyte migration on a type I collagen matrix. J. Cell Biol. 137, 1445–1457 10.1083/jcb.137.6.1445 (doi:10.1083/jcb.137.6.1445) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Donaldson D. J., Mahan J. T. 1988. Keratinocyte migration and the extracellular matrix. J. Invest. Dermatol. 90, 623–628 10.1111/1523-1747.ep12560762 (doi:10.1111/1523-1747.ep12560762) [DOI] [PubMed] [Google Scholar]
- 39.Pettet G. J., Chaplain M. A. J., McElwain D. L. S., Byrne H. M. 1996. On the role of angiogenesis in wound healing. Proc. R. Soc. Lond. B 263, 1487–1493 10.1098/rspb.1996.0217 (doi:10.1098/rspb.1996.0217) [DOI] [PubMed] [Google Scholar]
- 40.Yamamoto M., Yanaga H., Nishina H., Watabe S., Mamba K. 2005. Fibrin stimulates the proliferation of human keratinocytes through the autocrine mechanism of transforming growth factor-α and epidermal growth factor receptor. Tohoku J. Exp. Med. 207, 33–40 10.1620/tjem.207.33 (doi:10.1620/tjem.207.33) [DOI] [PubMed] [Google Scholar]
- 41.Varani J., Perone P., O'Brien Deming M., Warner R. L., Aslam M. N., Bhagavathula N., Dame M. K., Voorhees J. J. 2009. Impaired keratinocyte function on matrix metalloproteinase-1 (MMP-1) damaged collagen. Arch. Dertmatol. Res. 301, 497–506 10.1007/s00403-009-0948-4 (doi:10.1007/s00403-009-0948-4) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rubin J. S., et al. 1995. Keratinocyte growth factor. Cell Biol. Int. 19, 399–412 10.1006/cbir.1995.1085 (doi:10.1006/cbir.1995.1085) [DOI] [PubMed] [Google Scholar]
- 43.Brauchle M., Angermeyer K., Hubner G., Werner S. 1994. Large induction of keratinocyte growth factor expression by serum growth factors and pro-inflammatory cytokines in cultured fibroblasts. Oncogene 9, 3199–3204 [PubMed] [Google Scholar]
- 44.Hebda P. A. 1988. Stimulatory effects of transforming growth factor-beta and epidermal growth factor on epidermal cell outgrowth from porcine skin explant cultures. J. Invest. Dermatol. 91, 440–445 10.1111/1523-1747.ep12476480 (doi:10.1111/1523-1747.ep12476480) [DOI] [PubMed] [Google Scholar]
- 45.Postlethwaite A. E., Keski-Oja J., Moses H. L., Kang A. H. 1987. Stimulation of the chemotactic migration of human fibroblasts by transforming growth factor beta. J. Exp. Med. 165, 251–256 10.1084/jem.165.1.251 (doi:10.1084/jem.165.1.251) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schmidt J. A., Mizel S. B., Cohen D., Green I. 1982. Interleukin 1, a potential regulator of fibroblast proliferation. J. Immunol. 128, 2177–2182 [PubMed] [Google Scholar]
- 47.Sun T., Higham M., Layton C., Haycock J., Short R., MacNeil S. 2004. Developments in xenobiotic-free culture of human keratinocytes for clinical use. Wound Repair Regen. 12, 626–634 10.1111/j.1067-1927.2004.12609.x (doi:10.1111/j.1067-1927.2004.12609.x) [DOI] [PubMed] [Google Scholar]
- 48.Faler B. J., Macsata R. A., Plummer D., Mishra L., Sidawy A. N. 2006. Transforming growth factor-β and wound healing. Perspect. Vasc. Surg. Endovasc. Ther. 18, 55–62 10.1177/153100350601800123 (doi:10.1177/153100350601800123) [DOI] [PubMed] [Google Scholar]
- 49.Warriner R., Burrell R. 2005. Infection and the chronic wound: a focus on silver. Adv. Skin Wound Care 18, 2–12 [DOI] [PubMed] [Google Scholar]
- 50.Streit M., Beleznay Z., Braathen L. R. 2006. Topical application of the tumour necrosis factor-α antibody infliximab improves healing of chronic wounds. Int. Wound J. 3, 171–179 10.1111/j.1742-481X.2006.00233.x (doi:10.1111/j.1742-481X.2006.00233.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Welch M. P., Odland G. F., Clark R. A. F. 1990. Temporal relationships of F-actin bundle formation, collagen and fibronectin matrix assembly, and fibronectin receptor expression to wound contraction. J. Cell Biol. 110, 133–145 10.1083/jcb.110.1.133 (doi:10.1083/jcb.110.1.133) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Flegg J. A., McElwain D. L. S., Byrne H. M. 2010. Mathematical model of hyperbaric oxygen therapy applied to chronic diabetic wounds. Bull. Math. Biol. 72, 1867–1891 10.1007/s11538-010-9514-7 (doi:10.1007/s11538-010-9514-7) [DOI] [PubMed] [Google Scholar]
- 53.Tracqui P., Woodward D. E., Cruywagen G. C., Cook J., Murray J. D. 1995. A mechanical model for fibroblast-driven wound healing. J. Biol. Syst. 3, 1075–1084 10.1142/S0218339095000976 (doi:10.1142/S0218339095000976) [DOI] [Google Scholar]
- 54.Sen C. K. 2009. Wound healing essentials: let there be oxygen. Wound Repair Regen. 17, 1–18 10.1111/j.1524-475X.2008.00436.x (doi:10.1111/j.1524-475X.2008.00436.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mauviel A., Heino J., Kähäri V.-M., Hartmann D.-J., Loyau G., Pujol J.-P., Vuorio E. 1991. Comparative effects of interleukin-1 and tumor necrosis factor-α on collagen production and corresponding procollagen mRNA levels in human dermal fibroblasts. J. Invest. Dermatol. 96, 243–249 10.1111/1523-1747.ep12462185 (doi:10.1111/1523-1747.ep12462185) [DOI] [PubMed] [Google Scholar]
- 56.Ignotz R. A., Massagué J. 1986. Transforming growth factor-β stimulates the expression of fibronectin and collagen and their incorporation into the extracellular matrix. J. Biol. Chem. 261, 4337–4345 [PubMed] [Google Scholar]
- 57.Bienkowski R. S., Baum B. J., Crystal R. G. 1978. Fibroblasts degrade newly synthesised collagen within the cell before secretion. Nature 276, 413–416 10.1038/276413a0 (doi:10.1038/276413a0) [DOI] [PubMed] [Google Scholar]
- 58.McDougall S., Dallon J., Sherratt J., Maini P. 2006. Fibroblast migration and collagen deposition during dermal wound healing: mathematical modelling and clinical implications. Phil. Trans. R. Soc. A 364, 1385–1405 10.1098/rsta.2006.1773 (doi:10.1098/rsta.2006.1773) [DOI] [PubMed] [Google Scholar]
- 59.Barone E. J., Yager D. R., Pozez A. L., Olutoye O. O., Crossland M. C., Diegelmann R. F., Cohen I. K. 1998. Interleukin-α and collagenase activity are elevated in chronic wounds. Plast. Reconstr. Surg. 102, 1023–1027 [PubMed] [Google Scholar]
- 60.Luger T. A., Schwarz T. 1990. Evidence for an epidermal cytokine network. J. Invest. Dermatol. 95, S100–S104 (doi:10.1111/1523-1747.ep12874944) [DOI] [PubMed] [Google Scholar]
- 61.Folkman J. 1995. Angiogenesis in cancer, vascular, rheumatoid and other diseases. Nat. Med. 1, 1. 10.1038/nm0195-27 (doi:10.1038/nm0195-27) [DOI] [PubMed] [Google Scholar]
- 62.Staiano-Coico L., Krueger J. G., Rubin J. S., D'Limi S., Vallat V. P., Valentino L., Fahey T., Hawes A., Kingston G., Madden M. R. 1993. Human keratinocyte growth factor effects in a porcine model of epidermal wound healing. J. Exp. Med. 178, 865–878 10.1084/jem.178.3.865 (doi:10.1084/jem.178.3.865) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ferguson M. W. J, O'Kane S. 2004. Scar-free healing: From embryonic mechanisms to adult therapeutic intervention. Phil. Trans. R. Soc. Lond. B 359, 839–850 10.1098/rstb.2004.1475 (doi:10.1098/rstb.2004.1475) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lawrence D. A. 1996. Transforming growth factor-beta: a general review. Eur. Cytokine Netw. 7, 363–374 [PubMed] [Google Scholar]
- 65.Roberts A. B., Sporn M. B. 1996. Transforming growth factor-β. In The molecular and cellular biology of wound repair (ed. Clark R. A. F.), pp. 275–308 New York, NY: Plenum Press [Google Scholar]
- 66.Ghahary A., Shen Y. J., Nedelec B., Scott P. G., Tredget E. E. 1995. Enhanced expression of mRNA for insulin-like growth factor-1 in post-burn hypertrophic scar tissue and its fibrogenic role by dermal fibroblasts. Mol. Cell. Biochem. 148, 25–32 10.1007/BF00929499 (doi:10.1007/BF00929499) [DOI] [PubMed] [Google Scholar]
- 67.Wipff P.-J., Rifkin D. B., Meister J.-J., Hinz B. 2007. Myofibroblast contraction activates latent TGF-β1 from the extracellular matrix. J. Cell Biol. 179, 1311–1323 10.1083/jcb.200704042 (doi:10.1083/jcb.200704042) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Le Poole I. C., Boyce S. T. 1999. Keratinocytes suppress transforming growth factor-β1 expression by fibroblasts in cultured skin substitutes. Br. J. Dermatol. 140, 409–416 10.1046/j.1365-2133.1999.02700.x (doi:10.1046/j.1365-2133.1999.02700.x) [DOI] [PubMed] [Google Scholar]
- 69.Vande Berg J. S., Rudolph R., Poolman W. L., Disharoon D. R. 1989. Comparative growth dynamics and actin concentration between cultured human myofibroblasts from granulating wounds and dermal fibroblasts from normal skin. Lab. Invest. 61, 532–538 [PubMed] [Google Scholar]
- 70.Namy P., Ohayon J., Tracqui P. 2004. Critical conditions for pattern formation and in vitro tubulogenesis driven by cellular traction fields. J. Theor. Biol. 227, 103–120 10.1016/j.jtbi.2003.10.015 (doi:10.1016/j.jtbi.2003.10.015) [DOI] [PubMed] [Google Scholar]
- 71.Togawa A., Oppenheim J. J., Mizel S. B. 1979. Characterization of lymphocyte-activating factor (LAF) produced by human mononuclear cells: biochemical relationship of high and low molecular weight forms of LAF. J. Immunol. 122, 2112–2118 [PubMed] [Google Scholar]
- 72.Yang L., Qiu C. X., Ludlow A., Ferguson M. W. J., Brunner G. 1999. Active transforming growth factor-β in wound repair: determination using a new assay. Am. J. Pathol. 154, 105–111 10.1016/S0002-9440(10)65256-X (doi:10.1016/S0002-9440(10)65256-X) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bard J. B., Hay E. D. 1975. The behavior of fibroblasts from the developing avian cornea. Morphology and movement in situ and in vitro. J. Cell. Biol. 67, 400–418 10.1083/jcb.67.2.400 (doi:10.1083/jcb.67.2.400) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Chedid M., Rubin J. S., Csaky K. G., Aaronson S. A. 1994. Regulation of keratinocyte growth factor gene expression by interleukin 1. J. Biol. Chem. 269, 10 753–10 757 [PubMed] [Google Scholar]
- 75.Yang Y., Gil M., Byun S. M., Choi I., Pyun K. H., Ha H. 1996. Transforming growth factor-β1 Inhibits human keratinocyte proliferation by upregulation of a receptor-type tyrosine phosphatase R-PTP-κ gene expression. Biochem. Biophys. Res. Co. 228, 807–812 10.1006/bbrc.1996.1736 (doi:10.1006/bbrc.1996.1736) [DOI] [PubMed] [Google Scholar]
- 76.Postlethwaite A. E., Lachman L. B., Kang A. H. 1984. Induction of fibroblast proliferation by interleukin-1 derived from human monocytic leukemia cells. Arthritis Rheum. 27, 995–1001 10.1002/art.1780270905 (doi:10.1002/art.1780270905) [DOI] [PubMed] [Google Scholar]
- 77.Strutz F., Zeisberg M., Renziehausen A., Raschke B., Becker V., Van Kooten C., Müller G. A. 2001. TGF-β1 induces proliferation in human renal fibroblasts via induction of basic fibroblast growth factor (FGF-2). Kidney Int. 59, 579–592 10.1046/j.1523-1755.2001.059002579.x (doi:10.1046/j.1523-1755.2001.059002579.x) [DOI] [PubMed] [Google Scholar]
- 78.Eickelberg O., Köhler E., Reichenberger F., Bertschin S., Woodtli T., Erne P., Perruchoud A., Roth M. 1999. Extracellular matrix deposition by primary human lung fibroblasts in response to TGF-β1 and TGF-β3. Am. J. Physiol. Lung C 276, L814–L824. [DOI] [PubMed] [Google Scholar]
- 79.Gorospe M., Kumar S., Baglioni C. 1993. Tumor necrosis factor increases stability of interleukin-1 mRNA by activating protein kinase C. J. Biol. Chem. 268, 6214–6220 [PubMed] [Google Scholar]
- 80.Wang R., Ghahary A., Shen Q., Scott P. G., Roy K., Tredget E. E. 2000. Hypertrophic scar tissues and fibroblasts produce more transforming growth factor-β1 mRNA and protein than normal skin and cells. Wound Repair Regen. 8, 128–137 10.1046/j.1524-475x.2000.00128.x (doi:10.1046/j.1524-475x.2000.00128.x) [DOI] [PubMed] [Google Scholar]
- 81.Dale P. D. 1995 Time heals all wounds? Mathematical models of epithelial and dermal wound healing. PhD thesis, University of Oxford, Oxford, UK. [Google Scholar]
- 82.NAG 2009. The NAG Fortran library manual. Mark 22 Oxford, UK: The Numerical Algorithms Group [Google Scholar]
- 83.Ron D., Bottaro D. P., Finch P. W., Morris D., Rubin J. S., Aaronson S. A. 1993. Expression of biologically active recombinant keratinocyte growth factor. Structure/function analysis of amino-terminal truncation mutants. J. Biol. Chem. 268, 2984–2988 [PubMed] [Google Scholar]
- 84.Haslett C., Henson P. 1996. Resolution of inflammation. In The molecular and cellular biology of wound repair (ed. Clark R. A. F.), pp. 143–165 New York, NY: Plenum Press [Google Scholar]
- 85.Schreml S., Szeimies R. M., Prantl L., Karrer S., Landthaler1 M., Babilas P. 2010. Oxygen in acute and chronic wound healing. Br. J. Dermatol. 163, 257–268 10.1111/j.1365-2133.2010.09804.x (doi:10.1111/j.1365-2133.2010.09804.x) [DOI] [PubMed] [Google Scholar]
- 86.Siddiqui A., Galiano R., Connors D., Gruskin E., Wu L., Mustoe T. 1996. Differential effects of oxygen on human dermal fibroblasts: acute versus chronic hypoxia. Wound Repair Regen. 4, 211–218 10.1046/j.1524-475X.1996.40207.x (doi:10.1046/j.1524-475X.1996.40207.x) [DOI] [PubMed] [Google Scholar]
- 87.Sheffield P., Smith A. 2002. Physiological and pharmacological basis of hyperbaric oxygen therapy. In Hyperbaric surgery: perioperative care (eds Bakker D. J., Cramer F. S.), pp. 63–109 Flagstaff, AZ: Best Publishing Company [Google Scholar]
- 88.Doctor N., Pandya S., Supe A. 1992. Hyperbaric oxygen therapy in diabetic foot. J. Postgrad. Med. 38, 112–115 [PubMed] [Google Scholar]