

Summary
MicroRNAs are a class of short ~22 nucleotide RNAs predicted to regulate nearly half of all protein coding genes, including many involved in basal cellular processes and organismal development. Although a global reduction in miRNAs is commonly observed in various human tumors, complete loss has not been documented, suggesting an essential function for miRNAs in tumorigenesis. Here we present the finding that transformed or immortalized Dicer1-null somatic cells can be isolated readily in vitro, maintain the characteristics of DICER1-expressing controls and remain stably proliferative. Furthermore, Dicer1-null cells from a sarcoma cell line, though depleted of miRNAs, are competent for tumor formation. Hence, miRNA levels in cancer may be maintained in vivo by a complex stabilizing selection in the intratumoral environment.
Keywords: Dicer1, microRNAs, cancer, sarcoma, mesenchymal stem cells
Introduction
MicroRNAs (miRNAs) are short ~22 nucleotide RNAs that comprise an essential class of regulators predicted to repress over half of all genes post-transcriptionally (Bartel, 2009; Friedman et al., 2009). Consistent with computational predictions of widespread targeting, they have been implicated experimentally in a variety of fundamental cellular processes such as cell cycle (Wang et al., 2008), apoptosis (Chivukula and Mendell, 2008), and differentiation (Herranz and Cohen, 2010; Stefani and Slack, 2008). Given these broad roles, the relationship between miRNAs and cancer is understandably complex. At the level of individual miRNAs, either gains or losses may promote tumor formation. However, analysis of global miRNA levels in tumors suggests a surprisingly unidirectional relationship, with multiple human tumors showing decreased miRNA content (Gaur et al., 2007; Lu et al., 2005). In some cases, this downregulation may be directly achieved by decreased expression of DICER1 and DROSHA, key processing enzymes of miRNA production (Lin et al., 2010; Martello et al., 2010; Torres et al., 2011) or mutations in their binding partners (Melo et al., 2009).
Despite these trends towards decreased miRNA expression, a number of observations suggest that miRNAs may in fact be important for a variety of tumor types. For instance, while heterozygous somatic mutations in DICER1 can be found in tumor genotyping atlases, homozygous loss has not been reported in these databases (Kumar et al., 2009). Similarly, in rare cases of heterozygous germline DICER1 mutations, the pleuropulmonary blastomas to which patients are predisposed retain an intact DICER1 allele in tumor tissue (Hill et al., 2009). Somatic point mutations in DICER1 associated with nonepithelial ovarian cancers are hypomorphic, likely resulting in expression of a full-length protein but in loss of some specific miRNAs and retention of others, further suggesting a requirement for DICER1 and miRNA expression in tumors (Heravi-Moussavi et al., 2011). Mouse models of Dicer1 loss in cancer also suggest an advantage of retaining miRNA regulation. In a mouse model of Dicer1 deletion in the liver, tumors emerge several months after deletion following a period of hepatic repopulation by Dicer1-intact “escapers” (Sekine et al., 2009). In Dicer1-conditional mouse models of either soft tissue sarcoma or lung adenocarcinoma, haploinsufficiency of Dicer1 promotes tumor development but homozygous loss of Dicer1 is not observed (Kumar et al., 2009). Similarly, in both an Eμ-myc lymphoma model and a retinoblastoma model, viable tumors could not be identified following homozygous Dicer1 deletion (Arrate et al., 2010; Lambertz et al., 2010). These studies suggest that complete Dicer1 loss and the subsequent misregulation of gene expression are highly deleterious to tumor development.
To better understand how cancer cells respond to loss of miRNA expression, we characterized the effects of homozygous deletion of Dicer1-conditional alleles on the tumorigenicity of an established line of murine sarcoma cells and on the cellular phenotype of immortalized murine mesenchymal stem cells (MSCs).
Results
Dicer1-null cells derived from a mouse sarcoma proliferate indefinitely in vitro
Previously, we generated Dicer1-heterozygous tumors by injection of Adeno-Cre virus into the hindlimbs of KrasLSL-G12D;Trp53f/f;Dicer1f/f mice. The resultant tumors always retained at least one conditional Dicer1 allele (Kumar et al., 2009). From these KrasG12D;Trp53-/-;Dicer1f/- tumors, we established sarcoma cell lines and deleted the remaining allele of Dicer1 by transducing the cells with a retroviral construct encoding MSCV.CreERT2.puro and then activating recombination in vitro with tamoxifen treatment (Fig. 1A). A genotyping time course indicated efficient homozygous recombination (Fig. S1A). After multiple passages, however, genotyping PCR indicated the outgrowth of heterozygous cells, consistent with previous findings in both this sarcoma model and an Eμ-myc/Dicer1 lymphoma model (Arrate et al., 2010; Kumar et al., 2009).
Figure 1.
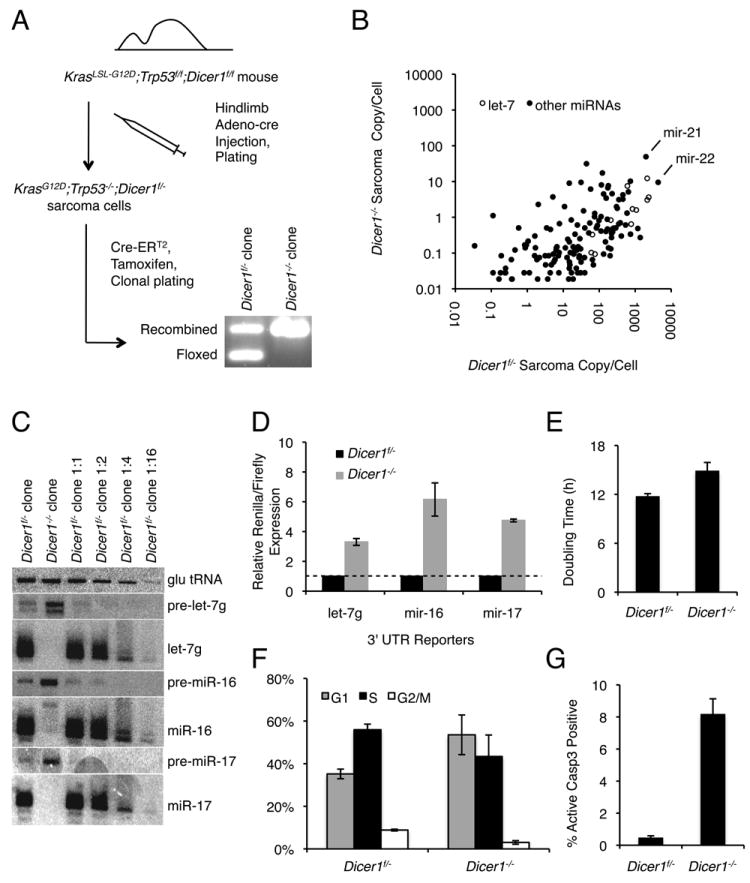
Characterization of KrasG12D;Trp53-/-;Dicer1-/- sarcoma cells. A) Derivation scheme for Dicer1-/- sarcoma cells. Hindlimb injection of Adeno-cre generates KrasG12D;Trp53-/-;Dicer1f/- tumors. Clones isolated following Cre-ER integration and tamoxifen treatment were genotyped by PCR to identify Dicer1-/- clones. B) miRNA expression (copies per cell). Per cell calculations are based on relative representation of each miRNA in Dicer1f/- and Dicer1-/- small RNA-seq libraries, normalized to quantitative Northern blot of miR-22 in Dicer1f/- cells (shown in Fig. S1C). miR-21, miR-22, and let-7 family members are indicated. C) Northern analysis for precursor and mature miRNAs. Glutamine tRNA was used to control for loading, and a dilution series of Dicer1f/- RNA (1:1 to 1:16) is provided for quantitation. D) Luciferase reporter assays for abundant miRNAs. The renilla luciferase reporter contains six bulged sites for the let-7 family, and two perfect sites for miR-16 and miR-17. Targeted renilla luciferase reporters were normalized to nontargeted firefly luciferase reporters. Renilla/firefly luciferase expression was normalized to expression in the Dicer1f/- sarcoma cell line. E) Proliferation assay. F) Cell cycle distribution determined by BrdU labeling. G) Apoptosis determined by caspase-3 cleavage assay.
All error bars represent the SEM (D-G). See also Figure S1 and Table S1.
To prevent the preferential outgrowth of DICER1-expressing cells, we isolated monoclonal populations by plating low-density cultures immediately after a 24-hour treatment with tamoxifen. The resulting clones appeared at comparable frequencies in tamoxifen-treated and control cultures, and were also morphologically similar to the parental cell lines. Genotyping PCR indicated that the majority of isolated clones had deleted the second allele of Dicer1 (Fig. 1A). We also confirmed recombination of the conditional Dicer1 allele at the protein level by Western blot against DICER1 (Fig. S1B). Once a KrasG12D;Trp53-/-;Dicer1-/- clonal line was established, we did not observe outgrowth of KrasG12D;Trp53-/-;Dicer1f/- cells, even after several months of continual passage. Hereafter, we will refer to the monoclonal homozygous KrasG12D;Trp53-/-;Dicer1-/- line as Dicer1-/- cells and the parental heterozygous KrasG12D;Trp53-/-;Dicer1f/- cell line as Dicer1f/- cells. These results suggest that sarcoma cells survive after homozygous Dicer1 deletion but have a growth disadvantage relative to cells retaining Dicer1 expression. To prevent outgrowth of Dicer1f/- sarcoma cells, all subsequent experiments were carried out with monoclonal Dicer1-/- sarcoma cell lines.
To determine whether Dicer1-/- clones lacked miRNAs, we carried out massively parallel sequencing of small RNAs (small RNA-seq), ~15-50 nucleotides in length, from Dicer1f/- and Dicer1-/- sarcoma cells. Both libraries contained comparable sequencing depths at 9.3 and 9.6 million reads, respectively. However, due to miRNA loss, sequence complexity was greater in Dicer1-/- cells, which contained 830,000 unique sequences, relative to 190,000 unique sequences in Dicer1f/- cells. Of all reads mapping to the genome with 0 or 1 mismatch, 58% correspond to mature miRNAs in Dicer1f/- cells in comparison to 0.8% in Dicer1-/- cells. Approximately 48% of mature miRNAs detected in Dicer1f/- cells became undetectable in Dicer1-/- cells, while the remainder of miRNAs underwent a median decrease of 111-fold, confirming the global loss of mature miRNAs with homozygous Dicer1 loss. By quantitative Northern blot, miR-22 was present at ~4,000 copies per cell in Dicer1f/- sarcoma cells (Fig. S1C). Based on the ratio of miRNA reads in the Dicer1f/- to Dicer1-/- small RNA-seq libraries normalized to the copy number of miR-22 in Dicer1f/- sarcoma cells, miR-22 is present at fewer than 10 copies per Dicer1-/- sarcoma cell. Similarly, based on normalization to miR-22, other abundant miRNAs such as individual let-7 family members are also expressed at fewer than ten copies per cell in Dicer1-/- cells, as compared to several thousand in Dicer1f/- cells (Fig. 1B). miR-451, a DICER1-independent miRNA processed by Ago2 and expressed abundantly in red blood cells, is not detectable in Dicer1f/- sarcoma cells and present at extremely low levels (0.4 copies/cell) in Dicer1-/- sarcoma cells (Cheloufi et al., 2010; Cifuentes et al., 2010). These results indicate near-complete loss of miRNAs upon Dicer1 deletion.
In Dicer1f/- cells, the five most abundant miRNAs were miR-22 (13%), let-7f (7%), let-7a (7%), let-7c (7%), and miR-21 (6%) (Table S1). Collapsing miRNAs by seeds, the dominant heptamer seed sequence corresponds to let-7, accounting for 31.6% of all miRNA reads. Let-7 dominance has been observed in other somatic tissues, such as embryonic fibroblasts and neural precursors (Marson et al., 2008). In addition, we observed miRNAs associated with tissue-specific expression and function, such as kidney-specific miR-196a and -196b (1.2% combined) (Landgraf et al., 2007) and miR-96 (2.2%), implicated in progressive hearing loss (Lewis et al., 2009), suggesting broader regulatory roles for these short RNAs. Reads from the miR-290-295 cluster, specific to embryonic stem cells, were negligible in number, distinguishing these cells as somatic. In total, our results establish let-7 as the dominant seed in Dicer1f/- sarcoma cells and confirm the loss of mature miRNAs following Dicer1 deletion.
As a confirmation of these sequencing results, we performed Northern analysis for let-7g, miR-16, and miR-17, all detected abundantly in our Dicer1f/- sequencing library. In contrast to Dicer1f/- cells, Dicer1-/- cells showed an absence of mature miRNAs and concomitant accumulation of precursors (Fig. 1C). Luciferase reporters containing six bulged sites for let-7g or one perfect site for either miR-16 or miR-17 were also derepressed 3-6 fold, consistent with functional loss (Fig. 1D). To evaluate proliferative differences, we measured doubling times for each genotype (Fig. 1E). Dicer1-/- cells divided more slowly (~15 hours) than the Dicer1f/- controls (~12 hours), but without obvious senescence or onset of crisis. Dicer1-/- sarcoma cells exhibited a delay in G1 phase relative to Dicer1f/- sarcoma cells (Figure 1F). Additionally, the Dicer1-/- sarcoma cells exhibited elevated levels of apoptosis (Figure 1G).
Dicer1-/- sarcoma cells retain tumorigenicity in vivo
Our findings indicate that genetic ablation of Dicer1 is tolerated in mouse sarcoma cells in vitro. However, in vivo mouse models and human patient data suggest that homozygous deletion of Dicer1 is not tolerated in tumors. To test whether proliferative defects in homozygous Dicer1-deleted tumors, and subsequent loss through competition in vivo by DICER1-expressing cells, accounts for these differences, we carried out tumor formation assays. Upon subcutaneous injection of 1 × 106 cells into the flanks of immune-compromised mice, Dicer1-/- cells were indeed tumorigenic, forming tumors at 7/18 sites within 24 days, as compared to 4/8 sites by day 14 for the original Dicer1f/- strain. To better evaluate the difference in tumor formation kinetics, we repeated this injection experiment with 2.5 × 104 cells. At this lower cell number, Dicer1-/- sarcoma cells began to develop tumors in ~45 days, as compared to 22 days for the parental Dicer1f/- sarcoma cell line (Fig. 2A). Pathologic analysis of either Dicer1-/- or Dicer1f/- tumors identified both as high grade sarcomas with pleomorphic nuclei and abnormal mitoses, consistent with previous reports of KRAS-driven sarcoma models (Fig. 2B) (Kirsch et al., 2007; Kumar et al., 2007). Sample genotypes could not be readily distinguished in a blinded analysis.
Figure 2.
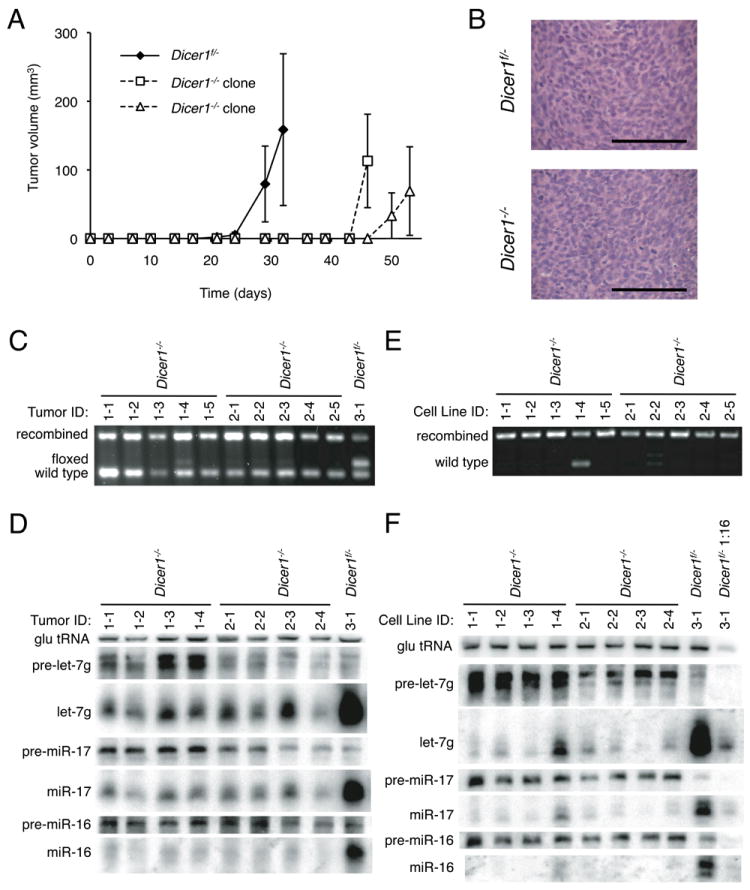
Tumorigenesis of KrasG12D;Trp53-/-;Dicer1-/- sarcoma cells in transplant assays. A) Injection of 2.5 × 104 Dicer1 f/- and Dicer1-/- sarcoma cells into the flanks of nude mice. Error bars represent the SEM. B) Hematoxylin and eosin section of Dicer1f/- and Dicer1-/- tumors. In C-F, each lane represents an independent tumor derived from one injection of the indicated Dicer1-/- sarcoma cell line. Scale bar, 100 μm. C) PCR genotyping of Dicer1-/- tumors. Recombined and floxed bands are derived from the injected tumor cells, while wild type bands derive from host tissue. D) Northern analysis of tumor tissue derived from sarcoma injections. E) PCR and F) Northern analysis following one round of in vitro passage of secondary tumors. In C-F, each sample ID contains a prefix identifying the injected sarcoma cell clone followed by a suffix identifying the tumor replicate (e.g. sample 1-3 corresponds to clone 1 and tumor replicate 3). See also Figure S2.
We also performed syngeneic injections into immunocompetent C57Bl6/SV129 F1 mice. As before, the rate was slower than the parental Dicer1f/- line, with the first tumors appearing 7 days after injection of the Dicer1f/- cells and 21 days after injection of the Dicer1-/- cells (Fig. S2). Thus, the absence of DICER1 impairs but does not preclude tumor formation, even in an immunocompetent background.
Although the sarcoma cells at the time of the injection were Dicer1-/-, it is possible that in vivo selection resulted in outgrowth of contaminating Dicer1f/- cells. Therefore, we genotyped DNA prepared from primary tumor tissue and confirmed a significant recombined band corresponding to the injected Dicer1-/- cells with an accompanying background wild-type band contributed by contaminating host tissue (Fig 2C). Northern analysis of primary tumors revealed accumulation of precursors as well as significant but incomplete depletion of mature miRNAs (Fig 2D).
To test if the residual miRNAs were a result of contaminating wild-type tissue, we generated cell lines from these tumors. PCR genotyping confirmed a depletion of the wild type tissue during this process (Fig. 2E). By Northern blotting, the level of mature miRNAs was lower than the detection limit of the blot, and this was again accompanied by enrichment in the pre-miRNA (Fig. 2F). The residual mature miRNA observed is likely due to host tissue contamination, as evidenced by the greatest miRNA signal in the tumor sample showing the greatest wild-type contaminant band by PCR (Fig 2E, 2F). Thus, injected Dicer1-/- cells survived and proliferated in vivo without recovery of miRNA processing. The earlier in vitro results extend to an in vivo setting, with sarcoma cells retaining the capacity to form phenotypically similar tumors, albeit more slowly, in the absence of DICER1 and miRNAs.
Mesenchymal stem cells were generated as an alternative model of somatic Dicer1 deletion
The viability of Dicer1-null sarcoma cells, which lack TRP53 and express oncogenic KRAS, may be a function of the strong oncogenic background required for rapid in vivo growth or may require additional genetic alterations that occur during tumor formation. Therefore, we tested whether Dicer1 loss could be tolerated in a defined immortalized cell model. Since sarcomas are thought to be mesenchymal in origin (Clark et al., 2005), we turned to mesenchymal stem cells (MSCs), a multipotent population of cells that can differentiate into osteoblasts, chondrocytes, adipocytes, or myocytes (Pittenger et al., 1999).
From a one year-old adult Dicer1f/f mouse, we prepared a primary culture of MSCs that was then immortalized with a retroviral vector encoding SV40 large T-antigen (Fig. 3A). Individual clones were isolated and analyzed by flow cytometry to confirm the expression of CD49e and CD106 (Fig 3B, left panels), surface markers associated with MSCs (Pittenger, 2008), and the absence of CD31, specific to endothelial cells, and CD45, a marker of hematopoietic stem cells (data not shown).
Figure 3.
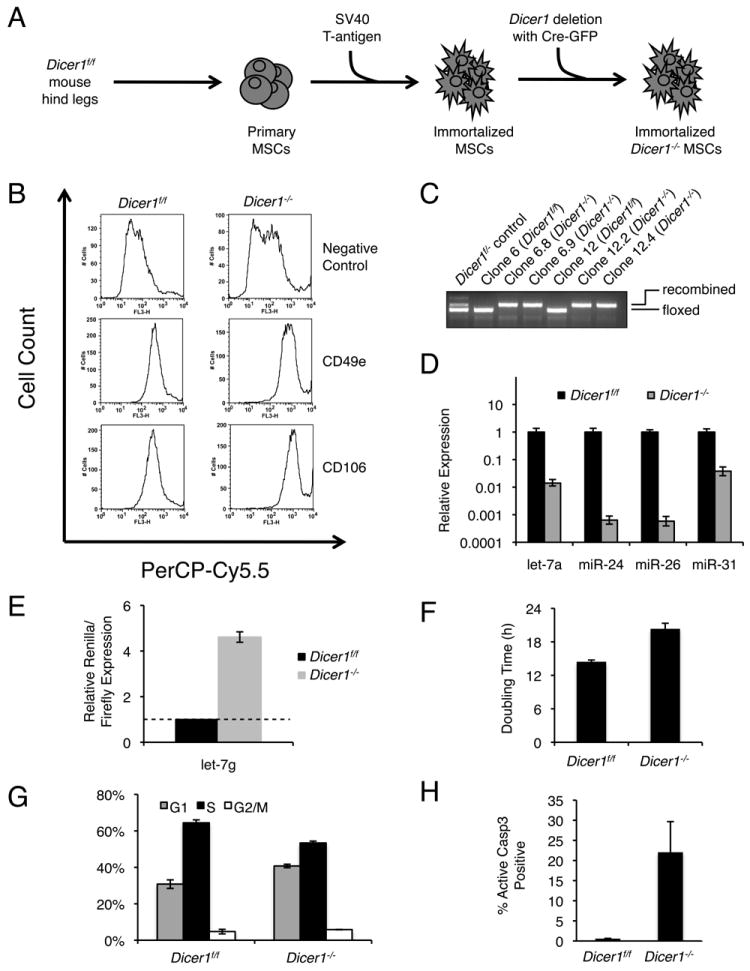
Derivation and characterization of Dicer1-/- mesenchymal stem cells. A) Schematic of MSC preparation. Primary MSC cultures were prepared from the tibia, femur, and pelvic bones of a one-year old Dicer1f/f mouse. The primary cells were then infected with retrovirus encoding SV40 large T-antigen. Monoclonal cultures were then isolated, infected with Adeno-Cre-GFP, sorted by FACS for GFP-positive cells, and plated at low density to isolate Dicer1-recombined clones. B) Cell surface marker expression in Dicer1f/f (left) and Dicer1-/- (right) MSCs. Cells were analyzed by flow cytometry with antibodies against CD49e and CD106. C) PCR genotyping of clonally isolated Dicer1f/f or Dicer1-/- MSCs. Clones 6.8 and 6.9 (lanes 3, 4) were derived from parental clone 6 (lane 2), and clones 12.2 and 12.4 (lanes 7, 8) were derived from parental clone 12 (lane 5). PCR genotyping of a Dicer1f/- sarcoma cell line was used as a heterozygous control (lane 1). D) Expression of miRNAs in Dicer1f/f and Dicer1-/- MSCs. Total RNA was analyzed with a Qiagen miScript qPCR assay for let-7a, mir-24, -26, and -31). A representative qPCR experiment is shown. Error bars indicate standard deviation. E) Luciferase reporter assay for let-7g. The reporter contains 6 bulged sites. Targeted renilla luciferase reporters were normalized to nontargeted firefly luciferase reporters. Renilla/firefly luciferase expression was normalized to expression in the Dicer1f/f MSC line. F) Proliferation assay. G) Cell cycle distribution determined by BrdU labeling. H) Apoptosis determined by caspase-3 cleavage assay.
For (E-H), error bars represent the SEM. See also Figure S3.
To delete Dicer1, we carried out Adeno-Cre-GFP infection and sorted the infected cells by GFP. This protocol enriched for Dicer1-/- cells, as seen by the predominance of the deletion-specific PCR product 6 days after sorting (Fig. S3A). This signal was accompanied by loss of DICER1 protein (Fig. S3B), as well as a decrease in mature miRNA levels by qPCR (Fig. S3C) and Northern blot at day 7 (Fig. S3D). However, as observed for the sarcoma cells, additional passage led to a decrease in the deletion-specific PCR band (Fig. S3A), suggesting outgrowth of DICER1-expressing cells.
To prevent competition by cells retaining Dicer1, we repeated the strategy used for the sarcoma cell lines and isolated clones. MSCs were infected with Adeno-Cre-GFP, sorted for GFP two days later, then plated at low-density after an additional ten days. Clones were then isolated, expanded, and PCR genotyped to confirm recombination (Fig. 3C). Of these clones, a majority (55%) had undergone homozygous deletion of Dicer1, indicating that immortalized MSCs can readily tolerate loss of Dicer1. Dicer1-/- clones were stably proliferative and remained Dicer1-null after multiple passages (>3 months) as determined by PCR genotyping (data not shown). All subsequent experiments were carried out with a monoclonal Dicer1-/- MSC line. Following loss of Dicer1, we observed a ~100-fold reduction of miRNA expression by qPCR analysis of abundant miRNAs (let-7a, miR-24, -26, and -31) (Fig. 3D). We also observed ~5-fold derepression of a let-7g luciferase reporter in Dicer1-/- cells (Fig. 3E). Dicer1-/- MSCs exhibited a proliferative lag, with a doubling time of ~20 hours relative to ~14 hours for Dicer1f/f cells (Fig. 3F). Dicer1-/- MSCs also exhibited a G1 delay in cell cycle (Figure 3G) and elevated levels of basal apoptosis (Figure 3H). Notably, Dicer1-/- MSCs remained positive for CD49e and CD106 (Fig. 3B, right panels) and negative for CD31 and CD45 (data not shown), suggesting a retention of cell identity in the absence of miRNAs.
Discussion
We have carried out homozygous Dicer1 deletion in a Dicer1-conditional KRAS-activated, Trp53-null sarcoma cell line and observed resultant loss of miRNA expression by small RNA sequencing. Relative to Dicer1f/- sarcoma cells, these miRNA-depleted cells proliferate more slowly, exhibit a cell cycle delay in G1 phase, and have a higher level of basal apoptosis. Additionally, we have generated a second in vitro model of homozygous Dicer1-deletion in murine mesenchymal stem cells, related in cell type to sarcomas, established from an adult Dicer1f/f mouse and immortalized in vitro. Similar to the sarcoma model, MSCs that had undergone homozygous Dicer1 deletion were readily isolatable but exhibited a reduction in proliferation, a delay in G1, and an increase in basal apoptosis.
Strikingly, Dicer1-/- sarcoma cells retain the ability, upon transplant, to form tumors in both immunocompromised and immunocompetent recipient mice, albeit at slower rates relative to Dicer1f/- sarcoma cells. We present three compelling lines of evidence excluding the possibility that these tumors arose from contaminating Dicer1f/- cells:
Clonal Dicer1-/- sarcoma cells were used for the tumor injection studies to preclude the possibility of Dicer1f/- outgrowth.
Tumors derived from Dicer1-/- injections exhibit strong PCR genotyping bands for (i) recombined Dicer1 and (ii) wild-type Dicer1, derived from host wild-type tissue associated with the tumors. In contrast, the control tumor derived from Dicer1f/- cells exhibits a third PCR band, representing unrecombined floxed Dicer1. This stark difference in genotype demonstrates that the tumors that arose from Dicer1-/- injections are composed predominantly of Dicer1-/- cells.
In vitro passage of cells derived from Dicer1-/- tumors resulted in depletion of wild-type primary host tissue and outgrowth of Dicer1-/- cells, as determined by PCR genotyping, and concomitant depletion in miRNA signal by Northern blot. Furthermore, in most tumor samples, miRNAs were present at levels less than 1/16th of the Dicer1f/- control, likely an overestimate given the low level of contaminating host-derived cells.
Our results stand in stark contrast to many published reports. The failure to observe Dicer1-null tumors from Dicer1-conditional in vivo mouse models of cancer have been interpreted to suggest that DICER1 and miRNAs are essential for tumor formation and, furthermore, may be required for tumor cell survival. Contrary to these observations, our results indicate that miRNAs are not essential for in vitro survival or proliferation. In vivo, Dicer1-null sarcoma cells, though depleted of miRNAs, are competent for tumor development. Furthermore, our observation of tumor growth in an immunocompetent background demonstrates that miRNAs are not essential for escape from immune surveillance. In addition, the histologic resemblance of Dicer1-/- sarcomas to Dicer1f/- sarcomas, as well as the retention of cell type-specific cell surface markers in Dicer1-/- MSCs, suggest that cellular identity is largely retained despite loss of miRNAs. Given the high frequency with which Dicer1-null cells can be isolated in both the sarcoma and MSC models, secondary mutational or other low frequency events beyond the initial immortalization are likely not necessary to tolerate DICER1 loss.
Based on the outgrowth of DICER1-expressing cells in vitro relative to Dicer1-/- cells, and the proliferative delay observed in monoclonal Dicer1-/- cells, we conclude that the absence of Dicer1-null cells in previously characterized mouse models of cancer is due in part to the preferential outgrowth, in vivo, of cells expressing DICER1. Notably, tumor genotyping analyses in published studies have typically been carried out in whole tumor samples and, as such, do not exclude the possibility that Dicer1-null cells comprise a subpopulation of the samples.
Furthermore, factors additional to proliferative capacity may contribute to the preferential outgrowth of DICER1-expressing cells in vivo. The viability of Dicer1-null transformed cells in our study indicates that total miRNA loss itself, and resultant genetic misregulation, is not intrinsically catastrophic, but rather triggers secondary signals that initiate changes in proliferation or cell death. Numerous studies have reported that miRNAs mediate stress responses (Hermeking, 2007; Leung and Sharp, 2010; Leung et al., 2011a) and loss of DICER1 in embryonic stem cells results in increased sensitivity to stress (Zheng et al., 2011). Similarly, we have observed that sarcoma cells and MSCs that lack DICER1 exhibit increased apoptosis. Given the intrinsically stressful nature of the in vivo tumor environment, the retention of a miRNA-mediated stress response may provide an additional growth advantage to tumor cells that retain at least one copy of Dicer1.
Notably, inactivation of TRP53 activity is a common feature in both the sarcoma and MSC models presented here and may facilitate or be required for viability in the absence of DICER1. This possibility is consistent with the observation that TRP53 loss allows primary MEFs to bypass an immediate senescence phenotype induced by DICER1 loss (Mudhasani et al., 2008). However, TRP53 loss alone only prolongs proliferative capacity of Dicer1-/- MEFs for a few additional passages (personal communication with S.N. Jones), suggesting that other events, such as activation of oncogenes or inactivation of additional tumor suppressor genes, are required.
In addition to expanding our understanding of DICER1 in tumorigenesis, the cell lines reported here may complement existing models such as the hypomorphic DICER1 HCT116 line, a human colorectal cancer line containing homozygous deletion of exon 5 in the DICER1 gene (Cummins et al., 2006). The availability of Dicer1-/- cancer cells will permit further characterization of somatic miRNA families through miRNA transfection and “add-back” cell culture experiments. Finally, the observations that DICER1 loss leads to a negative selective pressure in vitro and in vivo suggests that DICER1 activity could be a therapeutic target.
Experimental Procedures
Animal Work
All animal studies were performed with protocols approved by the NIH and the Massachusetts Institute of Technology Committee for Animal Care, and were consistent with the Guide for Care and Use of Laboratory Animals, National Research Council, 1996 (Institutional Animal Welfare Assurance No. A-3125-01).
Cell culture
Sarcoma cells were generated from hindlimb injection of Adeno-Cre into KrasLSL-G12D;Trp53f/f;Dicer1f/f mice as described previously (Kirsch et al., 2007). Sarcoma cells infected with MSCV.CreERT2.puro were treated with 250 nM 4-hydroxy tamoxifen for 24 hours to recombine the floxed Dicer1 allele and plated at clonal density. Primary MSC cultures were prepared from an adult Dicer1f/f mouse using a previously described protocol (Mukherjee et al., 2008), immortalized with SV40 Large T-antigen, and infected with Adeno-Cre-GFP to recombine the floxed alleles. Cells were genotyped as described previously (Calabrese et al., 2007).
Histology
Tumor-bearing animals were sacrificed with CO2 asphyxiation. Tumors were isolated, fixed in 4% paraformaldehyde, transferred to 70% ethanol, and embedded in paraffin. Tumors were then sectioned and stained with hematoxylin and eosin.
Tumor Injection
2.5 × 104, 105, or 106 Kras G12D;Trp53-/- sarcoma cells of Dicer1f/- or Dicer1-/- genotype were suspended in PBS and subcutaneously injected into the flanks of Rag2-/- or C57Bl6/SV129 F1 mice. Tumors were measured over time by calipers and volumes were assessed as described previously (Sage et al., 2000). DNA, RNA, and histological sections were prepared from tumors. Some tumors were trypsinized and replated for the development of secondary cell lines as described above.
Small RNA Northerns and Cloning
Small RNA Northern blots were performed using 20 μg total RNA on a 15% denaturing polyacrylamide gel. Following semi-dry transfer to a Hybond-N+ membrane, a DNA oligo probe for glutamine tRNA or LNA probes for let-7, mir-16, and mir-17 were used for visualization. Small RNA sequencing from sarcoma lines was carried out as described previously (Leung et al., 2011b). The sequencing data are available under GEO accession GSE34825.
Supplementary Material
Significance.
The nearly global decrease in miRNAs observed across a range of human tumors suggests that restoration of miRNA levels may have a valuable therapeutic role. Here we report that some tumor cells devoid of miRNA activity are viable and form tumors under certain conditions. However, the failure to detect human tumors with complete loss suggests that the opposite approach, namely a further decrease in these levels, may surprisingly also be beneficial. We explore this alternative in a well-characterized sarcoma model, demonstrating that miRNA depletion can indeed inhibit tumor growth rates through reduced proliferation and increased cell death. These findings suggest that the targeted inhibition of miRNA pathway elements, particularly DICER1, may be a potential therapy for the treatment of cancer.
Highlights.
Sarcoma cells remain stably proliferative in the absence of DICER1
Dicer1-null sarcoma cells form tumors in vivo, but with decreased kinetics
Immortalized mesenchymal stem cells can similarly tolerate DICER1 loss
MiRNA levels in tumors may result from a complex stabilizing selection
Acknowledgments
We thank members of the Sharp, Jacks, and Lees Labs for many helpful discussions. We are also specifically grateful to Margaret Ebert, Amy Seila, and Eliezer Calo for their generous contribution of reagents. We thank Allison Landman for providing assistance with mesenchymal stem cell isolation and cultures. We thank Roderick Bronson for analysis of histology slides. Massively parallel Illumina sequencing was carried out by the Biopolymers & Proteomics Core Facility in the Swanson Biotechnology Center at the David H. Koch Institute for Integrative Cancer Research at M.I.T. Bioinformatic analysis was carried out in the Bioinformatics & Computing division of the Swanson Biotechnology Center. This work was supported by a Fannie and John Hertz Foundation Fellowship (A.R.), a Leukemia and Lymphoma Society grant 5198-09 (A.M.G.), an NIH grant RO1-CA133404 (P.A.S.), an NCI grant PO1-CA42063 (P.A.S., T.J., J.A.L.), and partially by the NCI Cancer Center Support (core) grant P30-CA14051.
Footnotes
Author Contributions
A.R., A.M.G., and M.S.K. designed and performed the experiments. A.B. performed the informatics for the sequencing data. C.C. and V.L. performed experiments. A.R., A.M.G., and P.A.S. wrote the paper. J.A.L., T.J., and P.A.S. provided supervision and assisted with manuscript preparation. All authors reviewed and approved the manuscript.
Competing Financial Interests
The authors declare no competing financial interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Arrate MP, Vincent T, Odvody J, Kar R, Jones SN, Eischen CM. MicroRNA biogenesis is required for Myc-induced B-cell lymphoma development and survival. Cancer Res. 2010;70:6083–6092. doi: 10.1158/0008-5472.CAN-09-4736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136:215–233. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabrese JM, Seila AC, Yeo GW, Sharp PA. RNA sequence analysis defines Dicer’s role in mouse embryonic stem cells. Proc Natl Acad Sci U S A. 2007;104:18097–18102. doi: 10.1073/pnas.0709193104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheloufi S, Dos Santos CO, Chong MM, Hannon GJ. A dicer-independent miRNA biogenesis pathway that requires Ago catalysis. Nature. 2010;465:584–589. doi: 10.1038/nature09092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chivukula RR, Mendell JT. Circular reasoning: microRNAs and cell-cycle control. Trends Biochem Sci. 2008;33:474–481. doi: 10.1016/j.tibs.2008.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cifuentes D, Xue H, Taylor DW, Patnode H, Mishima Y, Cheloufi S, Ma E, Mane S, Hannon GJ, Lawson ND, et al. A novel miRNA processing pathway independent of Dicer requires Argonaute2 catalytic activity. Science. 2010;328:1694–1698. doi: 10.1126/science.1190809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark MA, Fisher C, Judson I, Thomas JM. Soft-tissue sarcomas in adults. N Engl J Med. 2005;353:701–711. doi: 10.1056/NEJMra041866. [DOI] [PubMed] [Google Scholar]
- Cummins JM, He Y, Leary RJ, Pagliarini R, Diaz LA, Jr, Sjoblom T, Barad O, Bentwich Z, Szafranska AE, Labourier E, et al. The colorectal microRNAome. Proc Natl Acad Sci U S A. 2006;103:3687–3692. doi: 10.1073/pnas.0511155103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman RC, Farh KK, Burge CB, Bartel DP. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009;19:92–105. doi: 10.1101/gr.082701.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaur A, Jewell DA, Liang Y, Ridzon D, Moore JH, Chen C, Ambros VR, Israel MA. Characterization of microRNA expression levels and their biological correlates in human cancer cell lines. Cancer Res. 2007;67:2456–2468. doi: 10.1158/0008-5472.CAN-06-2698. [DOI] [PubMed] [Google Scholar]
- Heravi-Moussavi A, Anglesio MS, Cheng SW, Senz J, Yang W, Prentice L, Fejes AP, Chow C, Tone A, Kalloger SE, et al. Recurrent Somatic DICER1 Mutations in Nonepithelial Ovarian Cancers. N Engl J Med. 2011 doi: 10.1056/NEJMoa1102903. [DOI] [PubMed] [Google Scholar]
- Hermeking H. p53 enters the microRNA world. Cancer Cell. 2007;12:414–418. doi: 10.1016/j.ccr.2007.10.028. [DOI] [PubMed] [Google Scholar]
- Herranz H, Cohen SM. MicroRNAs and gene regulatory networks: managing the impact of noise in biological systems. Genes Dev. 2010;24:1339–1344. doi: 10.1101/gad.1937010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill DA, Ivanovich J, Priest JR, Gurnett CA, Dehner LP, Desruisseau D, Jarzembowski JA, Wikenheiser-Brokamp KA, Suarez BK, Whelan AJ, et al. DICER1 mutations in familial pleuropulmonary blastoma. Science. 2009;325:965. doi: 10.1126/science.1174334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirsch DG, Dinulescu DM, Miller JB, Grimm J, Santiago PM, Young NP, Nielsen GP, Quade BJ, Chaber CJ, Schultz CP, et al. A spatially and temporally restricted mouse model of soft tissue sarcoma. Nat Med. 2007;13:992–997. doi: 10.1038/nm1602. [DOI] [PubMed] [Google Scholar]
- Kumar MS, Lu J, Mercer KL, Golub TR, Jacks T. Impaired microRNA processing enhances cellular transformation and tumorigenesis. Nat Genet. 2007;39:673–677. doi: 10.1038/ng2003. [DOI] [PubMed] [Google Scholar]
- Kumar MS, Pester RE, Chen CY, Lane K, Chin C, Lu J, Kirsch DG, Golub TR, Jacks T. Dicer1 functions as a haploinsufficient tumor suppressor. Genes Dev. 2009;23:2700–2704. doi: 10.1101/gad.1848209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambertz I, Nittner D, Mestdagh P, Denecker G, Vandesompele J, Dyer MA, Marine JC. Monoallelic but not biallelic loss of Dicer1 promotes tumorigenesis in vivo. Cell Death Differ. 2010;17:633–641. doi: 10.1038/cdd.2009.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landgraf P, Rusu M, Sheridan R, Sewer A, Iovino N, Aravin A, Pfeffer S, Rice A, Kamphorst AO, Landthaler M, et al. A mammalian microRNA expression atlas based on small RNA library sequencing. Cell. 2007;129:1401–1414. doi: 10.1016/j.cell.2007.04.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung AK, Sharp PA. MicroRNA functions in stress responses. Mol Cell. 2010;40:205–215. doi: 10.1016/j.molcel.2010.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung AK, Vyas S, Rood JE, Bhutkar A, Sharp PA, Chang P. Poly(ADP-ribose) regulates stress responses and microRNA activity in the cytoplasm. Mol Cell. 2011a;42:489–499. doi: 10.1016/j.molcel.2011.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung AK, Young AG, Bhutkar A, Zheng GX, Bosson AD, Nielsen CB, Sharp PA. Genome-wide identification of Ago2 binding sites from mouse embryonic stem cells with and without mature microRNAs. Nat Struct Mol Biol. 2011b;18:237–244. doi: 10.1038/nsmb.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis MA, Quint E, Glazier AM, Fuchs H, De Angelis MH, Langford C, van Dongen S, Abreu-Goodger C, Piipari M, Redshaw N, et al. An ENU-induced mutation of miR-96 associated with progressive hearing loss in mice. Nat Genet. 2009;41:614–618. doi: 10.1038/ng.369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin RJ, Lin YC, Chen J, Kuo HH, Chen YY, Diccianni MB, London WB, Chang CH, Yu AL. microRNA signature and expression of Dicer and Drosha can predict prognosis and delineate risk groups in neuroblastoma. Cancer Res. 2010;70:7841–7850. doi: 10.1158/0008-5472.CAN-10-0970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, Getz G, Miska EA, Alvarez-Saavedra E, Lamb J, Peck D, Sweet-Cordero A, Ebert BL, Mak RH, Ferrando AA, et al. MicroRNA expression profiles classify human cancers. Nature. 2005;435:834–838. doi: 10.1038/nature03702. [DOI] [PubMed] [Google Scholar]
- Marson A, Levine SS, Cole MF, Frampton GM, Brambrink T, Johnstone S, Guenther MG, Johnston WK, Wernig M, Newman J, et al. Connecting microRNA genes to the core transcriptional regulatory circuitry of embryonic stem cells. Cell. 2008;134:521–533. doi: 10.1016/j.cell.2008.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martello G, Rosato A, Ferrari F, Manfrin A, Cordenonsi M, Dupont S, Enzo E, Guzzardo V, Rondina M, Spruce T, et al. A MicroRNA targeting dicer for metastasis control. Cell. 2010;141:1195–1207. doi: 10.1016/j.cell.2010.05.017. [DOI] [PubMed] [Google Scholar]
- Melo SA, Ropero S, Moutinho C, Aaltonen LA, Yamamoto H, Calin GA, Rossi S, Fernandez AF, Carneiro F, Oliveira C, et al. A TARBP2 mutation in human cancer impairs microRNA processing and DICER1 function. Nat Genet. 2009;41:365–370. doi: 10.1038/ng.317. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Mudhasani R, Zhu Z, Hutvagner G, Eischen CM, Lyle S, Hall LL, Lawrence JB, Imbalzano AN, Jones SN. Loss of miRNA biogenesis induces p19Arf-p53 signaling and senescence in primary cells. J Cell Biol. 2008;181:1055–1063. doi: 10.1083/jcb.200802105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee S, Raje N, Schoonmaker JA, Liu JC, Hideshima T, Wein MN, Jones DC, Vallet S, Bouxsein ML, Pozzi S, et al. Pharmacologic targeting of a stem/progenitor population in vivo is associated with enhanced bone regeneration in mice. J Clin Invest. 2008;118:491–504. doi: 10.1172/JCI33102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pittenger MF. Mesenchymal stem cells from adult bone marrow. Methods Mol Biol. 2008;449:27–44. doi: 10.1007/978-1-60327-169-1_2. [DOI] [PubMed] [Google Scholar]
- Pittenger MF, Mackay AM, Beck SC, Jaiswal RK, Douglas R, Mosca JD, Moorman MA, Simonetti DW, Craig S, Marshak DR. Multilineage potential of adult human mesenchymal stem cells. Science. 1999;284:143–147. doi: 10.1126/science.284.5411.143. [DOI] [PubMed] [Google Scholar]
- Sage J, Mulligan GJ, Attardi LD, Miller A, Chen S, Williams B, Theodorou E, Jacks T. Targeted disruption of the three Rb-related genes leads to loss of G(1) control and immortalization. Genes Dev. 2000;14:3037–3050. doi: 10.1101/gad.843200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekine S, Ogawa R, Ito R, Hiraoka N, McManus MT, Kanai Y, Hebrok M. Disruption of Dicer1 induces dysregulated fetal gene expression and promotes hepatocarcinogenesis. Gastroenterology. 2009;136:2304–2315. e2301–2304. doi: 10.1053/j.gastro.2009.02.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefani G, Slack FJ. Small non-coding RNAs in animal development. Nat Rev Mol Cell Biol. 2008;9:219–230. doi: 10.1038/nrm2347. [DOI] [PubMed] [Google Scholar]
- Torres A, Torres K, Paszkowski T, Jodlowska-Jedrych B, Radomanski T, Ksiazek A, Maciejewski R. Major regulators of microRNAs biogenesis Dicer and Drosha are down-regulated in endometrial cancer. Tumour Biol. 2011;32:769–776. doi: 10.1007/s13277-011-0179-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Baskerville S, Shenoy A, Babiarz JE, Baehner L, Blelloch R. Embryonic stem cell-specific microRNAs regulate the G1-S transition and promote rapid proliferation. Nat Genet. 2008;40:1478–1483. doi: 10.1038/ng.250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng GX, Ravi A, Calabrese JM, Medeiros LA, Kirak O, Dennis LM, Jaenisch R, Burge CB, Sharp PA. A latent pro-survival function for the mir-290-295 cluster in mouse embryonic stem cells. PLoS Genet. 2011;7:e1002054. doi: 10.1371/journal.pgen.1002054. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.