

Abstract
Follistatin (FST)-type proteins are important antagonists of some members of the large TGF-β family of cytokines. These include myostatin, an important negative regulator of muscle growth, and the closely related activin A, which is involved in many physiological functions, including maintenance of a normal reproductive axis. FST-type proteins, including FST and FST-like 3 (FSTL3), differentially inhibit various TGF-β family ligands by binding each ligand with two FST-type molecules. In this study, we sought to examine features that are important for ligand antagonism by FST-type proteins. Previous work has shown that a modified construct consisting of the FST N-terminal domain (ND) followed by two repeating follistatin domains (FSD), herein called FST ND-FSD1-FSD1, exhibits strong specificity for myostatin over activin A. Using cell-based assays, we show that FST ND-FSD1-FSD1 is unique in its specificity for myostatin as compared with similar constructs containing domains from FSTL3 and that the ND is critical to its activity. Furthermore, we demonstrate that FSD3 of FST provides affinity to ligand inhibition and confers resistance to perturbations in the ND and FSD2, likely through the interaction of FSD3 of one FST molecule with the ND of the other FST molecule. Additionally, our data suggest that this contact provides cooperativity to ligand antagonism. Cross-linking studies show that this interaction also potentiates formation of 1:2 ligand-FST complexes, whereas lack of FSD3 allows formation of 1:1 complexes. Altogether, these studies support that domain differences generate FST-type molecules that are each uniquely suited ligand antagonists.
Follistatin (FST)-type proteins are multidomain regulatory factors that function by binding to and inhibiting certain members of the TGF-β family of signaling molecules (1). These include myostatin, a staunch inhibitor of muscle growth, and activin A, which is involved in a variety of physiological functions including reproduction, metabolism, and immunity (2–5). FST-type proteins include FST and FST-like 3 (FSTL3) and differ from one another in their architecture, binding characteristics, and specificity for TGF-β family ligands, leading to differences in physiological functions. FST, which includes FST288 and the isoform FST315, was originally identified as a suppressor of the release of FSH, whereas FSTL3 was identified as a factor involved in leukemia (6, 7). Regulation by FST is critical to life, because FST knockout in mice causes a multitude of defects that are fatal shortly after birth (8). FSTL3 knockout does not have as severe an effect yet leads to a variety of metabolic phenotypes (9, 10). FST contains an N-terminal domain (ND) followed by three FST domains (FSD1–3) with a heparin-binding sequence located in FSD1 (Fig. 1A) (7, 11). FSTL3 has a similar domain layout but lacks FSD3 and heparin binding (12).
Fig. 1.

Overview of ligand and FST-type structures. A, Domain layouts of FST and FSTL3, which are similar to one another. B, TGF-β ligands are dimers with four surfaces important for type I and type II receptor binding, as indicated. Myostatin is shown as an example. C, The structure of myostatin bound to FST288 [3HH2 (28)]. FST antagonizes signaling by completely surrounding a ligand with two FST molecules, blocking all receptor-binding sites. The ND of FST plugs the type I receptor-binding site, whereas FSD1 and FSD2 cover the type II site. There are additional contacts between the ND of one FST molecule and FSD3 of the other. FSD3 itself does not interact with ligand. D, The structure of myostatin bound to FSTL3 [3SEK (25)]. FSTL3 binds ligands similarly to FST but lacks the ND-FSD3 interaction. Structure figures were rendered using PyMOL (48).
TGF-β family ligands are secreted molecules that signal by binding transmembrane serine/threonine kinase receptors at the cell surface. These ligands are typically disulfide-bonded dimers that contain two concave surfaces for type I receptor binding and two convex surfaces for type II receptor binding (Fig. 1B) (13–21). Ligands can generally be divided into three main classes: activin/inhibin, bone morphogenetic protein (BMP)/growth and differentiation factor, and TGF-β (22). Both FST and FSTL3 are staunch inhibitors of the activin class of ligands, whereas only FST inhibits some members of the BMP class, although to a lesser extent (23–27). This is despite the fact that all TGF-β family ligands display a similar fold (Fig. 1B). Multiple crystal structures of different FST-type proteins in complex with either myostatin or the close family member activin A show that these antagonists inhibit ligand signaling by completely surrounding the ligand with two FST-type molecules, blocking all four receptor-binding sites (Fig. 1, C and D) (25, 28–31). These structures reveal that FSD1 and FSD2 cover the type II receptor binding site, whereas the ND plugs the type I receptor binding site. With FST, although FSD3 makes no contact with the ligand itself, there are additional interactions that occur between the ND of one FST molecule and FSD3 of the other. However, the contribution of the ND-FSD3 interaction to ligand antagonism remains largely unexplored. Despite the structural similarity between myostatin and activin A, FST and FSTL3 exhibit differential affinity for these ligands, with both binding more tightly to activin A (23, 25–27). Our lab and others have directed significant effort toward understanding this difference in ligand affinity and specificity.
Several studies have analyzed the contribution and importance of the different domains of FST-type proteins to ligand antagonism (30, 32–36). For example, we previously determined that single negative charges introduced into FSTL3 at the hydrophobic interface between the ND and ligand could be detrimental to binding, whereas similar mutations in FST had little effect (25). These types of studies are important to further our understanding of how TGF-β family members are regulated by FST-type molecules and, in particular, to elucidate how these antagonists specifically inhibit signaling of certain subsets of ligands. This knowledge has led to the design of modified FST-type proteins that have altered ligand specificity, including a FST construct consisting of the ND followed by two FSD1 (ND-FSD1-FSD1) that exhibits high affinity for myostatin but not activin A [(33, 37) Note that bold type indicates a FST domain, whereas italic type indicate a FSTL3 domain, and plain type signifies broad reference to a particular domain in both proteins.] This construct has shown promise in animal models in promoting the growth of muscle mass and, therefore, may have therapeutic potential in the treatment of muscle-wasting diseases such as muscular dystrophy (26). Structural data support that this affinity and specificity for myostatin may be due to unique interactions between the FST ND and myostatin that are absent with activin A (28). Here, we have built on this knowledge base by further characterizing the domains of FST-type proteins and their contribution to binding myostatin and activin A. We show that the FST ND-FSD1-FSD1 construct is unique in its ligand specificity as compared with other similar FST and FSTL3 domain swap constructs and that the ND is necessary but not sufficient for this property. We also demonstrate that FSD3 provides added affinity and stability to ligand inhibition, likely through its interaction with the ND of its partner FST molecule when bound to ligand. We furthermore provide evidence that this interaction confers cooperativity to ligand antagonism and also potentiates the formation of 1:2 complexes, whereas 1:1 complexes can form in the absence of FSD3. These findings are an important step toward further modification and design of highly specific TGF-β family ligand inhibitors.
Materials and Methods
Production and purification of protein constructs
Human FST288 and FSTL3 were cloned into the pcDNA3.1/myc-His expression vector (Invitrogen, Grand Island, NY) as described previously (35, 36). FSTΔD3 was generated by amplifying FST ND-FSD1-FSD2 from wild-type FST with primers that added EcoRI and XhoI sites to the DNA ends and then ligating this fragment into the pcDNA3.1/myc-His expression vector (Invitrogen). The FST ND-FSD1-FSD1 construct was similarly generated by amplifying this fragment from FST ND-FSD1-FSD1-FSD3 (35). The ND-FSD1-FSD1 construct was made by first amplifying ND-FSD1 from ND-FSD1-FSD2 (30) with primers that added EcoRI and XhoI sites to the DNA ends and then ligating the fragment into the pcDNA vector. FSD1 was amplified from wild-type FSTL3 while adding XhoI and XbaI sites onto the ends and then ligated into this construct. Site-directed mutagenesis was then used to remove the XhoI site from between the two FSD1. The ND-FSD1-FSD1 construct was made by first introducing an EcoRV site between the two FSD1 of ND-FSD1-FSD1 and then removing ND-FSD1 using EcoRI and EcoRV. ND-FSD1 was then amplified from wild-type FSTL3 while introducing EcoRI and EcoRV sites onto the DNA ends. This fragment was then introduced into the previous construct using EcoRI and EcoRV, after which the EcoRV site was removed from between the two FSD1. The ND-FSD1-FSD1 construct was similarly generated by first introducing an EcoRV site between the two FSD1 of FST ND-FSD1-FSD1 and then removing ND-FSD1 using EcoRI and EcoRV. ND-FSD1 was then amplified from ND-FSD1-FSD2 while adding EcoRI and EcoRV sites onto the ends. This product was then introduced into the previous construct using EcoRI and EcoRV, after which the EcoRV site was removed from between the two FSD1. Resulting constructs were used as templates for site-directed mutagenesis to generate point mutants. Wild-type and altered myc-His-tagged FST-type proteins were produced and purified as previously described (30). Briefly, HEK293F cells were transiently transfected with expression vectors containing the FST variants. Conditioned media were harvested after 6 d and applied to nickel-nitriloacetic acid His-affinity resin (His Bind; EMD Millipore, Darmstadt, Germany). The column was washed with a buffer of 500 mm NaCl, 20 mm Tris (pH 8.0) to remove nonspecific interactions, and the myc-His-tagged FST proteins were eluted with a buffer of 500 mm NaCl, 500 mm imidazole, 20 mm Tris (pH 8.0). Fractions containing FST variants were verified by anti-myc Western blotting and pooled. Proteins were quantified by a solution phase assay directed toward the C-terminal myc tag (36). Briefly, a radiolabeled synthetic peptide containing the myc epitope was captured with a polyclonal anti-myc antibody and quantified by a scintillation counter. FST-type proteins were quantified by comparing the samples to a standard curve generated by the competition of the labeled myc peptide with increasing concentrations of unlabeled peptide from 0.3–100 nm. For cross-linking experiments, activin A and FST288 were produced and purified as previously described (18, 31). In each case, CHO cells that stably express each protein were used. FST288 was purified from conditioned medium by binding to a heparin-Sepharose column and then a cation exchange column as previously described (31). Activin A was purified from conditioned medium by binding to a HiTrap N-hydroxysuccinimide-activated HP column (GE Healthcare, Piscataway, NJ) coupled with FST288 and eluting with a buffer containing 150 mm NaCl, 50 mm glycine (pH 2.5) with 0.03% Tween 80.
Luciferase reporter assays
Luciferase reporter assays with purified protein were performed as previously described using our HEK293 (CAGA)12 stable cell line (25). Assays with transiently expressed constructs were performed using the same cell line in a 24-well plate format. Cells were plated at 0.6 × 105 cells per well on a plate (TPP, St. Louis, MO) coated with poly-d-lysine (BD Biosciences, San Diego, CA) and grown approximately 24 h. Cells were then transfected with increasing amounts of the appropriate FST-type construct using TransIT-LT1 transfection reagent (Mirus, Madison, WI), and empty pcDNA3 was used to normalize the total DNA transfected to 750 ng/well. Approximately 24 h later, cells were treated with either myostatin or activin A at 8 ng/well and incubated 18–20 h. The medium was then removed, and the cells were lysed in 100 μl passive lysis buffer (Promega, Madison, WI). Twenty microliters of lysate were mixed with 40 μl luciferase assay substrate (Promega), and luminescence was recorded immediately in a GloMax 20/20 luminometer (Promega). Each condition was performed in triplicate for each experiment, and each experiment was performed two or more times. Figures represent the combined data from all experiments. Data for cooperativity experiments was analyzed using GraphPad Prism version 5 and nonlinear regression. Parameters selected for graphing were least-squares curve fit and inhibition with normalized dose-response and variable slope. Hill slope values were calculated by comparing independent fits with a global fit where the Hill slope was shared. The Hill slope was found to be the same with each experiment performed three times in triplicate, and this analysis was reported (see Fig. 7).
Fig. 7.

Luciferase reporter assays support that FSD3 confers cooperativity to antagonism by FST. Myostatin and activin A were mixed with FSTL3, FST, or FSTΔD3 and applied to HEK293-CAGA cells. Data were analyzed using GraphPad Prism version 5. The table represents the average of data from three independent experiments for each complex. One representative experiment is shown in each graph. Error bars represent ±sem. Hill slope values close to −1 suggest no cooperativity, whereas those much less than −1 reveal cooperative inhibition.
Radiolabel binding assay
Binding of altered FST-type proteins to labeled activin A was determined by competition assay as described (38). Relative potencies were calculated by comparison of half-maximal inhibition of labeled activin A binding by altered and wild-type FSTL3 or FST.
Cross-linking experiments
For cross-linking experiments, activin A and FSTL3 were dialyzed into a buffer of 150 mm NaCl, 20 mm HEPES (pH 7.5); FST was in a buffer of 150 mm NaCl, 50 mm 2-(N-morpholino)ethanesulfonic acid (pH 5); and FSTΔD3 was in PBS. Activin A was mixed with the appropriate FST-type protein in a buffer of 500 mm NaCl and 20 mm HEPES (pH 7.5) and incubated 30 min. Proteins were cross-linked with 1% glutaraldehyde for 20 min, and then 200 mm Tris was used to quench the reaction. Western blotting was performed on the resulting products.
Results
FST ND-FSD1-FSD1 is a unique, specific myostatin antagonist
Previous studies have shown that a FST construct consisting of the ND followed by two FSD1 (ND-FSD1-FSD1) shows high specificity and affinity for myostatin as compared with the closely related activin A (26, 37). We sought to determine whether or not similar domain swap constructs, namely FSTL3 ND-FSD1-FSD1, ND-FSD1-FSD1, and ND-FSD1-FSD1, also exhibited high affinity and/or specificity for myostatin and activin A. To begin to address this question, we transiently expressed these FST-type constructs in a luciferase reporter assay and tested their ability to antagonize myostatin and activin A signaling. For this and all subsequent luciferase reporter assays, we used HEK293 cells that stably express luciferase under the control of the (CAGA)12 promoter (39). This promoter is activated by activin A and myostatin ligands through phosphorylation of Smad3 by the type I TGF-β family receptors Alk4 or Alk5. As shown in Fig. 2, ND-FSD1-FSD1 serves as a potent, specific myostatin antagonist, as previously demonstrated. In contrast, none of the other three similar domain swap constructs exhibited antagonism for either myostatin or activin A (Fig. 2). This was somewhat surprising given the high affinity of wild-type FSTL3 for these ligands, because ND-FSD1-FSD1 differs from wild-type only by the replacement of FSD2 with FSD1. However, in this transient assay, it is possible that the different constructs were expressed at varying levels, leading to differences in apparent ligand inhibition. To control for this, we expressed, purified, and quantified these proteins and tested their activity in a luciferase reporter assay that we have optimized into a semi-high-throughput format (25). Results of this assay confirmed those of the transient assays, because only ND-FSD1-FSD1 again showed strong and specific antagonism of myostatin over activin A (Fig. 3). This is consistent with previous data showing that deleting either the ND or FSD1 from full-length FST significantly decreases myostatin antagonism (33). Altogether, this demonstrates that it is neither FST ND nor FSD1 alone but the unique combination of the two that creates a highly specific, effective myostatin antagonist.
Fig. 2.
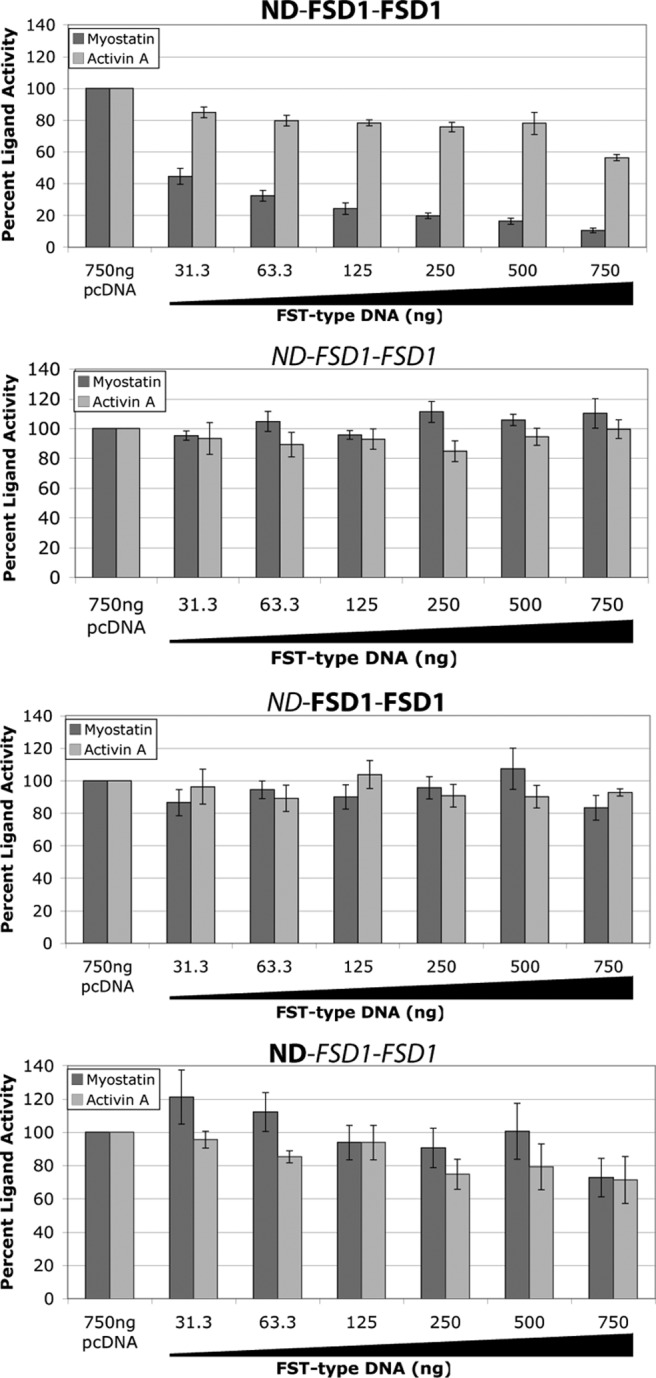
FST ND-FSD1-FSD1 is a unique, specific myostatin antagonist as shown through transient expression in luciferase reporter assays. Increasing amounts of FST-type DNA were transfected into HEK293-CAGA cells, which were later treated with exogenous ligands. Total DNA was normalized using empty vector. Each condition was performed in triplicate for each experiment, and each experiment was performed twice. Figures represent the combined data from all experiments. Error bars represent ±sem.
Fig. 3.
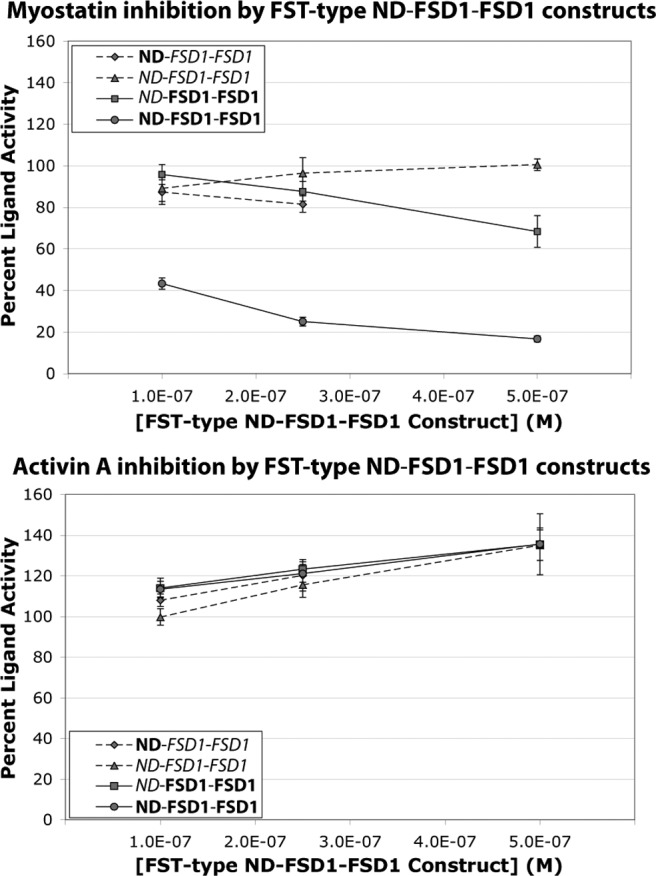
Unique specificity of FST ND-FSD1-FSD1 is confirmed in luciferase reporter assays using purified proteins. FST-type proteins at various concentrations were mixed with ligands and applied to HEK293-CAGA cells. Limited protein levels restricted analysis of the highest concentration of ND-FSD1-FSD1. Each condition was performed in triplicate for each experiment, and each experiment was performed three to four times. Figures represent the combined data from all experiments. Error bars represent ±sem.
Point mutations in the ND of FST ND-FSD1-FSD1 can abrogate myostatin inhibition
Structural and biochemical evidence has suggested that the ND of FST-type proteins are important for antagonist specificity and ligand inhibition. Previously, we began to investigate the hydrophobic interface between the ND of FST-type molecules and ligands through single point mutations in the ND. We found that the introduction of a single negatively charged residue into this interface on FST had little effect on activity but on FSTL3 was sufficient to significantly disrupt binding and ligand antagonism (25). Here, we used similar ND mutations to likewise investigate this interface on FST ND-FSD1-FSD1. Single and double point mutations of F47 and F52 to aspartate residues were tested by transient expression in our luciferase reporter assay. Interestingly, single point mutations alone were adequate to greatly abrogate myostatin inhibition (Figs. 2 and 4A). Again, luciferase reporter assays with purified proteins supported the results of the transient assays (Fig. 4B). This together with our domain swap analysis demonstrates that the ND of FST ND-FSD1-FSD1 is critical to, but not sufficient for, myostatin antagonism. This also highlights that ligand inhibition by FST ND-FSD1-FSD1 is somehow significantly different from that of full-length FST. These findings are important, because mutations and permutations (e.g. ND-FSD1-FSD1) of this modified construct have not been previously investigated, and they begin to reveal details of how FST ND-FSD1-FSD1 specificity and affinity is achieved.
Fig. 4.
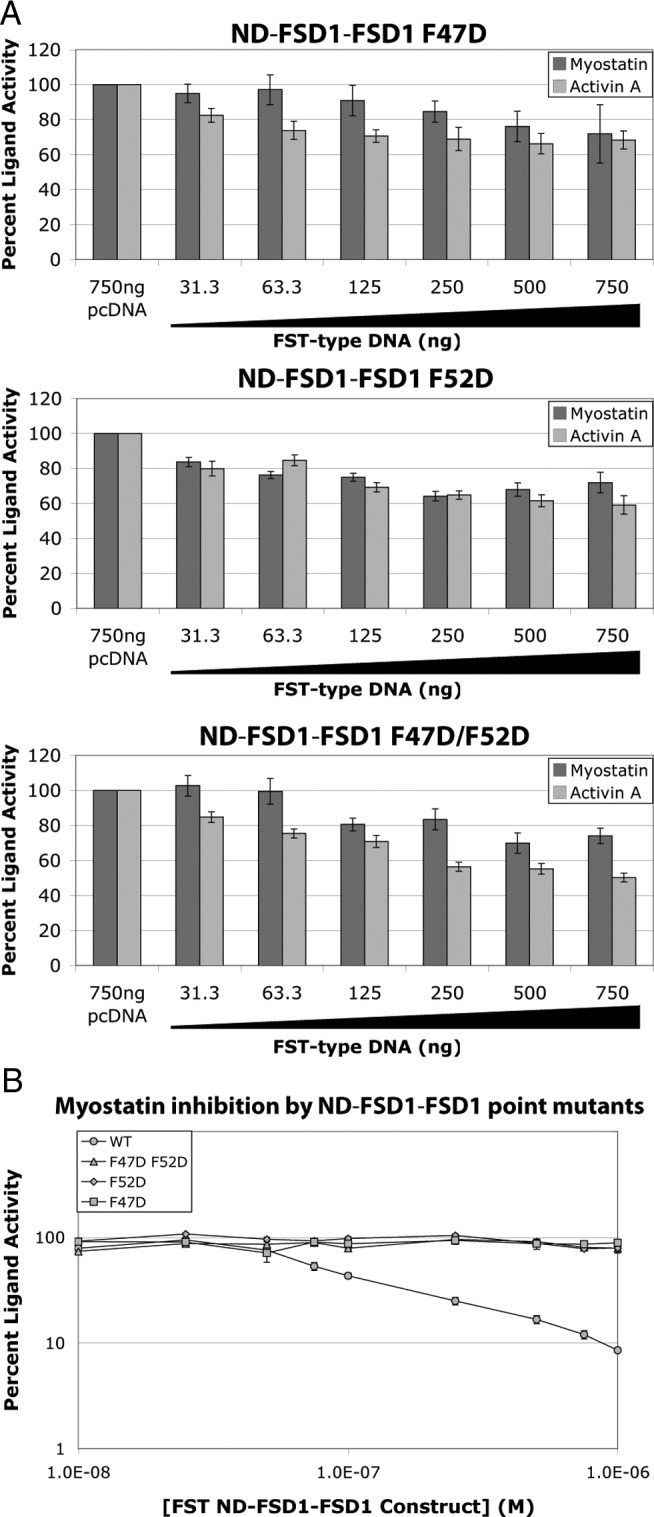
Point mutations in the ND of FST ND-FSD1-FSD1 can abrogate myostatin inhibition. A, Increasing amounts of FST ND-FSD1-FSD1 construct DNA were transfected into HEK293-CAGA cells, which were later treated with exogenous ligands. Total DNA was normalized using empty vector. Each condition was performed in triplicate for each experiment, and each experiment was performed two times. Figures represent the combined data from all experiments. B, Purified FST ND-FSD1-FSD1 proteins at various concentrations were mixed with myostatin and applied to HEK293-CAGA cells. Each condition was performed in triplicate for each experiment, and each experiment was performed two to three times. Figures represent the combined data from all experiments. Error bars represent ±sem.
Double point mutation F47D/F52D in the ND reduces inhibition by FST, especially in the absence of FSD3
It is interesting to consider why full-length FST, but not FST ND-FSD1-FSD1, is resistant to single point mutations at the hydrophobic ND-ligand interface. It is possible that multiple mutations in FST may be required to see an effect on ligand inhibition. To test this possibility, we produced and purified the full-length FST double point mutant F47D/F52D and tested its activity against myostatin and activin A in our luciferase reporter assay. Indeed, this double point mutation greatly reduced ligand inhibition by nearly an order of magnitude for myostatin and more so for activin A (Fig. 5). However, even when combined, these mutations still do not eliminate antagonism of full-length FST as they do with FST ND-FSD1-FSD1 (Fig. 4).
Fig. 5.
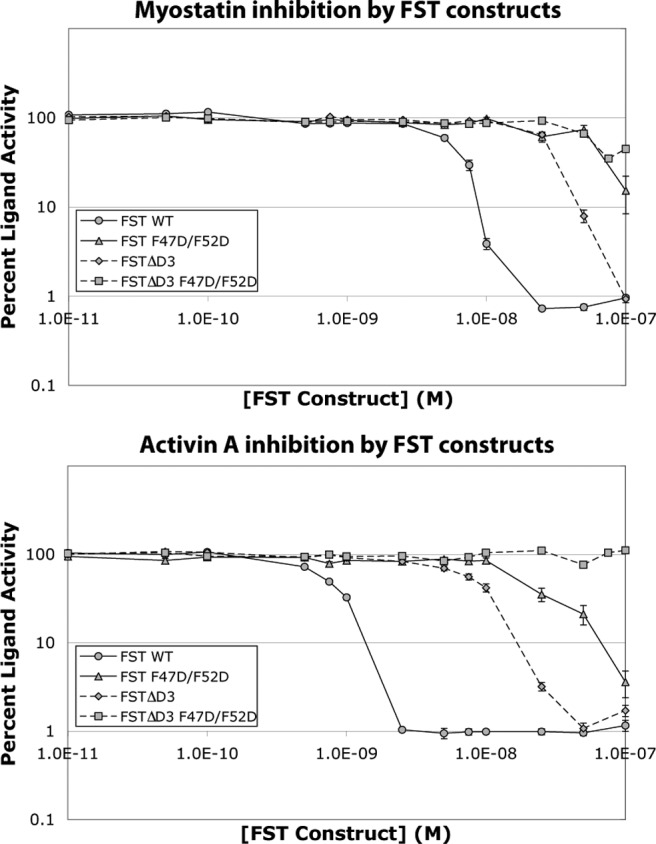
The double point mutation F47D/F52D negatively affects inhibition by FST, especially in the absence of FSD3. Purified proteins expressed from the corresponding FST-type constructs were mixed with ligands and applied to HEK293-CAGA cells. Each condition was performed in triplicate for each experiment, and each experiment was performed three to four times. Figures represent the combined data from all experiments. Error bars represent ±sem.
We next sought to determine what accounts for this difference in resistance to ND point mutations at the hydrophobic interface. One variation between the two constructs is the absence of FSD3. Although FSD3 does not interact with ligand itself, FSD3 of one FST molecule does interact with the ND of the partner FST molecule when bound to ligand, which may enhance the affinity of the complex. Indeed, it has been shown that a FST construct without FSD3 (FSTΔD3) exhibits reduced activin A binding and inhibition (35). Moreover, FSTL3, which lacks a third FST domain, is susceptible to single ND point mutations that are similar to those that have little effect on full-length FST (25). These observations led us to investigate the contribution of FSD3 to FST binding. We first tested inhibition of myostatin and activin A by FSTΔD3 in our luciferase reporter assay, and as expected, antagonism was reduced as compared with full-length FST (Fig. 5). We next tested FSTΔD3 F47D/F52D and found that myostatin inhibition was greatly impaired, whereas activin A inhibition was eliminated (Fig. 5). This suggests that although the double mutation F47D/F52D may disrupt the ND-ligand interface, the interaction between FST F47D/F52D and ligand is stabilized by the interaction between FSD3 of one FST molecule and the ND of the other. Therefore, lack of FSD3 leaves FST susceptible to point mutations at the hydrophobic interface between the ND and ligand.
Mutations in FSD2 are also more detrimental in the absence of FSD3
Another difference between full-length FST and FST ND-FSD1-FSD1 is the absence of FSD2, which, in full-length FST, makes extensive contacts with ligands. To begin to investigate the importance of FSD2, we created a series of FSD2 single point mutations in FST and FSTL3 and tested binding of these constructs to activin A (Table 1). FSTL3 R199 and FST R192 participate in salt bridge interactions with ligands, whereas the other chosen residues form part of a hydrophobic interface between FSD2 and ligands. As shown in Table 1, mutation of any of these FSTL3 residues to negatively charged ones, thereby introducing either an oppositely charged residue or a charged residue into a hydrophobic interface, greatly reduced or eliminated activin A binding. In contrast, mutation of these residues in full-length FST had a range of effects on activity, with Y159E having no effect, V161D having a slight effect (2.1-fold reduction in binding), and V151D and R192E having a somewhat larger effect (7.7- and 3.7-fold reduction, respectively) on activin A binding. This demonstrates that in addition to ND point mutations, FSTL3 is also relatively more susceptible to point mutations at the FSD2-ligand interface. We furthermore confirmed the particular importance of FSD2 to FSTL3 by testing the effect of a less disruptive mutation, R199A, on FSTL3 activity in our luciferase reporter assay (Fig. 6). This point mutation greatly reduced inhibition of activin A and eliminated inhibition of myostatin. Overall, this supports the complete lack of ligand inhibition by ND-FSD1-FSD1, as a functional FSD2 seems to be critical to antagonism by FSTL3.
Table 1.
Comparative radiolabeled activin A binding activity of FSD2 point mutants
| FSTL3 | FST | FSTΔD3 | FST/FSTΔD3b | ||
|---|---|---|---|---|---|
| WT | 1.00 | WT | 1.00 | 0.40 | 2.5 |
| V157D | <0.01 | V151D | 0.13 ± 0.03 | ||
| H165E | 0.05a | Y159E | 0.97 ± 0.08 | 0.04a | 24 |
| V167D | <0.01 | V161D | 0.47 ± 0.13 | <0.01 | >47 |
| R199E | <0.01 | R192E | 0.27 ± 0.02 | <0.01 | >27 |
Potencies relative to respective full-length wild-type (WT) proteins. Mutated residues in FSTL3 correspond to those in FST. Values are represented as mean ± sem.
Estimate from nonparallel binding inhibition curve.
The last column represents the fold decrease in binding activity of each point mutant in FSTΔD3 compared with that mutant in full-length FST.
Fig. 6.
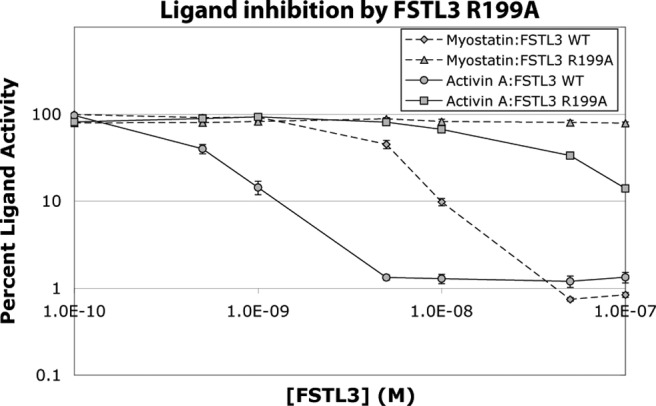
Mutation R199A drastically reduces or eliminates FSTL3 activity. Purified FSTL3 R199A was mixed with ligands and applied to HEK293-CAGA cells. Each condition was performed in triplicate for each experiment, and each experiment was performed three times. Figures represent the combined data from all experiments. Error bars represent ±sem.
This relative resistance of FST to point mutations, this time in FSD2, could again be conferred by FSD3. To examine this, we measured the effects of these FST point mutations in the absence of FSD3 and compared them to those in full-length FST (Table 1). FSTΔD3 itself exhibits a 2.5-fold decrease in activin A binding as compared with full-length FST. As compared with the effects of the mutations in full-length FST, the effects of mutations in FSTΔD3 were substantially exaggerated with another 24- to more than 47-fold decrease in activin A binding. In fact, the binding potencies of FSTΔD3 point mutants are at levels comparable with those of FSTL3 point mutants. In other words, removing FSD3 creates an FST molecule that is equally as sensitive to point mutations as FSTL3. Unfortunately, we could not test these FSTΔD3 point mutants in our luciferase reporter assay because protein yields were low, and the luciferase reporter assay requires significantly more protein than the binding assay.
Evidence supports that FSD confers cooperativity to antagonism by FST
Thus far, we have shown that FST-type proteins lacking an FSD3 (FST ND-FSD1-FSD1, FSTΔD3, and FSTL3) are more sensitive to point mutations in the ND and FSD2 (if applicable) than full-length FST, suggesting that the FST ND-FSD3 interaction somehow contributes added affinity to ligand antagonism. Structures of ligand-FST complexes show that although FSD3 does not directly contact ligand, there are interactions between the ND of one FST molecule and FSD3 of the other that may actually provide cooperativity to ligand inhibition. However, to our knowledge, cooperativity in FST binding has not been demonstrated. We therefore wanted to test for cooperativity in antagonism by FST-type proteins with and without FSD3 by using our luciferase reporter assay. In general, cooperativity can be characterized by fitting the Hill equation (40) to ligand-binding data and examining the resulting Hill slope value. Values close to −1 indicate that binding is not cooperative, whereas values much less than −1, visualized by a steeper inhibition curve, indicate cooperative antagonism, meaning the binding of one molecule strengthens the binding of subsequent molecules. In this assay, FSTL3 and FST were titrated in with ligands, and Hill slope values were calculated from the resulting data using GraphPad (Fig. 7). Hill slope values for FSTL3 inhibition of myostatin and activin A were −1.0 and −1.5, respectively, whereas those for FST were −3.1 and −2.6, respectively, suggesting that cooperativity is prevalent in antagonism by FST but not FSTL3. To further investigate whether this cooperativity is conferred by the ND-FSD3 interaction, we examined cooperativity in inhibition by FSTΔD3. This construct shows Hill slope values of −1.9 and −1.5 in myostatin and activin A antagonism, respectively, implying loss of cooperative inhibition as compared with full-length FST. Altogether, these data suggest that whereas full-length FST exhibits cooperative antagonism, FST-type proteins lacking FSD3, and thus the ND-FSD3 interaction, also lack cooperativity.
FSD3 potentiates the formation of 1:2 complexes, whereas 1:1 complexes can form in the absence of FSD3
Structural studies have shown that both FST and FSTL3 bind TGF-β family ligands at a stoichiometric ratio of one ligand to two FST-type molecules (1:2), and this seems to be the biological complex (25, 28–32). However, if binding of one FST-type molecule is completely independent of the other binding, as it likely is with FSTL3, it seems logical that at substoichiometric ratios, 1:1 complexes would also form. With FST, 1:2 complex formation may be driven under this condition by the ND-FSD3 interaction, because one FST bound to ligand would provide an additional interaction surface for the second FST molecule to bind. To establish whether or not FST-type molecules can form 1:1 complexes with ligands, we titrated increasing amounts of activin A into either FST or FSTL3, cross-linked the resulting complexes with glutaraldehyde, and analyzed the sizes of FST-type molecule-containing species by SDS-PAGE and Western blotting. As shown in Fig. 8, FSTL3 formed both 1:1 and 1:2 complexes with activin A, with 1:1 complexes becoming more prevalent as increasing amounts of activin A were added. In contrast, FST formed only 1:2 complexes even when there was enough activin A present (2.0 μg) such that there was very little free FST. To determine whether or not this tendency for FST to form only 1:2 complexes was conferred by the ND-FSD3 interaction, we performed this cross-linking experiment with FSTΔD3. Similar to FSTL3, FSTΔD3 formed both 1:1 and 1:2 complexes with activin A. Collectively, these data suggest that the interaction between the ND of one FST molecule and FSD3 of the other provides cooperativity to ligand antagonism, which in turn potentiates the formation of 1:2 over 1:1 ligand-FST complexes.
Fig. 8.

FSD3 potentiates the formation of 1:2 ligand-FST complexes. Activin A was titrated into FST-type proteins, mixes were cross-linked, and SDS-PAGE and Western blotting were performed on the resulting products. Arrow with 1 indicates 1:1 complexes whereas arrow with 2 indicates 1:2 complexes.
Discussion
Because FST antagonists bind with varying affinities to a number of TGF-β family ligands, it is of significant interest to understand the basis of this specificity. It was recently discovered that the FST ND-FSD1-FSD1 construct exhibited specificity for myostatin over activin A, although the affinity for myostatin is much weaker than that for the native FST protein (26, 33). Our previous structural studies have provided an explanation for this specificity and affinity difference (28, 31). The loss of affinity is likely due to replacing FSD2 with FSD1. FSD2 forms significant contacts with the ligand that would not be maintained with FSD1, because the FSD2 residues at the interface are not conserved within FSD1. The myostatin-FST288 structure also suggested that the ND forms additional contacts with myostatin that are unique and might be responsible for the specificity difference (28). The results of this study further explore the basis of this specificity.
We have herein demonstrated that the FST ND-FSD1-FSD1 construct is unique in its specificity and affinity for myostatin as compared with other similar domain swap constructs containing FSTL3 components. It is the unique combination of FST ND and FSD1 that confer these properties, because ND-FSD1-FSD1 and ND-FSD1-FSD1 are not effective myostatin antagonists. These data were unexpected, because structural evidence suggests that the FST ND interacts well with myostatin, especially in comparison with activin A (28), implying that ND-FSD1-FSD1 would also exhibit myostatin antagonism. Because FSD1 is also required, it is possible that FSD1 flexibility is important in properly positioning the FST ND on ligand, because FST FSD1 exhibits higher temperature factors and likely more flexibility than FSTL3 FSD1 (30, 31). Overall, each domain within a given FST-type molecule may simply be specialized to work in concert with the other domains of that molecule.
Through our data presented here, we have shown that FSD3 provides added affinity to ligand inhibition, because FSTL3 and FSTΔD3 are much more sensitive to point mutations in the ND and FSD2 than full-length FST. Our data underline the importance of each domain to antagonism by FSTL3. FSD2 is crucial to FSTL3 function, because FSTL3 ND-FSD1-FSD1 is inactive and FSD2 single point mutations in full-length FSTL3 greatly reduce or eliminate binding. However, even with FSD2, ND point mutations can still be detrimental to FSTL3 binding. The affinity for ligands therefore seems to be spread out over both the ND and FSD2. This multidomain distribution of affinity also seems to occur with FSTΔD3. Removing FSD3 from FST makes it equally susceptible to these point mutations in the ND or FSD2, implying that the ND-FSD3 interaction is a mechanism of overcoming this sensitivity and adding affinity, which may furthermore also confer more broad antagonism to FST. Lack of interaction of a domain or part of a domain with a particular ligand may be compensated for by the ND-FSD3 interaction, which would be somewhat ligand independent. This is upheld in that FST ND-FSD1-FSD1, although specific for myostatin, exhibits greatly reduced affinity as compared with FST or FSTL3, because most of the interaction interface is likely contained in the ND and first FSD1. In contrast to FST, FSTL3 appears to be a very specialized molecule, because it is also a more specific ligand antagonist. Altogether, these points are supported in that a construct of ND-FSD1-FSD2 has greatly reduced activin A affinity as compared with wild-type FSTL3 (>60-fold), whereas adding FSD3 to this construct increases its affinity 150-fold (30).
Our data suggest that the ND-FSD3 interaction provides FST with cooperative antagonism. This cooperativity may in turn promote the formation of 1:2 FST complexes, whereas 1:1 FSTL3 complexes can form at substoichiometric ratios. This is consistent with a previous study showing that a FST construct consisting of FSD1-FSD2 could form a 1:1 complex with ligand (32). In a biological context, this could actually render FSTL3 a more potent antagonist, because a 1:1 complex would still have two open receptor-binding sites, each on different ends of the ligand, that could hypothetically bind one type I and one type II receptor but be incapable of generating a signal (Fig. 9). In support of this, it has been shown that with TGF-β class ligands, a signal is initiated only by receptors that bind on the same end of the ligand and that BMP signaling can be blocked when only one of the two type II receptor sites is blocked (41–43). In other words, FSTL3 could sequester not only ligands but also receptors, forming inactive complexes and thereby exerting two levels of antagonism. Because FST tends to act more locally due to its affinity for cell-surface-bound heparan, it may be important for FST to bind cooperatively and avoid this property of receptor antagonism. Because TGF-β family ligands are dimers with two identical surfaces, cooperativity may be an important theme in TGF-β family regulation in general. Many antagonists have been shown to bind these ligands symmetrically, with either one antagonist dimer or two antagonist molecules (25, 28–32, 44–46).
Fig. 9.

Schematic of potential function of 1:1 ligand-FSTL3 complexes. A, Model of a ternary myostatin signaling complex, with type I and type II receptors labeled. BMPRIA and ActRII from a ternary BMP2 complex were superimposed onto myostatin [2GOO (13)]. B, Model of a hypothetical 1:1 ligand-FSTL3 complex bound to one type I and one type II receptor in a conformation that would not likely generate a signal. Structure figures were rendered using PyMOL (48).
In addition to antagonism, cooperativity has also been suggested to be involved in ligand signaling. Crystal structures of activin A alone or in complex with type II receptors show the ligand in multiple orientations, all of which display a distorted type I receptor binding site (18, 32, 47). It has been postulated that binding to the membrane-bound type II receptor would immobilize activin A and stabilize the conformation of the ligand, facilitating formation of the type I receptor binding site and subsequent type I receptor binding (47). This would be cooperativity conferred though a conformation change induced by a binding partner. Another type of cooperativity has been shown to be very important for TGF-β class ligand signaling, because contact between the extracellular portions of the type I and type II receptors is essential for type I receptor binding (19). Cooperativity in FST binding could be of either type. Binding of the first FST molecule could hold the flexible ligand in a more ordered conformation, facilitating binding of the second FST molecule. Alternatively or in conjunction, cooperativity could be conferred through binding of the first FST molecule providing an additional interaction interface for the second, thus potentiating its binding. If cooperativity was related to a conformational change, removing FSD3 should not affect it, because FSD3 does not contact ligand. Because removing FSD3 does affect cooperativity, we believe that cooperative inhibition by FST is conferred through the ND-FSD3 interaction.
Accumulating structural and biochemical work is beginning to clarify the features of FST-type molecules that are important for affinity and specificity in ligand binding. This information is significant to our understanding of how to rationally design highly specific, effective inhibitors of select TGF-β family ligands that will be valuable to the therapeutic treatment of the multitude of diseases caused by aberrant TGF-β family signaling. Toward this goal, future work should also continue to explore ligand characteristics that confer these properties to antagonist binding (34). This is the first time that the mechanism of action of the FST ND-FSD1-FSD1 construct has been investigated or its ligand affinity altered. Previously, myostatin was shown to form different interactions with the FST ND as compared with activin A that could be taken advantage of and optimized (28). Collectively, this work supports the possibility of engineering the ND of FST ND-FSD1-FSD1 to enhance the affinity of this construct for myostatin, effectively creating a better antagonist.
Acknowledgments
This work was supported by research grants from the National Institutes of Health GM R01 (GM084186) and Muscular Dystrophy Association (93887) to T.B.T. J.N.C. was supported by an American Heart Association predoctoral fellowship.
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- BMP
- Bone morphogenetic protein
- FSD
- FST domain
- FST
- follistatin
- FSTL3
- FST-like 3
- ND
- N-terminal domain.
References
- 1. Nakamura T, Takio K, Eto Y, Shibai H, Titani K, Sugino H. 1990. Activin-binding protein from rat ovary is follistatin. Science 247:836–838 [DOI] [PubMed] [Google Scholar]
- 2. Matzuk MM, Kumar TR, Bradley A. 1995. Different phenotypes for mice deficient in either activins or activin receptor type II. Nature 374:356–360 [DOI] [PubMed] [Google Scholar]
- 3. Xia Y, Schneyer AL. 2009. The biology of activin: recent advances in structure, regulation and function. J Endocrinol 202:1–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lee SJ. 2004. Regulation of muscle mass by myostatin. Annu Rev Cell Dev Biol 20:61–86 [DOI] [PubMed] [Google Scholar]
- 5. McPherron AC, Lawler AM, Lee SJ. 1997. Regulation of skeletal muscle mass in mice by a new TGF-β superfamily member. Nature 387:83–90 [DOI] [PubMed] [Google Scholar]
- 6. Hayette S, Gadoux M, Martel S, Bertrand S, Tigaud I, Magaud JP, Rimokh R. 1998. FLRG (follistatin-related gene), a new target of chromosomal rearrangement in malignant blood disorders. Oncogene 16:2949–2954 [DOI] [PubMed] [Google Scholar]
- 7. Ueno N, Ling N, Ying SY, Esch F, Shimasaki S, Guillemin R. 1987. Isolation and partial characterization of follistatin: a single-chain Mr 35,000 monomeric protein that inhibits the release of follicle-stimulating hormone. Proc Natl Acad Sci USA 84:8282–8286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Matzuk MM, Lu N, Vogel H, Sellheyer K, Roop DR, Bradley A. 1995. Multiple defects and perinatal death in mice deficient in follistatin. Nature 374:360–363 [DOI] [PubMed] [Google Scholar]
- 9. Brown ML, Bonomi L, Ungerleider N, Zina J, Kimura F, Mukherjee A, Sidis Y, Schneyer A. 2011. Follistatin and follistatin like-3 differentially regulate adiposity and glucose homeostasis. Obesity 19:1940–1949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mukherjee A, Sidis Y, Mahan A, Raher MJ, Xia Y, Rosen ED, Bloch KD, Thomas MK, Schneyer AL. 2007. FSTL3 deletion reveals roles for TGF-β family ligands in glucose and fat homeostasis in adults. Proc Natl Acad Sci USA 104:1348–1353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hashimoto O, Nakamura T, Shoji H, Shimasaki S, Hayashi Y, Sugino H. 1997. A novel role of follistatin, an activin-binding protein, in the inhibition of activin action in rat pituitary cells. Endocytotic degradation of activin and its acceleration by follistatin associated with cell-surface heparan sulfate. J Biol Chem 272:13835–13842 [DOI] [PubMed] [Google Scholar]
- 12. Sidis Y, Schneyer AL, Keutmann HT. 2005. Heparin and activin-binding determinants in follistatin and FSTL3. Endocrinology 146:130–136 [DOI] [PubMed] [Google Scholar]
- 13. Allendorph GP, Vale WW, Choe S. 2006. Structure of the ternary signaling complex of a TGF-β superfamily member. Proc Natl Acad Sci USA 103:7643–7648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Greenwald J, Groppe J, Gray P, Wiater E, Kwiatkowski W, Vale W, Choe S. 2003. The BMP7/ActRII extracellular domain complex provides new insights into the cooperative nature of receptor assembly. Mol Cell 11:605–617 [DOI] [PubMed] [Google Scholar]
- 15. Keller S, Nickel J, Zhang JL, Sebald W, Mueller TD. 2004. Molecular recognition of BMP-2 and BMP receptor IA. Nat Struct Mol Biol 11:481–488 [DOI] [PubMed] [Google Scholar]
- 16. Kirsch T, Sebald W, Dreyer MK. 2000. Crystal structure of the BMP-2-BRIA ectodomain complex. Nat Struct Biol 7:492–496 [DOI] [PubMed] [Google Scholar]
- 17. Weber D, Kotzsch A, Nickel J, Harth S, Seher A, Mueller U, Sebald W, Mueller TD. 2007. A silent H-bond can be mutationally activated for high-affinity interaction of BMP-2 and activin type IIB receptor. BMC Struct Biol 7:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Thompson TB, Woodruff TK, Jardetzky TS. 2003. Structures of an ActRIIB:activin A complex reveal a novel binding mode for TGF-β ligand:receptor interactions. EMBO J 22:1555–1566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Groppe J, Hinck CS, Samavarchi-Tehrani P, Zubieta C, Schuermann JP, Taylor AB, Schwarz PM, Wrana JL, Hinck AP. 2008. Cooperative assembly of TGF-β superfamily signaling complexes is mediated by two disparate mechanisms and distinct modes of receptor binding. Mol Cell 29:157–168 [DOI] [PubMed] [Google Scholar]
- 20. Hart PJ, Deep S, Taylor AB, Shu Z, Hinck CS, Hinck AP. 2002. Crystal structure of the human TβR2 ectodomain-TGF-β3 complex. Nat Struct Biol 9:203–208 [DOI] [PubMed] [Google Scholar]
- 21. Kotzsch A, Nickel J, Seher A, Sebald W, Müller TD. 2009. Crystal structure analysis reveals a spring-loaded latch as molecular mechanism for GDF-5-type I receptor specificity. EMBO J 28:937–947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Innis CA, Shi J, Blundell TL. 2000. Evolutionary trace analysis of TGF-β and related growth factors: implications for site-directed mutagenesis. Protein Eng 13:839–847 [DOI] [PubMed] [Google Scholar]
- 23. Sidis Y, Mukherjee A, Keutmann H, Delbaere A, Sadatsuki M, Schneyer A. 2006. Biological activity of follistatin isoforms and follistatin-like-3 is dependent on differential cell surface binding and specificity for activin, myostatin, and bone morphogenetic proteins. Endocrinology 147:3586–3597 [DOI] [PubMed] [Google Scholar]
- 24. Iemura S, Yamamoto TS, Takagi C, Uchiyama H, Natsume T, Shimasaki S, Sugino H, Ueno N. 1998. Direct binding of follistatin to a complex of bone-morphogenetic protein and its receptor inhibits ventral and epidermal cell fates in early Xenopus embryo. Proc Natl Acad Sci USA 95:9337–9342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cash JN, Angerman EB, Kattamuri C, Nolan K, Zhao H, Sidis Y, Keutmann HT, Thompson TB. 2012. The structure of myostatin:follistatin-like 3: N-terminal domains of follistatin-type molecules exhibit alternate modes of binding. J Biol Chem 287:1043–1053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Nakatani M, Takehara Y, Sugino H, Matsumoto M, Hashimoto O, Hasegawa Y, Murakami T, Uezumi A, Takeda S, Noji S, Sunada Y, Tsuchida K. 2008. Transgenic expression of a myostatin inhibitor derived from follistatin increases skeletal muscle mass and ameliorates dystrophic pathology in mdx mice. FASEB J 22:477–487 [DOI] [PubMed] [Google Scholar]
- 27. Takehara-Kasamatsu Y, Tsuchida K, Nakatani M, Murakami T, Kurisaki A, Hashimoto O, Ohuchi H, Kurose H, Mori K, Kagami S, Noji S, Sugino H. 2007. Characterization of follistatin-related gene as a negative regulatory factor for activin family members during mouse heart development. J Med Invest 54:276–288 [DOI] [PubMed] [Google Scholar]
- 28. Cash JN, Rejon CA, McPherron AC, Bernard DJ, Thompson TB. 2009. The structure of myostatin:follistatin 288: insights into receptor utilization and heparin binding. EMBO J 28:2662–2676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lerch TF, Shimasaki S, Woodruff TK, Jardetzky TS. 2007. Structural and biophysical coupling of heparin and activin binding to follistatin isoform functions. J Biol Chem 282:15930–15939 [DOI] [PubMed] [Google Scholar]
- 30. Stamler R, Keutmann HT, Sidis Y, Kattamuri C, Schneyer A, Thompson TB. 2008. The structure of FSTL3.activin A complex. Differential binding of N-terminal domains influences follistatin-type antagonist specificity. J Biol Chem 283:32831–32838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Thompson TB, Lerch TF, Cook RW, Woodruff TK, Jardetzky TS. 2005. The structure of the follistatin:activin complex reveals antagonism of both type I and type II receptor binding. Dev Cell 9:535–543 [DOI] [PubMed] [Google Scholar]
- 32. Harrington AE, Morris-Triggs SA, Ruotolo BT, Robinson CV, Ohnuma S, Hyvönen M. 2006. Structural basis for the inhibition of activin signalling by follistatin. EMBO J 25:1035–1045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Schneyer AL, Sidis Y, Gulati A, Sun JL, Keutmann H, Krasney PA. 2008. Differential antagonism of activin, myostatin and growth and differentiation factor 11 by wild-type and mutant follistatin. Endocrinology 149:4589–4595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Harrison CA, Chan KL, Robertson DM. 2006. Activin-A binds follistatin and type II receptors through overlapping binding sites: generation of mutants with isolated binding activities. Endocrinology 147:2744–2753 [DOI] [PubMed] [Google Scholar]
- 35. Keutmann HT, Schneyer AL, Sidis Y. 2004. The role of follistatin domains in follistatin biological action. Mol Endocrinol 18:228–240 [DOI] [PubMed] [Google Scholar]
- 36. Sidis Y, Schneyer AL, Sluss PM, Johnson LN, Keutmann HT. 2001. Follistatin: essential role for the N-terminal domain in activin binding and neutralization. J Biol Chem 276:17718–17726 [DOI] [PubMed] [Google Scholar]
- 37. Nakatani M, Kokubo M, Ohsawa Y, Sunada Y, Tsuchida K. 2011. Follistatin-derived peptide expression in muscle decreases adipose tissue mass and prevents hepatic steatosis. Am J Physiol Endocrinol Metab 300:E543–E553 [DOI] [PubMed] [Google Scholar]
- 38. Schneyer AL, Rzucidlo DA, Sluss PM, Crowley WF., Jr 1994. Characterization of unique binding kinetics of follistatin and activin or inhibin in serum. Endocrinology 135:667–674 [DOI] [PubMed] [Google Scholar]
- 39. Dennler S, Itoh S, Vivien D, ten Dijke P, Huet S, Gauthier JM. 1998. Direct binding of Smad3 and Smad4 to critical TGF β-inducible elements in the promoter of human plasminogen activator inhibitor-type 1 gene. EMBO J 17:3091–3100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hill AV. 1910. Proceedings of the Physiological Society: January 22, 1910. J Physiol 40:iv-vii [Google Scholar]
- 41. Knaus P, Sebald W. 2001. Cooperativity of binding epitopes and receptor chains in the BMP/TGFβ superfamily. Biol Chem 382:1189–1195 [DOI] [PubMed] [Google Scholar]
- 42. Huang T, David L, Mendoza V, Yang Y, Villarreal M, De K, Sun L, Fang X, López-Casillas F, Wrana JL, Hinck AP. 2011. TGF-β signalling is mediated by two autonomously functioning TβRI:TβRII pairs. EMBO J 30:1263–1276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Isaacs MJ, Kawakami Y, Allendorph GP, Yoon BH, Izpisua Belmonte JC, Choe S. 2010. Bone morphogenetic protein-2 and -6 heterodimer illustrates the nature of ligand-receptor assembly. Mol Endocrinol 24:1469–1477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Shi M, Zhu J, Wang R, Chen X, Mi L, Walz T, Springer TA. 2011. Latent TGF-β structure and activation. Nature 474:343–349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Groppe J, Greenwald J, Wiater E, Rodriguez-Leon J, Economides AN, Kwiatkowski W, Affolter M, Vale WW, Belmonte JC, Choe S. 2002. Structural basis of BMP signalling inhibition by the cystine knot protein Noggin. Nature 420:636–642 [DOI] [PubMed] [Google Scholar]
- 46. Zhang JL, Qiu LY, Kotzsch A, Weidauer S, Patterson L, Hammerschmidt M, Sebald W, Mueller TD. 2008. Crystal structure analysis reveals how the Chordin family member crossveinless 2 blocks BMP-2 receptor binding. Dev Cell 14:739–750 [DOI] [PubMed] [Google Scholar]
- 47. Greenwald J, Vega ME, Allendorph GP, Fischer WH, Vale W, Choe S. 2004. A flexible activin explains the membrane-dependent cooperative assembly of TGF-β family receptors. Mol Cell 15:485–489 [DOI] [PubMed] [Google Scholar]
- 48. DeLano WL. 2002. The PyMOL molecular graphics system. Palo Alto, CA: DeLano Scientific [Google Scholar]