

Abstract
Although vitamin D has been implicated in cardiovascular protection, few studies have addressed the role of vitamin D receptor (VDR) in atherosclerosis. Here we investigate the effect of inactivation of the VDR signaling on atherogenesis and the antiatherosclerotic mechanism of vitamin D. Low density lipoprotein receptor (LDLR)−/−/VDR−/− mice exhibited site-specific accelerated atherogenesis, accompanied by increases in adhesion molecules and proinflammatory cytokines in the aorta and cholesterol influx in macrophages. Macrophages showed marked renin up-regulation in the absence of VDR, and inhibition of renin by aliskiren reduced atherosclerosis in LDLR−/−/VDR−/− mice, suggesting that the renin-angiotensin system (RAS) promotes atherosclerosis in the absence of VDR. LDLR−/− mice receiving LDLR−/−/VDR−/− BMT developed larger lesions than LDLR−/− BMT controls. Moreover, LDLR−/− mice receiving Rag-1−/−/VDR−/− BMT, which were unable to generate functional T and B lymphocytes, still had more severe atherosclerosis than Rag-1−/− BMT controls, suggesting a critical role of macrophage VDR signaling in atherosclerotic suppression. Aliskiren treatment eliminated the difference in lesions between Rag-1−/−/VDR−/− BMT and Rag-1−/− BMT recipients, indicating that local RAS activation in macrophages contributes to the enhanced atherogenesis seen in Rag-1−/−/VDR−/− BMT mice. Taken together, these observations provide evidence that macrophage VDR signaling, in part by suppressing the local RAS, inhibits atherosclerosis in mice.
Atherosclerotic lesion formation involves the interactions of modified lipoproteins, endothelial cells, macrophages, T cells, and vascular smooth muscle cells in the arterial wall (1, 2). Atherosclerosis is usually initiated by elevated plasma lipids such as low-density lipoprotein (LDL), which infiltrates into the vascular wall and, after being modified, promotes local inflammation and macrophage lipid loading. This chain of events ultimately leads to complex vascular lesions. Modified LDL particles have multiple activities that promote atherosclerosis (1), including inducing adhesion molecules (3–5) and promoting monocyte infiltration (6, 7) and foam cell formation (8, 9). Thus hyperlipidemia is a major risk factor for atherosclerosis.
Macrophages play an essential role in atherogenesis (8–10). Macrophages internalize modified lipoproteins (e.g. oxidized LDL) in an unregulated fashion, leading to formation of foam cells (11). Macrophages and foam cells induce adhesion molecules in endothelial cells and secrete chemokines and cytokines to promote monocyte recruitment and differentiation. As antigen-presenting cells, macrophages process peptides and lipids from the modified lipoproteins to present to T lymphocytes, and activated T cells augment the inflammatory response mediated by secreted cytokines, further driving atherosclerotic lesion progression (2). Macrophages also produce metalloproteinase and type 1 plasminogen activator inhibitor, which can destabilize atherosclerotic plaques (1, 12), leading to plaque rupture.
Macrophages express all components of the renin-angiotensin system (RAS), a potent promoter of atherosclerosis (13, 14). Angiotensin (Ang) II production is increased during monocyte to macrophage differentiation (15, 16). Specific deletion of renin in macrophages leads to significant reduction in atherosclerosis without altering circulating renin concentrations, which indicates the importance of the macrophage RAS in atherosclerosis (17).
Vitamin D deficiency is a global health problem affecting more than one billion people worldwide (18). The biological activity of the vitamin D hormone, 1,25-dihydroxvitamin D, is mediated by the vitamin D receptor, a member of nuclear receptor superfamily (19). There is overwhelming epidemiological and clinical evidence that links vitamin D deficiency to cardiovascular disease (20), including coronary stenoses, coronary artery calcification and carotid atherosclerosis (21–23); however, our understanding of vitamin D in vascular disease remains very limited. Recent studies suggested that vitamin D is able to inhibit foam cell formation by suppressing cholesterol uptake in macrophages from type 2 diabetes patients (24) and decrease atherosclerosis by regulating T lymphocyte functions (25). No studies have directly addressed the role of VDR in atherogenesis. In this study we present evidence to suggest that the macrophage VDR signaling inhibits atherosclerosis, in part, by targeting the local RAS in these cells.
Materials and Methods
Animal models and treatment
The animal studies were approved by the Institutional Animal Care and Use Committee at The University of Chicago. LDLR−/− and Rag-1−/− mice in C57BL/6 background were obtained from The Jackson Laboratory (Bar Harbor, ME). VDR−/− mice in C57BL/6 background were reported previously (26). LDLR−/−/VDR−/− mice were generated by crossing LDLR−/− and VDR−/− mice. Rag-1−/−/VDR−/− mice were generated by crossing Rag-1−/− and VDR−/− mice. Atherosclerosis was induced by placing animals on a high fat-high cholesterol (HFHC) diet (Harlan Teklad, Madison, WI; TD88137; 42% calories from fat, 0.2% cholesterol) for 8 or 12 wk. To prevent hypocalcemia in LDLR−/−/VDR−/− mice, the animals were weaned on a high-calcium diet (Harlan Teklad TD96348; 20% Lactose, 2% Ca, 1.25% P) until 8 wk of age when they were fed the HFHC diet and drinking water containing 2 mm CaCl2 to induce atherosclerosis. In some experiments, mice were treated with 25 mg/kg aliskiren (dissolved in PBS), ip three times per wk during HFHC dietary induction of atherosclerosis. Blood pressure was determined using the carotid artery cannulation method as described previously (27).
BMT
Bone marrow (BM) cells were isolated from femurs and tibias of 2-month-old donor mice, suspended in DMEM containing 2% fetal bovine serum (FBS) and washed in cold PBS. Red blood cells were lysed with ACK buffer containing 10 mm NH4Cl (pH 7.4). Recipient mice were transplanted with 1–5 × 106 BM cells via introorbital injection 6 h after receiving lethal γ-irradiation (1050 rads at 200 rads/min). After 6 wk recovery, the transplanted mice were used for atherosclerosis studies.
Lesion assessment
Mice were perfused with 4% paraformaldehyde/5% sucrose, after which the heart and aorta, including the innominate artery (IA), left carotid, and left subclavian, were removed en bloc and embedded in optimal cutting temperature compound. Atherosclerotic lesions were assessed at four vascular sites: IA, ascending aorta (AA), descending aorta (DA), and the aortic root (AR) of the heart according to a published method (28). A section of liver was included in each block, which allowed identification of the aortic root sinuses. Aortas were cut at 10 μm serial sections using a cryostat microtome. Selected slides were stained with oil red-O, 3× Gill's hematoxylin, and fast green, and the size of the lesions was quantified from the internal elastic lamina to their projection into the lumen of the artery using Openlab software (28).
Serum and plasma parameters
Plasma cholesterol and triglyceride concentrations were determined using commercial assay kits as described previously (29). Serum calcium levels were measured using a Calcium Liquicolor kit (Stanbio Laboratory, Boerne, TX), and PTH levels measured using a commercial ELISA kit (Immutopics, San Clemente, CA) as described elsewhere (27).
Cholesterol transport
For cholesterol influx assays, peritoneal macrophages were incubated with 25 μg/ml acLDL and 3 μCi/ml [3H]cholesterol in DMEM supplemented with 1% FBS for 8 or 24 h. After wash with PBS, lipids were extracted with hexane-isopropanol (3:2). The solvent was allowed to evaporate overnight in a fume hood, and radioactivity was determined by liquid scintillation counting normalized to cellular protein content that was extracted with NaOH. For cholesterol efflux assays, peritoneal macrophages were incubated in DMEM supplemented with 1% FBS containing 25 μg/ml acLDL, 2 μg/ml Sandoz ACAT (acyl-coA cholesterol acyltransferas) inhibitor (Sigma), and 3 μCi/ml [3H]cholesterol for 24 h. After labeling, the cells were washed with PBS and incubated with 0.2% BSA and 2 μg/ml ACAT inhibitor with or without the ATP-binding cassette transporter A1 (ABCA1) inducer TO901317 (10 μm) in serum-free DMEM for 12–16 h. Lipid efflux was initiated by incubation with human recombinant apolipoprotein AI (16 μg/ml) for 4 h. The media were harvested and extracted with ethanol and hexane. The organic phase was harvested, and the solvent evaporated overnight before the radioactivity was measured by scintillation counting. The cellular lysate radioactivity was measured by extraction with hexane-isopropanol (3:2) followed by liquid scintillation counting. Percent efflux was calculated based on the formula [medium/(medium + lysate)], normalized to cell protein content.
Immunostaining
Frozen aorta sections were fixed with cold 100% methanol, rehydrated in PBS, and blocked in 2% normal goat serum in PBS before being probed with primary antibodies [anti-renin or anti-monocyte macrophage (MOMA-2)] (Santa Cruz Biotechnology, Inc., Santa Cruz, CA) overnight at 4 C. After wash, the samples were incubated for 1 h at room temperature with Cy3 or fluorescein isothiocyanate-conjugated secondary antibodies. The slides were imaged using an Olympus microscope Fluoview confocal system with distinct emission spectra for Cy3 or fluorescein isothiocyanate fluorescence.
RT-PCR
Total RNA were extracted from aorta or peritoneal macrophages using TRIzol reagent (Invitrogen, Carlsbad, CA). First-strand cDNA were synthesized from total RNA using mouse mammary leukemia virus reverse transcriptase (Invitrogen) and hexanucleotide random primers. Regular RT-PCR was performed using a Bio-Rad DNA Engine (Bio-Rad Laboratories, Hercules, CA). Real-time RT-PCR was carried out in an Applied Biosystems 7900 Real-Time PCR System using a SYBR green PCR reagent kit (Applied Biosystems, Foster City, CA) as described previously (30). Primer sequences are available upon request.
Statistical analysis
Data values were presented as means ± sem. Statistical comparisons were carried out using unpaired two-tailed Student's t test or AVOVA as appropriate, with P < 0.05 being considered statistically significant.
Results
Accelerated atherosclerosis in LDLR−/−/VDR−/− mice despite lower plasma lipid levels
We generated LDLR−/−/VDR−/− mice by crossing LDLR−/− and VDR−/− mice. Mice ablated for VDR develop hypocalcemia (26). To prevent hypocalcemia, LDLR−/− and LDLR−/−/VDR−/− pups were weaned on a high-calcium diet until 8 wk of age, at which time they were placed on a HFHC diet with drinking water containing 2 mm CaCl2 for 8 or 12 wk before being euthanized for assessment of atherosclerosis. Under this condition there was no significant difference in serum calcium and PTH levels between LDLR−/− and LDLR−/−/VDR−/− mice (Supplemental Fig. 1A published on The Endocrine Society's Journals Online web site at http://mend.endojournals.org). On the HFHC diet, both LDLR−/− and LDLR−/−/VDR−/− mice developed dramatic hyperlipidemia; however, plasma cholesterol levels were significantly lower (by >30%) in LDLR−/−/VDR−/− mice compared with LDLR−/− mice at wk 8 and wk 12 (triglyceride levels were also significantly lower at wk 8) (Fig. 1A). In fact, VLDL, LDL, and high density lipoprotein (HDL) cholesterols were all lower in LDLR−/−/VDR−/− mice (Supplemental Fig. 1B). Interestingly, food intake (Supplemental Fig. 1C), intestinal lipid absorption (Supplemental Fig. 1D), and VLDL production (Supplemental Fig. 1E) were not different between LDLR−/− and LDLR−/−/VDR−/− mice. Thus, VDR regulates plasma cholesterol levels by an unknown mechanism.
Fig. 1.

Effects of VDR deletion on atherosclerosis. A, Plasma total cholesterol (left panel) and triglyceride (TG, right panel) levels in LDLR−/− and LDLR−/−/VDR−/− mice fed the HFHC diet for 8 or 12 wk (W). B, Atherosclerotic lesion sizes at the four vascular sites (IA, AA, DA, and AR) at wk 8 of HFHC diet. C, Lesion morphology at the IA site at wk 8. D, Lesion sizes in the three sinuses of the AR at 8 wk. E, Lesion sizes at the four vascular sites at wk 12. *, P < 0.05; ***, P < 0.001 vs. LDLR−/−; n = 8–10.
We assessed atherogenesis by quantifying the lesion size at four sites of the vascular tree: IA, AA, DA, and AR of the heart. Despite their lower plasma lipid levels, known to be the major risk factor for atherosclerosis (2), LDLR−/−/VDR−/− mice developed significantly larger lesions at the IA site at wk 8 compared with LDLR−/− mice (P < 0.05) (Fig. 1, B and C). Within the AR site, we stratified the lesion by sinus location: near the right coronary artery (RC), not associated with a coronary artery (NC), and near the left coronary artery (LC), and found that LDLR−/−/VDR−/− NC sinus lesions were significantly larger than LDLR−/− NC sinus (P < 0.05) (Fig. 1D). At wk 12, the lesion sizes were indistinguishable between LDLR−/− and LDLR−/−/VDR−/− mice (Fig. 1E) in the context of lower plasma cholesterol levels in LDLR−/−/VDR−/− mice (Fig. 1A). These observations indicate that VDR deletion accelerates atherogenesis in the early course, suggesting that VDR inhibits the initiation of atherosclerosis more than its progression. Thus we focused on the early phase of atherosclerosis in later studies. In view of the lower plasma cholesterol levels, however, the inhibitory role of VDR in atherogenesis might well be underestimated in the LDLR−/−/VDR−/− model.
Up-regulation of adhesion molecules, inflammation, and macrophage cholesterol uptake in LDLR−/−/VDR−/− mice
To understand the mechanism underlying the accelerated atherosclerotic phenotype seen in LDLR−/−/VDR−/− mice, we measured adhesion molecules, inflammation, and cholesterol transport in the aorta and peritoneal macrophages. After 4 wk on the HFHC diet, LDLR−/−/VDR−/− mice exhibited significantly higher intercellular adhesion molecule (ICAM)-1 and E-selectin expression in the aorta than LDLR−/− mice (Fig. 2, A and B). Peritoneal macrophages isolated from LDLR−/−/VDR−/− mice also showed significantly higher ICAM-1 and P-selectin expression compared with LDLR−/− mice at baseline (Fig. 2C) or after being stimulated with acetylated LDL (acLDL) (Fig. 2D). Incubation with acLDL was used to mimic the oxidized LDL insult on macrophages under the hyperlipidemic condition in vivo. Monocyte chemoattractant protein (MCP)-1, a chemokine that plays a key role in monocyte recruitment into the intima of vascular wall, was markedly up-regulated in the aorta of LDLR−/−/VDR−/− mice after 4-wk HFHC diet feeding (Fig. 2, E and F). MCP-1, IL-1β, interferon-γ, and matrix metalloproteinase 9 were also elevated in LDLR−/−/VDR−/− peritoneal macrophages at baseline (Fig. 2G) or when stimulated with acLDL (Fig. 2H) compared with LDLR−/− counterparts. Increased adhesion molecules and proinflammatory factors in the aorta and macrophages are predicted to promote lesion initiation.
Fig. 2.
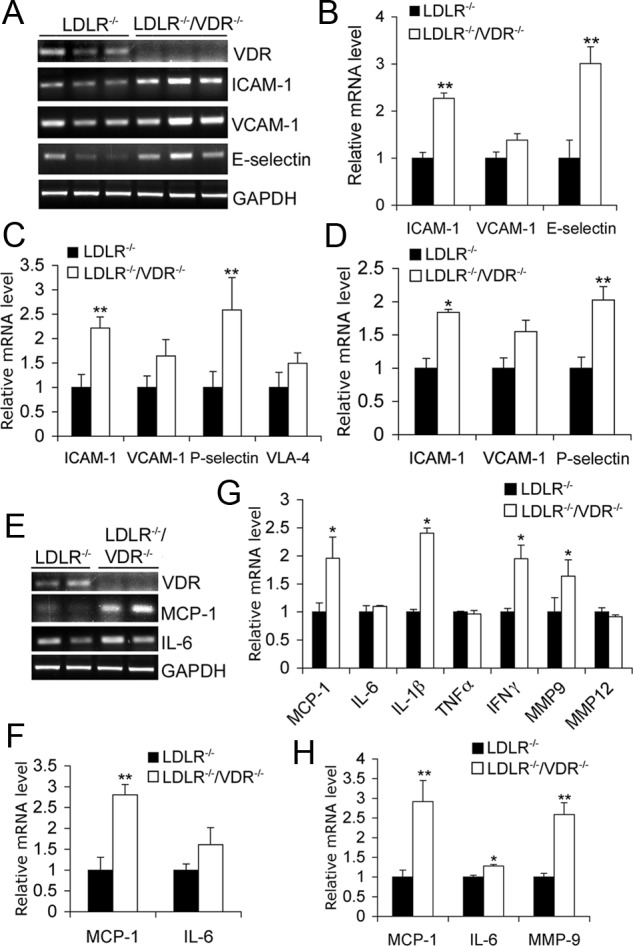
Expression of adhesion molecules and inflammatory cytokines in aorta and macrophages. A and B, RT-PCR amplification (A) and quantitation (B) of adhesion molecule transcripts in the aorta isolated from LDLR−/− and LDLR−/−/VDR−/− mice fed the HFHC diet for 4 wk. C and D, Real-time RT-PCR quantitation of adhesion molecules in LDLR−/− and LDLR−/−/VDR−/− peritoneal macrophages at baseline (C) or stimulated with 50 μg/ml acLDL for 24 h (D). E and F, RT-PCR amplification (E) and quantitation (F) of MCP-1 and IL-6 levels in aorta isolated from 4-wk HFHC diet-treated LDLR−/− and LDLR−/−/VDR−/− mice. G and H, Real-time RT-PCR quantitation of cytokines and chemokines in LDLR−/− and LDLR−/−/VDR−/− peritoneal macrophages at baseline (G) or after being stimulated with acLDL (H). *, P < 0.05 vs. LDLR−/−; n = 5–10. GAPDH, Glyceraldehyde-3-phosphate dehydrogenase; IFN, interferon; MMP, matrix metalloproteinase; VCAM, vascular cell adhesion molecule.
Macrophage conversion to foam cells is a critical part of the atherogenic process. We therefore measured cholesterol influx and efflux in peritoneal macrophages. As shown in Fig. 3, LDLR−/−/VDR−/− macrophages accumulated significantly more [3H]cholesterol in the presence of acLDL compared with LDLR−/− macrophages at 8 and 24 h of incubation (Fig. 3A). Consistent with the increased cholesterol uptake, SR-A1 and CD36, membrane proteins that mediate cholesterol influx, were markedly up-regulated in LDLR−/−/VDR−/− macrophages relative to LDLR−/−macrophages at baseline (Fig. 3B) or after being stimulated with acLDL (Fig. 3C). SR-A1 elevation was also detected in the aorta isolated from LDLR−/−/VDR−/− mice after 4-wk HFHC diet feeding (Fig. 3, D and E). Cholesterol accumulation was also increased in VDR−/− macrophages (Supplemental Fig. 2A), suggesting that the VDR regulates macrophage cholesterol influx independent of the LDLR. In contrast, we did not detect a significant difference in cholesterol efflux between wild-type (WT) and VDR−/− macrophages (Supplemental Fig. 2B) or between LDLR−/− and LDLR−/−/VDR−/− macrophages (Supplemental Fig. 2C) either at baseline or after stimulation with apolipoprotein AI. Consistent with this observation, transcript levels of the gene products involved in the control of macrophage cholesterol efflux (peroxisomal proliferator-activated receptor-γ, liver X receptor-α, ABCA1, and ABCG1) were not significantly different between LDLR−/− and LDLR−/−/VDR−/− macrophages (Supplemental Fig. 2D). These data suggest that the VDR signaling inhibits foam cell formation by suppressing cholesterol influx in macrophages. Taken together, it appears that macrophage dysfunction plays a key role in promoting and accelerating atherosclerosis in LDLR−/−/VDR−/− mice.
Fig. 3.

Enhanced cholesterol influx in macrophages with VDR deletion. A, Cholesterol uptake assays. [3H]cholesterol accumulation in the presence of acLDL in LDLR−/− and LDLR−/−/VDR−/− peritoneal macrophages at 8 and 24 h of incubation. B and C, Real-time RT-PCR quantitation of SR-A1 and CD36 in peritoneal macrophages at baseline (B) or with acLDL stimulation (C). D and E, RT-PCR assessment of SR-A1 and CD36 expression in aorta from 4-wk HFHC diet-fed mice (D) and quantitation (E). *, P < 0.05 vs. LDLR−/−, n = 5–6. GAPDH, Glyceraldehyde-3-phosphate dehydrogenase.
VDR−/− bone marrow-derived cells promote atherosclerosis in the presence or absence of mature T and B lymphocytes
To address the contribution of BM-derived cells to the enhanced atherosclerotic lesion development seen in LDLR−/−/VDR−/− mice, we compared LDLR−/− recipient mice transplanted with LDLR−/− BM or LDLR−/−/VDR−/− BM. After 8-wk HFHC dietary treatment, plasma cholesterol and triglyceride levels were not significantly different between LDLR−/− BM-transplanted (BMT) and LDLR−/−/VDR−/− BMT mice (Fig. 4A), but the lesions were significantly larger at the AA site in LDLR−/−/VDR−/− BMT mice (P < 0.05), whereas the lesions at the other three sites were not significantly different between these two BMT mice (Fig. 4B).
Fig. 4.

Role of leukocyte VDR signaling in atherogenesis. A and B, Effects of VDR-deleted BMT on atherosclerosis. LDLR−/− mice were transplanted with LDLR−/− BM (LDLR−/− BMT) or with LDLR−/−/VDR−/− BM (LDLR−/−/VDR−/− BMT). Atherosclerosis was induced by 8-wk HFHC feeding. A, Plasma total cholesterol (Chol) and triglyceride (TG) levels. B, Atherosclerotic lesion sizes at the four vascular sites (IA, AA, DA, and AR). C and D, Effect of VDR-deleted, T and B cell-deficient BMT on plasma lipids (C) and atherosclerosis (D). LDLR−/− mice transplanted with Rag-1−/− BM (Rag-1−/− BMT) or with Rag-1−/−/VDR−/− BM (Rag-1−/−/VDR−/− BMT) were fed the HFHC diet for 8 wk.
To explore further the role of leukocyte VDR signaling in atherogenesis, we performed another BM transplant experiment in which LDLR−/− recipients were transplanted with BM derived from Rag-1−/− or Rag-1−/−/VDR−/− mice. Because Rag-1−/− mice do not form mature T and B lymphocytes (31), the resulting Rag-1−/− BMT and Rag-1−/−/VDR−/− BMT mice would be deficient in functional T and B cells. After 8-wk HFHC diet feeding, no significant difference was detected in plasma cholesterol and triglyceride levels between Rag-1−/− BMT and Rag-1−/−/VDR−/− BMT mice (Fig. 4C); however, atherosclerotic lesions at the IA and AA sites were significantly larger in Rag-1−/−/VDR−/− BMT mice (P < 0.05) (Fig. 4D). These data strongly suggest that leukocyte VDR signaling, likely macrophage VDR, plays a key role in regulating atherogenesis.
Up-regulation of local RAS in atherosclerotic lesions and macrophages in the absence of VDR
Because renin and angiotensinogen (AGT) are directly targeted by 1,25-dihydroxyvitamin D3 (27, 32, 33), we assessed the local RAS status in the aorta and macrophages. In peritoneal macrophages isolated from VDR−/− mice, renin expression was markedly elevated at both mRNA (Fig. 5A) and protein (Fig. 5B) levels relative to the WT counterpart. Western blot showed that the protein levels of AGT and AT1 receptor were also elevated in VDR−/− macrophages (Fig. 5B). Similarly, the up-regulation of renin, AGT, and AT1 receptor were also detected in peritoneal macrophages obtained from LDLR−/−/VDR−/− mice at baseline (Fig. 5C) or after being stimulated with acLDL (Fig. 5D) compared with LDLR−/− mice. Renin receptor was also increased in stimulated LDLR−/−/VDR−/− macrophages (Fig. 5D). In the aorta obtained from 4-wk HFHC diet-fed LDLR−/−/VDR−/− mice, renin was also dramatically up-regulated relative to the LDLR−/− control (Fig. 5, E and F). Similar renin up-regulation was also seen in the aorta from VDR−/− mice (Supplemental Fig. 3, A and B). Moreover, immunostaining with antirenin and anti-MOMA-2 (macrophage-specific) antibodies showed colocalization of marked renin elevation with macrophages within the atherosclerotic lesion in LDLR−/−/VDR−/− mice (Fig. 5G). Together these data indicate that in the absence of VDR signaling, the local RAS is intrinsically up-regulated in macrophages and aorta. We speculate that increased local RAS activation within the aorta promotes lesion progression.
Fig. 5.

Effects of VDR deletion on the local RAS in macrophages and aorta. A and B, Assessment of RAS components in peritoneal macrophages isolated from WT and VDR(−/−) mice by RT-PCR (A) and Western blotting (B). C and D, Real-time RT-PCR quantitation of RAS components in peritoneal macrophages isolated from LDLR−/− and LDLR−/−/VDR−/− mice at baseline (C) or after acLDL stimulation (D). E and F, RT-PCR amplification (E) and quantitation (F) of renin transcripts in the aorta from LDLR−/− and LDLR−/−/VDR−/− mice. G, Atherosclerotic lesions from LDLR−/− and LDLR−/−/VDR−/− mice stained with anti-MOMA-2 (red) and anti-renin (green) antibodies. Note that renin is colocalized with macrophages (yellow), and LDLR−/−/VDR−/− macrophages have higher renin levels compared with LDLR−/− macrophages. ACE, Angiotensin-converting enzyme; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; Re R, renin receptor.
Inhibition of renin ameliorates the increment of atherogenesis in the absence of VDR
To address whether renin up-regulation indeed contributes to the increased atherogenesis seen in LDLR−/−/VDR−/− mice, we treated the mice with aliskiren, a specific renin inhibitor, for 8 wk. Plasma cholesterol levels remained lower in the treated LDLR−/−/VDR−/− mice (Fig. 6A), but in contrast to the untreated mice (see Fig. 1), the sizes of atherosclerotic lesions at the four vascular sites were not significantly different between LDLR−/− and LDLR−/−/VDR−/− mice (Fig. 5B). Compared with untreated mice, aliskiren significantly reduced the lesion sizes at the IA and AA sites in LDLR−/− mice (Fig. 6C), consistent with a previous report that aliskiren reduced atherosclerosis (17).
Fig. 6.
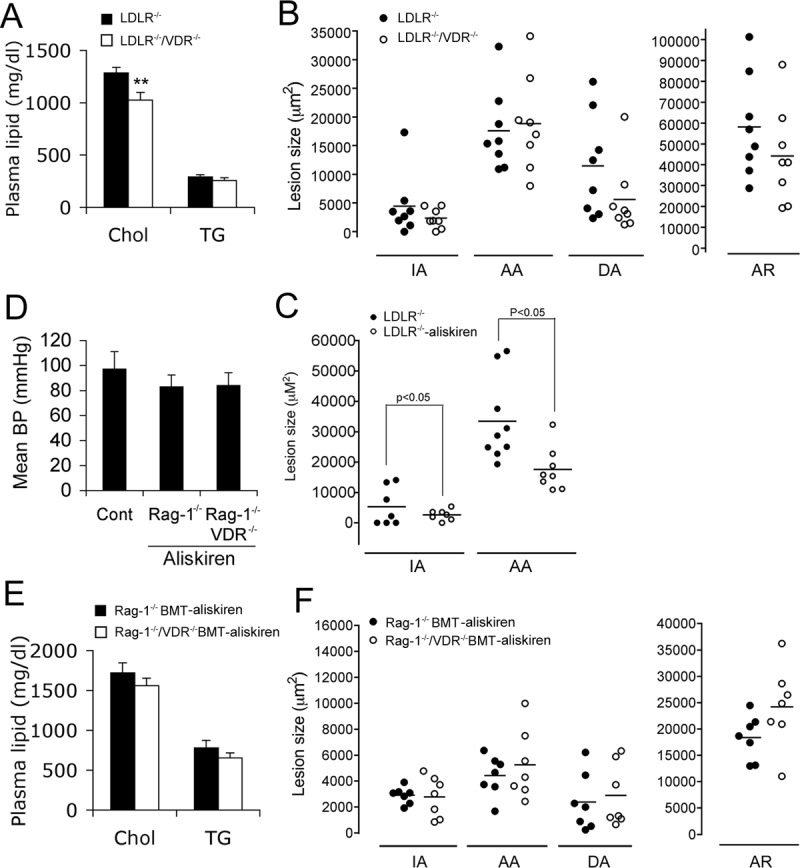
Effect of renin inhibition with aliskiren on atherosclerosis. A, Plasma cholesterol (Chol) and triglyceride (TG) levels in LDLR−/− and LDLR−/−/VDR−/− mice treated with aliskiren on 8-wk HFHC diet. B, Atherosclerotic lesion sizes at the four vascular sites in LDLR−/− and LDLR−/−/VDR−/− mice treated with aliskiren. C, Comparison of atherosclerotic lesion sizes on 8 wk HFHC diet in LDLR−/− mice with or without aliskiren treatment. D, Mean blood pressure (BP) of Rag-1−/− BMT and Rag-1−/−/VDR−/− BMT mice treated with aliskiren (n = 6 each phenotype), measured by the carotid artery cannulation method. E, Plasma cholesterol and triglyceride levels in Rag-1−/− BMT and Rag-1−/−/VDR−/− BMT mice treated with aliskiren on 8-wk HFHC diet. F, Atherosclerotic lesion sizes at the four vascular sites of the aortic tree in Rag-1−/− BMT and Rag-1−/−/VDR−/− BMT mice treated with aliskiren for 8 wk. Cont., Control.
To address further the role of local leukocyte RAS activation in atherogenesis, we treated Rag-1−/− BMT and Rag-1−/−/VDR−/− BMT mice on the HFHC diet with aliskiren for 8 wk. Aliskiren had similar inhibitory effects on the blood pressure of Rag-1−/− BMT and Rag-1−/−/VDR−/− BMT mice, but the change in blood pressure did not reach significance (P > 0.05) compared with the untreated control mice (Fig. 6D). Plasma cholesterol and triglyceride levels were not affected in the aliskiren-treated mice (Fig. 6E), but aliskiren completely eliminated the difference in atherosclerotic lesion size between Rag-1−/− BMT and Rag-1−/−/VDR−/− BMT mice (Fig. 6F). These data strongly suggest that activation of the local RAS in leukocytes, most likely in macrophages, plays a key role in promoting atherosclerosis in the absence of vitamin D-VDR signaling.
Discussion
Epidemiological studies have documented that serum 25-hydroxyvitamin D levels are inversely correlated with the prevalence of cardiovascular disease and its risk factors such as hypertension, diabetes, obesity, and hyperlipidemia in the general US adult population (34–36). Low vitamin D status is associated with congestive heart failure (37) and higher risk of myocardial infarction even after adjusting for factors known to be associated with coronary artery disease (38). Significant reduction in cardiovascular mortality has been noticed in many retrospective observational studies that report reduced mortality in chronic kidney disease patients receiving vitamin D therapy (see references in Ref. 39). These observations implicate a role for vitamin D in cardiovascular protection (20). Few studies, however, have directly addressed the role of the VDR signaling on vascular disease. In this study we demonstrated that the VDR signaling, most probably macrophage VDR signaling, inhibits atherogenesis in mice.
We took several approaches to assess the role of VDR in atherosclerosis. We first examined the impact of VDR deletion in LDLR-null mice. LDLR−/−/VDR−/− mice showed accelerated atherosclerotic lesion development in the IA and AR despite lower plasma lipid levels compared with LDLR−/− controls. At the other vascular sites the increase in lesions was not statistically significant. It is possible that a larger sample size is needed to detect significant changes at these sites. It is also possible that the difference in sheer stress and blood flow at these sites contributes to the results. In LDLR−/− mice plasma cholesterol levels rose dramatically after HFHC diet feeding because of the lack of LDLR to clear cholesterol from plasma. This dramatic increase in plasma cholesterol is the major cause promoting atherosclerosis in the animals. In LDLR−/−/VDR−/− mice, however, the rise in plasma cholesterol was significantly dampened as a result of VDR deletion, with both VLDL and LDL cholesterol levels being markedly suppressed. Because plasma cholesterol is a predominant driving force of atherogenesis, the inhibitory effect of VDR is likely underestimated in the LDLR−/−/VDR−/− model.
Despite the negative impact on the rise of plasma cholesterol, which does not favor atherogenesis, several lines of evidence suggest that VDR deletion renders mice prone to atherosclerosis. The aorta from LDLR−/−/VDR−/− mice exhibits up-regulated adhesion molecules (e.g. ICAM-1) and chemokines (e.g. MCP-1) early in the course of atherosclerosis. Macrophages with VDR deletion are more inflammatory in terms of cytokine/chemokine production. Moreover, VDR−/− macrophages show increased cholesterol uptake due to up-regulation of SR-A1 and CD36. Because cholesterol efflux is unaltered, increased influx will lead to intracellular cholesterol accumulation, suggesting that VDR signaling inhibits foam cell formation. This is consistent with an early report showing that 1,25-dihydroxyvitamin D suppresses cholesterol uptake in macrophages from type 2 diabetes patients (24).
In an attempt to limit the confounding effect resulting from different plasma cholesterol levels, we transplanted LDLR−/−/VDR−/− BM into LDLR−/− recipients. In this experiment no differences in plasma lipid levels were observed at 8 wk after transplantation regardless of the presence or absence of the VDR in the BM-derived cells. Yet the absence of the VDR in these donor cells was still associated with an increment in atherosclerosis in selected vascular sites. In a second BM transplantation experiment, VDR−/− BM lacking T and B cells from Rag-1−/−/VDR−/− donors was transplanted into LDLR−/− recipients. Here too the increased atherosclerosis phenotype was reproduced (statistically significant increase in lesions at the IA and AA). This points to leukocytes, probably monocytes/macrophages, as the major and most probable site of action of vitamin D signaling in modulating atherogenesis; however, more studies are still required to assign with absolute confidence the antiatherosclerotic role to macrophage VDR signaling. These observations argue that the VDR influences atherogenesis independent of its effect on T regulatory cell functions, in contrast to an explanation offered for the antiatherogenic influence of exogenous 1,25-dihydroxyvitamin D treatment (25). Consistent with an antiatherosclerotic role of macrophage VDR, we found that 1,25-dihydroxvitamin D markedly induced VDR expression at both mRNA and protein levels in peritoneal macrophages, whereas acLDL, an inducer of atherosclerosis, markedly suppressed VDR expression in these cells (Supplemental Fig. 4). It is speculated, therefore, that high levels of modified LDL are able to diminish the protective effect of VDR in leukocytes or macrophages, allowing atherogenesis to proceed.
There are many potential mechanisms whereby VDR signaling inhibits atherosclerosis. In this study we focused on the local RAS in macrophages. The RAS is known to play a crucial role in atherogenesis. All RAS components are found in the vascular wall (40–44) and in atherosclerotic lesions (45, 46). These RAS components colocalize with macrophages, particularly under hypercholesterolemic conditions (14, 47), suggesting that exposure to high levels of lipids activates the macrophage RAS leading to increased local Ang II generation in atherosclerotic lesions. Ang II promotes atherosclerosis at all stages of the atherosclerosis course independent of its effect on blood pressure (47, 48), including induction of endothelial dysfunction and vascular inflammation (49) and LDL oxidation (50), and promotion of monocyte recruitment (51–54), foam cell formation (55), vascular smooth muscle cell proliferation (56–58), and plaque instability (59, 60). Here we observed marked up-regulation of the RAS components (renin, AGT, and AT1R) in peritoneal macrophages, the aorta, and macrophages/foam cells within the atherosclerotic lesions in the absence of VDR. Importantly, blockade of renin by aliskiren ameliorated atherosclerosis not only in LDLR−/−/VDR−/− mice, but also in Rag-1−/−/VDR−/− BMT mice, suggesting that the local RAS in VDR−/− leukocytes is most likely responsible for the increase in atherosclerosis. That macrophage renin promotes atherosclerosis has been well documented in a prior elegant study (17). Based on these findings, we infer that VDR signaling in leukocytes, likely macrophages, inhibits atherosclerosis in part by suppressing the local RAS in macrophages. This conclusion, however, does not exclude the VDR signaling from influencing atherosclerosis by other mechanisms, such as regulation of calcium/phosphate homeostasis, PTH, fibroblast growth factor 23, atherosclerotic lesion calcification, inflammatory mediators/cytokines, and plasma lipid metabolism. The regulation of these biological processes by VDR signaling warrants further investigations.
In this study we observed that, in the absence of VDR signaling, plasma lipid levels were reduced. Unlike the situation with atherosclerosis, this does not appear to be attributable to a regulation by BM-derived cells, because the same reduction in plasma lipids was not observed in the BM transplantation protocols. The mechanism for the effect of VDR signaling on plasma lipids remains unclear and merits further studies. Two aspects of lipid metabolism seem to be excluded: no differential effects of VDR presence or absence were observed on lipid absorption or VLDL production. This tends to point to the catabolic arm of lipid and lipoprotein homeostasis. Because LDLR is absent in most of the models studied here, regulation of this receptor, which normally plays a major role in the catabolism of Apo-B containing lipoproteins, is not implicated. Consistently, VDR−/− mice also show reduced plasma cholesterol levels (29), confirming that the regulation of cholesterol metabolism is independent of LDLR. A plausible mechanism is the direct down-regulation of hepatic cholesterol 7α-hydroxylase (Cyp7A1), the rate-limiting enzyme involved in bile acid synthesis, by vitamin D signaling (61). Another possible mechanism is vitamin D up-regulation of intestinal fibroblast growth factor 15, an intestine-derived hormone that acts on the liver to repress hepatic Cyp7A1 (62). Both mechanisms could explain how vitamin D inhibits hepatic cholesterol 7α-hydroxylase, implying that VDR inactivation could lead to increased Cyp7A1 enzymatic activity, which promotes cholesterol catabolism and thus reduces plasma cholesterol levels.
In summary, in this study we provide strong evidence that supports an antiatherosclerotic role of the VDR signaling in leukocytes/macrophages, and at least part of the antiatherosclerotic mechanism is to block the activation of the local RAS in macrophages and within the atherosclerotic lesion. The knowledge obtained from this work will guide further investigations to explore the potentials of vitamin D and its analogs in regulation of atherosclerosis and lipoprotein homeostasis in both preclinical and clinical settings.
Supplementary Material
Acknowledgments
This work was supported in part by National Institutes of Health grant HL085793, a research grant from the Center for D-Receptor Activation Research, and Molecular and Cellular Biology Training Grant (T32 GM007183–35).
Disclosure Summary: R.T. received a research grant support from Abbott Laboratories. All other authors declare no conflict of interest.
NURSA Molecule Pages†:
Nuclear Receptors: VDR.
Annotations provided by Nuclear Receptor Signaling Atlas (NURSA) Bioinformatics Resource. Molecule Pages can be accessed on the NURSA website at www.nursa.org.
- AA
- Ascending aorta
- acLDL
- acetylated LDL
- ABCA1
- ATP-binding cassette transporter A1
- AGT
- angiotensinogen
- Ang II
- angiotensin II
- AR
- aortic root
- BM
- bone marrow
- BMT
- BM transplant
- Cyp7A1
- cholesterol 7α-hydroxylase
- DA
- descending aorta
- HFHC
- high fat high cholesterol
- IA
- innominate artery
- ICAM
- intercellular adhesion molecule
- LDL
- low-density lipoprotein
- LDLR
- LDL receptor
- MCP-1
- monocyte chemoattractant protein-1
- NC
- not associated with a coronary artery
- RAS
- renin-angiotensin system
- VDR
- vitamin D receptor
- WT
- wild type.
References
- 1. Glass CK, Witztum JL. 2001. Atherosclerosis. the road ahead. Cell 104:503–516 [DOI] [PubMed] [Google Scholar]
- 2. Ross R. 1999. Atherosclerosis–an inflammatory disease. N Engl J Med 340:115–126 [DOI] [PubMed] [Google Scholar]
- 3. Hynes RO, Wagner DD. 1996. Genetic manipulation of vascular adhesion molecules in mice. J Clin Invest 98:2193–2195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Galkina E, Ley K. 2007. Vascular adhesion molecules in atherosclerosis. Arterioscler Thromb Vasc Biol 27:2292–2301 [DOI] [PubMed] [Google Scholar]
- 5. Eriksson EE. 2003. Leukocyte recruitment to atherosclerotic lesions, a complex web of dynamic cellular and molecular interactions. Curr Drug Targets Cardiovasc Haematol Disord 3:309–325 [DOI] [PubMed] [Google Scholar]
- 6. Dong ZM, Chapman SM, Brown AA, Frenette PS, Hynes RO, Wagner DD. 1998. The combined role of P- and E-selectins in atherosclerosis. J Clin Invest 102:145–152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Collins RG, Velji R, Guevara NV, Hicks MJ, Chan L, Beaudet AL. 2000. P-Selectin or intercellular adhesion molecule (ICAM)-1 deficiency substantially protects against atherosclerosis in apolipoprotein E-deficient mice. J Exp Med 191:189–194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rajavashisth T, Qiao JH, Tripathi S, Tripathi J, Mishra N, Hua M, Wang XP, Loussararian A, Clinton S, Libby P, Lusis A. 1998. Heterozygous osteopetrotic (op) mutation reduces atherosclerosis in LDL receptor-deficient mice. J Clin Invest 101:2702–2710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Smith JD, Trogan E, Ginsberg M, Grigaux C, Tian J, Miyata M. 1995. Decreased atherosclerosis in mice deficient in both macrophage colony-stimulating factor (op) and apolipoprotein E. Proc Natl Acad Sci USA 92:8264–8268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Qiao JH, Tripathi J, Mishra NK, Cai Y, Tripathi S, Wang XP, Imes S, Fishbein MC, Clinton SK, Libby P, Lusis AJ, Rajavashisth TB. 1997. Role of macrophage colony-stimulating factor in atherosclerosis: studies of osteopetrotic mice. Am J Pathol 150:1687–1699 [PMC free article] [PubMed] [Google Scholar]
- 11. Pennings M, Meurs I, Ye D, Out R, Hoekstra M, Van Berkel TJ, Van Eck M. 2006. Regulation of cholesterol homeostasis in macrophages and consequences for atherosclerotic lesion development. FEBS Lett 580:5588–5596 [DOI] [PubMed] [Google Scholar]
- 12. Daugherty A, Webb NR, Rateri DL, King VL. 2005. Thematic review series: the immune system and atherogenesis. Cytokine regulation of macrophage functions in atherogenesis. J Lipid Res 46:1812–1822 [DOI] [PubMed] [Google Scholar]
- 13. Daugherty A, Rateri DL, Lu H, Inagami T, Cassis LA. 2004. Hypercholesterolemia stimulates angiotensin peptide synthesis and contributes to atherosclerosis through the AT1A receptor. Circulation 110:3849–3857 [DOI] [PubMed] [Google Scholar]
- 14. Potter DD, Sobey CG, Tompkins PK, Rossen JD, Heistad DD. 1998. Evidence that macrophages in atherosclerotic lesions contain angiotensin II. Circulation 98:800–807 [DOI] [PubMed] [Google Scholar]
- 15. Iwai N, Inagami T, Ohmichi N, Kinoshita M. 1996. Renin is expressed in rat macrophage/monocyte cells. Hypertension 27(3 Pt 1):399–403 [DOI] [PubMed] [Google Scholar]
- 16. Okamura A, Rakugi H, Ohishi M, Yanagitani Y, Takiuchi S, Moriguchi K, Fennessy PA, Higaki J, Ogihara T. 1999. Upregulation of renin-angiotensin system during differentiation of monocytes to macrophages. J Hypertens 17:537–545 [DOI] [PubMed] [Google Scholar]
- 17. Lu H, Rateri DL, Feldman DL, Charnigo RJ, Jr, Fukamizu A, Ishida J, Oesterling EG, Cassis LA, Daugherty A. 2008. Renin inhibition reduces hypercholesterolemia-induced atherosclerosis in mice. J Clin Invest 118:984–993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Holick MF. 2007. Vitamin D deficiency. N Engl J Med 357:266–281 [DOI] [PubMed] [Google Scholar]
- 19. Haussler MR, Whitfield GK, Haussler CA, Hsieh JC, Thompson PD, Selznick SH, Dominguez CE, Jurutka PW. 1998. The nuclear vitamin D receptor: biological and molecular regulatory properties revealed. J Bone Miner Res 13:325–349 [DOI] [PubMed] [Google Scholar]
- 20. Li YC. 2011. Molecular mechanism of vitamin D in the cardiovascular system. J Investig Med 59:868–871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lai H, Fishman EK, Gerstainblith G, Brinker JA, Tong W, Bhatia S, Detrick B, Lai S. 2011. Vitamin D deficiency is associated with significant coronary stenoses in asymptomatic African American chronic cocaine users. Int. J Cardiol. 10.1016/j.ijcard.2011.01.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Reis JP, von Mühlen D, Michos ED, Miller ER, III, Appel LJ, Araneta MR, Barrett-Connor E. 2009. Serum vitamin D, parathyroid hormone levels, and carotid atherosclerosis. Atherosclerosis 207:585–590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. de Boer IH, Kestenbaum B, Shoben AB, Michos ED, Sarnak MJ, Siscovick DS. 2009. 25-Hydroxyvitamin D levels inversely associate with risk for developing coronary artery calcification. J Am Soc Nephrol 20:1805–1812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Oh J, Weng S, Felton SK, Bhandare S, Riek A, Butler B, Proctor BM, Petty M, Chen Z, Schechtman KB, Bernal-Mizrachi L, Bernal-Mizrachi C. 2009. 1,25(OH)2 vitamin D inhibits foam cell formation and suppresses macrophage cholesterol uptake in patients with type 2 diabetes mellitus. Circulation 120:687–698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Takeda M, Yamashita T, Sasaki N, Nakajima K, Kita T, Shinohara M, Ishida T, Hirata K. 2010. Oral administration of an active form of vitamin D3 (calcitriol) decreases atherosclerosis in mice by inducing regulatory T cells and immature dendritic cells with tolerogenic functions. Arterioscler Thromb Vasc Biol 30:2495–2503 [DOI] [PubMed] [Google Scholar]
- 26. Li YC, Pirro AE, Amling M, Delling G, Baron R, Bronson R, Demay MB. 1997. Targeted ablation of the vitamin D receptor: an animal model of vitamin D-dependent rickets type II with alopecia. Proc Natl Acad Sci USA 94:9831–9835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Li YC, Kong J, Wei M, Chen ZF, Liu SQ, Cao LP. 2002. 1,25-Dihydroxyvitamin D3 is a negative endocrine regulator of the renin-angiotensin system. J Clin Invest 110:229–238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Reardon CA, Blachowicz L, White T, Cabana V, Wang Y, Lukens J, Bluestone J, Getz GS. 2001. Effect of immune deficiency on lipoproteins and atherosclerosis in male apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol 21:1011–1016 [DOI] [PubMed] [Google Scholar]
- 29. Wong KE, Szeto FL, Zhang W, Ye H, Kong J, Zhang Z, Sun XJ, Li YC. 2009. Involvement of the vitamin D receptor in energy metabolism: regulation of uncoupling proteins. Am J Physiol Endocrinol Metab 296:E820–E828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zhang Z, Sun L, Wang Y, Ning G, Minto AW, Kong J, Quigg RJ, Li YC. 2008. Renoprotective role of the vitamin D receptor in diabetic nephropathy. Kidney Int 73:163–171 [DOI] [PubMed] [Google Scholar]
- 31. Mombaerts P, Iacomini J, Johnson RS, Herrup K, Tonegawa S, Papaioannou VE. 1992. RAG-1-deficient mice have no mature B and T lymphocytes. Cell 68:869–877 [DOI] [PubMed] [Google Scholar]
- 32. Yuan W, Pan W, Kong J, Zheng W, Szeto FL, Wong KE, Cohen R, Klopot A, Zhang Z, Li YC. 2007. 1,25-Dihydroxyvitamin D3 suppresses renin gene transcription by blocking the activity of the cyclic AMP response element in the renin gene promoter. J Biol Chem 282:29821–29830 [DOI] [PubMed] [Google Scholar]
- 33. Deb DK, Chen Y, Zhang Z, Zhang Y, Szeto FL, Wong KE, Kong J, Li YC. 2009. 1,25-Dihydroxyvitamin D3 suppresses high glucose-induced angiotensinogen expression in kidney cells by blocking the NF-κB pathway. Am J Physiol Renal Physiol 296:F1212–F1218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kendrick J, Targher G, Smits G, Chonchol M. 2009. 25-Hydroxyvitamin D deficiency is independently associated with cardiovascular disease in the Third National Health and Nutrition Examination Survey. Atherosclerosis 205:255–260 [DOI] [PubMed] [Google Scholar]
- 35. Scragg R, Sowers M, Bell C. 2007. Serum 25-hydroxyvitamin D, ethnicity, and blood pressure in the Third National Health and Nutrition Examination Survey. Am J Hypertens 20:713–719 [DOI] [PubMed] [Google Scholar]
- 36. Martins D, Wolf M, Pan D, Zadshir A, Tareen N, Thadhani R, Felsenfeld A, Levine B, Mehrotra R, Norris K. 2007. Prevalence of cardiovascular risk factors and the serum levels of 25-hydroxyvitamin D in the United States: data from the Third National Health and Nutrition Examination Survey. Arch Intern Med 167:1159–1165 [DOI] [PubMed] [Google Scholar]
- 37. Zittermann A, Schleithoff SS, Tenderich G, Berthold HK, Körfer R, Stehle P. 2003. Low vitamin D status: a contributing factor in the pathogenesis of congestive heart failure? J Am Coll Cardiol 41:105–112 [DOI] [PubMed] [Google Scholar]
- 38. Giovannucci E, Liu Y, Hollis BW, Rimm EB. 2008. 25-hydroxyvitamin D and risk of myocardial infarction in men: a prospective study. Arch Intern Med 168:1174–1180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kovesdy CP. 2010. Survival benefits with vitamin D receptor activation: new insights since 2003. Clin J Am Soc Nephrol 5:1704–1709 [DOI] [PubMed] [Google Scholar]
- 40. Iwai N, Izumi M, Inagami T, Kinoshita M. 1997. Induction of renin in medial smooth muscle cells by balloon injury. Hypertension 29:1044–1050 [DOI] [PubMed] [Google Scholar]
- 41. Campbell DJ, Habener JF. 1986. Angiotensinogen gene is expressed and differentially regulated in multiple tissues of the rat. J Clin Invest 78:31–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hellmann W, Suzuki F, Ohkubo H, Nakanishi S, Ludwig G, Ganten D. 1988. Angiotensinogen gene expression in extrahepatic rat tissues: application of a solution hybridization assay. Naunyn Schmiedebergs Arch Pharmacol 338:327–331 [DOI] [PubMed] [Google Scholar]
- 43. Falkenhahn M, Franke F, Bohle RM, Zhu YC, Stauss HM, Bachmann S, Danilov S, Unger T. 1995. Cellular distribution of angiotensin-converting enzyme after myocardial infarction. Hypertension 25:219–226 [DOI] [PubMed] [Google Scholar]
- 44. Fernandez-Alfonso MS, Martorana PA, Licka I, van Even P, Trobisch D, Schölkens BA, Paul M. 1997. Early induction of angiotensin I-converting enzyme in rat carotid artery after balloon injury. Hypertension 30:272–277 [DOI] [PubMed] [Google Scholar]
- 45. Diet F, Pratt RE, Berry GJ, Momose N, Gibbons GH, Dzau VJ. 1996. Increased accumulation of tissue ACE in human atherosclerotic coronary artery disease. Circulation 94:2756–2767 [DOI] [PubMed] [Google Scholar]
- 46. Ohishi M, Ueda M, Rakugi H, Naruko T, Kojima A, Okamura A, Higaki J, Ogihara T. 1997. Enhanced expression of angiotensin-converting enzyme is associated with progression of coronary atherosclerosis in humans. J Hypertens 15:1295–1302 [DOI] [PubMed] [Google Scholar]
- 47. Daugherty A, Cassis L. 2004. Angiotensin II-mediated development of vascular diseases. Trends Cardiovasc Med 14:117–120 [DOI] [PubMed] [Google Scholar]
- 48. Daugherty A, Manning MW, Cassis LA. 2000. Angiotensin II promotes atherosclerotic lesions and aneurysms in apolipoprotein E-deficient mice. J Clin Invest 105:1605–1612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Sata M, Fukuda D. 2010. Crucial role of renin-angiotensin system in the pathogenesis of atherosclerosis. J Med Invest 57:12–25 [DOI] [PubMed] [Google Scholar]
- 50. Harrison DG. 1997. Cellular and molecular mechanisms of endothelial cell dysfunction. J Clin Invest 100:2153–2157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Pueyo ME, Gonzalez W, Nicoletti A, Savoie F, Arnal JF, Michel JB. 2000. Angiotensin II stimulates endothelial vascular cell adhesion molecule-1 via nuclear factor-κB activation induced by intracellular oxidative stress. Arterioscler Thromb Vasc Biol 20:645–651 [DOI] [PubMed] [Google Scholar]
- 52. Pastore L, Tessitore A, Martinotti S, Toniato E, Alesse E, Bravi MC, Ferri C, Desideri G, Gulino A, Santucci A. 1999. Angiotensin II stimulates intercellular adhesion molecule-1 (ICAM-1) expression by human vascular endothelial cells and increases soluble ICAM-1 release in vivo. Circulation 100:1646–1652 [DOI] [PubMed] [Google Scholar]
- 53. Tummala PE, Chen XL, Sundell CL, Laursen JB, Hammes CP, Alexander RW, Harrison DG, Medford RM. 1999. Angiotensin II induces vascular cell adhesion molecule-1 expression in rat vasculature: A potential link between the renin-angiotensin system and atherosclerosis. Circulation 100:1223–1229 [DOI] [PubMed] [Google Scholar]
- 54. Hernández-Presa M, Bustos C, Ortego M, Tuñon J, Renedo G, Ruiz-Ortega M, Egido J. 1997. Angiotensin-converting enzyme inhibition prevents arterial nuclear factor-κ B activation, monocyte chemoattractant protein-1 expression, and macrophage infiltration in a rabbit model of early accelerated atherosclerosis. Circulation 95:1532–1541 [DOI] [PubMed] [Google Scholar]
- 55. Keidar S, Heinrich R, Kaplan M, Hayek T, Aviram M. 2001. Angiotensin II administration to atherosclerotic mice increases macrophage uptake of oxidized ldl: a possible role for interleukin-6. Arterioscler Thromb Vasc Biol 21:1464–1469 [DOI] [PubMed] [Google Scholar]
- 56. Han Y, Runge MS, Brasier AR. 1999. Angiotensin II induces interleukin-6 transcription in vascular smooth muscle cells through pleiotropic activation of nuclear factor-κ B transcription factors. Circ Res 84:695–703 [DOI] [PubMed] [Google Scholar]
- 57. Brasier AR, Jamaluddin M, Han Y, Patterson C, Runge MS. 2000. Angiotensin II induces gene transcription through cell-type-dependent effects on the nuclear factor-κB (NF-κB) transcription factor. Mol Cell Biochem 212:155–169 [PubMed] [Google Scholar]
- 58. Ikeda U, Ikeda M, Oohara T, Oguchi A, Kamitani T, Tsuruya Y, Kano S. 1991. Interleukin 6 stimulates growth of vascular smooth muscle cells in a PDGF-dependent manner. Am J Physiol 260:H1713–H1717 [DOI] [PubMed] [Google Scholar]
- 59. Galis ZS, Khatri JJ. 2002. Matrix metalloproteinases in vascular remodeling and atherogenesis: the good, the bad, and the ugly. Circ Res 90:251–262 [PubMed] [Google Scholar]
- 60. Chen J, Li D, Schaefer RF, Mehta JL. 2004. Inhibitory effect of candesartan and rosuvastatin on CD40 and MMPs expression in apo-E knockout mice: novel insights into the role of RAS and dyslipidemia in atherogenesis. J Cardiovasc Pharmacol 44:446–452 [DOI] [PubMed] [Google Scholar]
- 61. Han S, Chiang JY. 2009. Mechanism of vitamin D receptor inhibition of cholesterol 7α-hydroxylase gene transcription in human hepatocytes. Drug Metab Dispos 37:469–478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Schmidt DR, Holmstrom SR, Fon Tacer K, Bookout AL, Kliewer SA, Mangelsdorf DJ. 2010. Regulation of bile acid synthesis by fat-soluble vitamins A and D. J Biol Chem 285:14486–14494 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.