

Abstract
Although indolone-N-oxide (INODs) genereting long-lived radicals possess antiplasmodial activity in the low-nanomolar range, little is known about their mechanism of action. To explore the molecular basis of INOD activity, we screened for changes in INOD-treated malaria-infected erythrocytes (Pf-RBCs) using a proteomics approach. At early parasite maturation stages, treatment with INODs at their IC50 concentrations induced a marked tyrosine phosphorylation of the erythrocyte membrane protein band 3, whereas no effect was observed in control RBCs. After INOD treatment of Pf-RBCs we also observed: (i) accelerated formation of membrane aggregates containing hyperphosphorylated band 3, Syk kinase, and denatured hemoglobin; (ii) dose-dependent release of microvesicles containing the membrane aggregates; (iii) reduction in band 3 phosphorylation, Pf-RBC vesiculation, and antimalarial effect of INODs upon addition of Syk kinase inhibitors; and (iv) correlation between the IC50 and the INOD concentrations required to induce band 3 phosphorylation and vesiculation. Together with previous data demonstrating that tyrosine phosphorylation of oxidized band 3 promotes its dissociation from the cytoskeleton, these results suggest that INODs cause a profound destabilization of the Pf-RBC membrane through a mechanism apparently triggered by the activation of a redox signaling pathway rather than direct oxidative damage.
Keywords: Red blood cells, Tyrosine phosphorylation, Plasmodium falciparum, Band 3, Oxidative damage, Free radicals
Because of the emergence of drug-resistant strains of Plasmodium falciparum, the incidence of malaria is increasing in many parts of the world. Although combinatorial therapies involving artemisinin offer a first line of treatment for uncomplicated P. falciparum infections, localized episodes of partial resistance are now emerging [1], emphasizing the need for new antimalarial drugs.
We recently developed a series of new antiplasmodial compounds generating stable radical species, 3-oxo-3H-indole-1-oxides (INODs)2, that display antiplasmodial activities in both murine models of P. berghei infection and human cell culture models of P. falciparum parasitemia. Because INODs have been shown to act at nanomolar concentrations and to cause little toxicity [2], they have been proposed as promising candidates for future clinical trials.
Unfortunately, little is known regarding the mechanism of action of INODs. Bunney and Hooper [54] have noted that INODs exhibit redox potentials and generate long-lived free radicals comparable to those of 1,4-quinones, suggesting that their biological activities may be related to their abilities to create an oxidizing environment within the parasitized cell. Indeed, the pharmacophore (i.e., the conjugated system between the nitrone and the ketone functions that is essential for antimalarial activity) contains all of the oxidative properties of this family of antimalarial drugs [2]. The fact that mutant erythrocytes that display a natural resistance to P. falciparum (e.g., cells with glucose-6-phosphate dehydrogenase deficiency, sickle cell anemia, or β-thalassemia) share a common predisposition to oxidative stress adds strength to the conjecture that oxidative overload may contribute to INODs’ antimalarial activity [3–6]. Not surprisingly, artemisinin, the most common antimalarial drug in use today, is also distinguished by its redox activities and ability to induce an oxidative stress in its target cell [2]. For unknown reasons the parasite itself triggers significant oxidative stress during the infection process.
Intraerythrocyte P. falciparum cell cycle is characterized by a 48-h development. Merozoites invade circulating erythrocytes and, within 12–24 h, the cytoplasm expands (ring forms) and further matures to the trophozoite stage. At late stages of maturation the parasite undergoes cellular segmentation and differentiation to form roughly 16–18 merozoite cells. At the end of the cycle, the erythrocyte membrane is rapidly destroyed and merozoites burst from the red blood cell to infect other erythrocytes. Soon after parasite invasion, a rapid decrease in intracellular reduced glutathione and concomitant rise in oxidative damage to the host cell are observed [3–5,7–12]. The prominent formation of denatured hemoglobin products, their binding to the membrane, the oxidation and clustering of band 3 [8,10], and the peroxidation of membrane lipids [13] provide further evidence for a parasite-induced oxidative milieu. The membrane damage appears magnified in mutant erythrocytes [3–6]; therefore an inadequate adaptive response of the host cells to the oxidative stress exerted by the intracellular parasite seems to play a central role in the mechanism of protection conferred by various mutations. In this already stressed condition, it can be hypothesized that any additional oxidative stress imposed by INOD drugs could force an oxidative overload that is simply too intense for the host–parasite system to survive.
We and others have noted that oxidative environments of the sort described above can stimulate tyrosine phosphorylation of band 3, i.e., the major integral protein of the erythrocyte membrane. This increase in band 3 tyrosine phosphorylation, which derives from both an oxidative activation of Syk kinase [14–16] and an oxidative inhibition of a major tyrosine phosphatase [17], leads to significant changes in membrane properties, altering both rates of glucose metabolism and multiple membrane protein interactions [18–20]. We recently demonstrated that band 3 tyrosine phosphorylation may have a role in the regulation of the structural stability of the red cell membrane causing the transition of band 3 molecules from a state of restricted lateral mobility to a state characterized by large mobility through the plane of the membrane. In accordance with this result we observed that phosphorylated band 3 greatly decreases its affinity to ankyrin, inducing membrane blebbing and vesiculation [21]. The fact that similar phosphorylation of band 3 and membrane destabilization are observed in G6PD-deficient erythrocytes that exhibit resistance to parasite habitation [22] raises the question whether redox activation of band 3 phosphorylation and its biological sequelae might play a role in INOD suppression of parasitemia.
In this paper, we conduct a comprehensive analysis of the host–parasite system’s oxidative and phosphorylation changes that occur during P. falciparum red blood cell (Pf-RBC) invasion in the presence and absence of INODs; INOD-1 was one of the most effective compounds of the INOD series in inducing band 3 hyperphosphorylation. Using a proteomics approach, we demonstrate that band 3 tyrosine phosphorylation constitutes the most prominent change in Pf-RBC after INOD exposure at nanomolar concentrations. We then offer a mechanism whereby this activity can provide a plausible pathway for INOD-mediated suppression of parasitemia.
Experimental procedures
Unless otherwise stated, all materials were obtained from Sigma–Aldrich (St. Louis, MO, USA).
Cultivation and preparation of P. falciparum-RBC membranes
Freshly drawn blood (Rh+) from healthy adults of both sexes was used after informed consent in all studies. Blood anticoagulated with heparin was stored in citrate–phosphate–dextrose with adenine before its use. RBCs were separated from plasma and leukocytes by three washes in wash medium (RPMI 1640 medium containing 2 mM glutamine, 24 mM NaHCO3, 25 mM Hepes, 20 mM glucose, and 32 mg/ml gentamicin, pH 6.80). P. falciparum strain Palo Alto (mycoplasma free) was cultured at a hematocrit of 0.5% and synchronized as previously described [23]. P. falciparum-infected cells were studied at defined parasite maturation stages, i.e., 14 to 18 h (ring stage) or 34 to 38 h (trophozoite stage). To assess total parasitemia and the relative numbers of ring- and trophozoite-stage cells, slides were prepared from cultures at the indicated times and stained with Diff-Quik stain before analysis of 400 to 1000 cells by microscopy. The parasitemia of cell populations used in this study was routinely 25–40%. Standard hypotonic membranes were prepared at 4 °C on ice as follows: 150 μl of packed RBCs was diluted into 1.5 ml of cold hemolysis buffer (5 mmol/L sodium phosphate, 1 mmol/L EDTA, pH 8.0) containing a protease and phosphatase inhibitor cocktail (Sigma–Aldrich) and then washed up to four more times in the same buffer in a refrigerated Eppendorf microfuge at 25,000 g. The preparations were stored frozen at −80 °C until use. Membrane protein content was quantified using the DC Protein Assay (Bio-Rad, Hercules, CA, USA).
Treatment of RBCs
RBCs and Pf-RBCs were washed 3× in phosphate-buffered saline (137 mM NaCl, 2.7 mM KCl, 8.1 mM K2HPO4, 1.5 mM KH2PO4, pH 7.4) in the presence of 5 mM glucose (PBS-glucose) to obtain packed RBCs. RBCs were then resuspended at 30% hematocrit in PBS-glucose and incubated at 37 °C in the presence and absence of various concentrations of INOD-1 and for various incubation times. When necessary for kinase inhibitor studies, infected and uninfected RBCs were treated with 10 μM Syk inhibitors II and IV (Calbiochem) for 1 h at 37 °C in the dark.
Assay for denatured hemoglobin products
Denatured hemoglobin products were quantified by measuring heme absorbance at 560, 577, and 630 nm [55] and expressed as nmol/ml of solubilized membranes.
Assay for GSH
GSH estimations were performed using 5,5′-dithiobis-(2-nitro-benzoic acid) [16].
Microvesicle isolation
Control RBCs, 20-h mature ring-stage RBCs, and 32-h mature trophozoite-stage RBCs were washed 3× in RPMI 1640–Hepes medium and then resuspended at 10% hematocrit in the same medium containing a protease inhibitor cocktail (Sigma–Aldrich). Cells were then treated for the desired times with various concentrations of INODs, after which the samples were pelleted at 800 g for 10 min at room temperature. Cell pellets were immediately used to prepare membranes. Supernatants were collected and centrifuged at 25,000 g for 10 min at 4 °C to eliminate spontaneously formed red cell ghosts. Supernatants, after the addition of phosphatase inhibitors, were centrifuged for 3 h at 100,000 g in a refrigerated ultracentrifuge (Beckman) to isolate microvesicles.
FACS analysis
Packed cells were resuspended at a 30% hematocrit in 2 mM PBS-glucose, treated with the desired concentration of INOD-1, and incubated for 2.5 h at 37 °C under gentle shaking. Ten microliters of RBC suspension was diluted 1:200 in PBS-glucose for FACS analysis. RBC vesicles were quantified using a FACSCalibur cytometer (BD Biosciences) and analyzed with CellQuest software (BD Biosciences). Vesicles were selected by forward and side light scatter (FSC and SSC signals set to logarithmic amplification) and a total of ~40,000 events were analyzed.
One-dimensional (1-DE) and two-dimensional electrophoresis (2-DE)
To perform 1-DE and 2-DE, RBC or Pf-RBC membrane proteins were solubilized and separated as described [24].
Immunoblot analysis and Infrared fluorescence detection
Proteins obtained from 1-DE or 2-DE were transferred onto nitro-cellulose membranes and further stained with either fluorescein-5′-maleimide (0.25 mg/ml; Pierce, Rockford, IL, USA) or anti-phosphotyrosine antibody (Santa Cruz Biotechnology, Santa Cruz, CA, USA) and anti-Syk (Santa Cruz) antibodies and with anti-band 3 (Sigma–Aldrich) and then detected using an 800-nm laser scanner (Odyssey, Li-Cor, USA) as described [24]. To ascertain the specificity of anti-phosphotyrosine staining, proteins were dephosphorylated before gel electrophoresis by incubating the samples for 20 min at 30 °C with 6 μl (400 units) λ phosphatase in 50 mM Tris buffer, pH 7.5, 0.1 mM Na2EDTA, 5 mM dithiothreitol, and 2 mM MnCl2.
Gel staining and image analysis
Proteins separated by one-dimensional SDS–PAGE were stained with blue colloidal Coomassie, and proteins separated on two-dimensional gels were stained with a mass spectrometry-compatible silver stain. 2-DE Western blot images were digitized with a GS800 scanner (Bio-Rad) and matched with corresponding stained gels using PDQuest software (Bio-Rad).
Membrane proteins separated on Sepharose CL-6B
RBCs and Pf-RBCs were treated in the presence or absence of 300 nM INOD-1, and membranes were prepared and fractionated as previously described [25].
Preparation of cells for immunofluorescence
Infected and uninfected RBCs were pelleted and rinsed twice in PBS, pH 7.4, containing 5 mM glucose and then fixed for 5 min in 0.5% acrolein in PBS. Fixed cells were fixed and permeabilized as previously described [21]. Staining was performed using a rabbit anti-band 3 antibody (raised against purified recombinant cdb3) diluted in blocking buffer. Cells were incubated with primary antibody for 45 min with motion and rinsed three times in blocking buffer. Band 3 was visualized with Cy3-conjugated anti-rabbit antibody (Jackson ImmunoResearch, West Grove, PA, USA) diluted in blocking buffer for 45 min and rinsed twice in blocking buffer and then once in plain PBS. Cells were resuspended in Syto 16 nuclear acid stain (Invitrogen) diluted to 500 nM in PBS. Cells were incubated in the counter-stain for 20 min with motion and then rinsed three times before observation. After being labeled the RBCs were allowed to attach to coverslips coated with poly-L-lysine, which were then mounted onto glass slides using Aqua-Mount (Lerner Laboratories, New Haven, CT, USA). Samples were imaged in sequential mode to avoid bleed over with an Olympus FluoView FV1000 (Tokyo, Japan) confocal microscope equipped with a 60×1.4 numerical aperture oil immersion lens. For easy visualization of color, the stains were computer-assigned blue for DNA and green for band 3 using the Olympus FV10-ASW software.
Peptide preparation for MS analysis
Bands and spots were excised from 1-DE and 2-DE gels, respectively, and proteins were digested with trypsin. Bands and spots were processed as described [16].
Peptide mass fingerprinting by MALDI-TOF MS
Samples were loaded onto MALDI target using 1 μl of tryptic digest mixed 1:1 with a solution of α-cyano-4-hydroxycinnamic acid (10 mg/ml in ACN/trifluoroacetic acid 0.1%, 40/60). MS analysis of peptides from 2-DE gel spots was performed as described [16].
Results
Analysis of INOD effects on proteomics of the host–parasite system
To perform a comprehensive study of the protein changes induced by INODs in Pf-RBC, we treated control and ring- and trophozoite-stage RBCs with 100 nM INOD-1, i.e., a representative INOD derivative (IC50 ~75 nM) chosen to represent this class of molecules [2]. We chose 100 nM INOD-1 concentration to obtain a better resolution of the 2-DE protein separations; at higher concentrations INOD-1 caused some smearing and proteolysis possibly due to exceeding parasite toxicity. Membranes were prepared by hypotonic lysis and extracted proteins were separated by 2-DE. A semiquantitative measure of the abundance of each protein was then obtained by silver nitrate staining (Figs. 1A and D), and the degree to which each polypeptide was altered by oxidation or phosphorylation was assessed by staining with fluorescein-5-maleimide (Figs. 1B and E), anti-phosphotyrosine (Figs. 1C and F), or anti-phosphoserine antibodies (data not shown). As seen by comparison of Figs. 1C and F (spot 3), INOD-1 induces a significant elevation of tyrosine phosphorylation of band 3 in ring-infected RBCs, constituting the only statistically significant INOD-induced change detected in these Pf-RBCs (p<0.01). To substantiate that band 3 tyrosine hyperphosphorylation is the only significant variation, Fig. 2 shows the levels of protein phosphorylation corresponding to Fig. 1. The protein spots numbered in Fig. 1 and identified by mass spectrometry are listed in Table 1. In contrast, INOD-1 did not impose any measurable changes in phosphorylation of uninfected or trophozoite-infected RBCs (data not shown), nor did it induce any significant changes in gels stained with anti-phosphoserine antibodies and fluorescein maleimide or silver nitrate. To add evidence to the lack of overall oxidant effect (protein thiol oxidation) of low INOD-1 concentrations, we measured GSH and GSSG levels after INOD-1 treatment without observing any significant change (data not shown).
Fig. 1.

Proteomics analysis of ring-infected RBCs after INOD-1 treatment. 2-DE maps of (A, B, C) untreated ring-infected RBCs (Ring) and (D, E, F) ring-infected RBCs treated with 100 nM INOD-1 (Ring+INOD-1) were prepared. 100 μg membrane protein was loaded and separated by isoelectric focusing on a 3–10 IPG strip before further separation by 10% SDS–PAGE. Gels were silver stained (A, D) or blotted onto nitrocellulose and then stained with either fluorescein-5-maleimide (F5M; 0.25 mg/ml; B, E) or anti-phosphotyrosine antibody (anti-pTyr; C, F). The arrows in (C) and (F) indicate band 3 protein. Images were acquired by scanning densitometry (Image Master VDS; Pharmacia) (A, B, D, E) or fluorescence detection (Odyssey IR; Li-Cor, USA) (C, F). Differential analysis of untreated compared to treated cells was conducted using PD Quest (Bio-Rad). The proteins numbered in (B), (C), (E), and (F) were identified by mass spectrometry (MALDI-TOF Micro MX; Micromass, Manchester, UK) and are listed in Table 1.
Fig. 2.
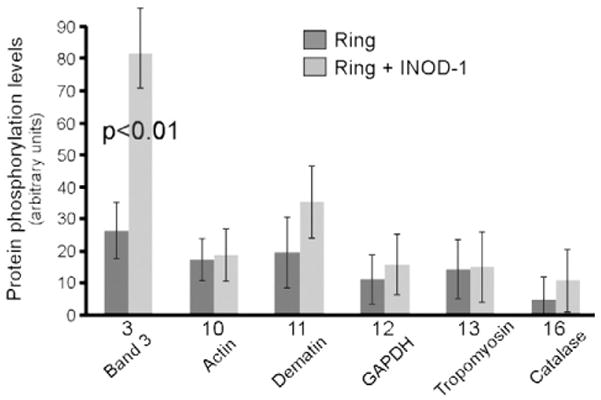
Quantification of protein phosphorylation levels of ring-infected RBC protein separated by 2-DE. Quantitative densitometry of protein phosphorylation levels corresponding to phosphoproteins numbered in Figs. 1C and 1F was performed using a laser IR fluorescence detector (Odyssey, Li-Cor) with Odyssey version 3.0 and expressed as arbitrary fluorescence units. Results are averages±SD of three separate experiments.
Table 1.
Erythrocyte membrane proteins identified by MALDI-TOF analysis.
| Spot | Protein | Mass | Accession No. | Score | Matched peptide |
|---|---|---|---|---|---|
| 1 | α-Spectrin | 281 | P02549 | 2792 | 63 |
| 2 | β-Spectrin | 246 | P11277 | 2280 | 55 |
| 3 | Band 3 | 102 | P02730 | 301 | 17 |
| 4 | Protein 4.1 | 97 | P11171 | 1386 | 42 |
| 5 | Band 3 fragment | 102 | P02730 | 251 | 8 |
| 6 | P55 | 52 | Q00013 | 332 | 19 |
| 7 | α-Spectrin | 281 | P02549 | 158 | 4 |
| 8 | Flotillin 1 | 47 | O75955 | 212 | 6 |
| 9 | Band 3 cyt domain | 42 | P02730 | 345 | 21 |
| 10 | Actin-β | 41 | P60709 | 354 | 16 |
| 11 | Dematin (protein 4.9) | 46 | Q08495 | 299 | 7 |
| 12 | GAPDH | 36 | P04406 | 312 | 12 |
| 13 | Tropomyosin 3 | 29 | P06753 | 372 | 18 |
| 14–15 | Band 3 cyt domain | 42 | P02730 | 345 | 21 |
| 16 | Catalase | 59 | P04040 | 300 | 15 |
Spot numbering corresponds to the spot numbers in the fluorescein-5-maleimide and anti-phosphotyrosine antibody 2-DE gels in Fig. 1.
Characterization of INOD-induced changes in band 3 tyrosine phosphorylation
To enable better quantitation of band 3 tyrosine phosphorylation, we also examined INOD-induced changes in tyrosine phosphorylation by SDS–PAGE followed by anti-phosphotyrosine Western blotting. As shown in Fig. 3A, INOD-1 promoted a significant increase (p<0.01) in ring-stage band 3 tyrosine phosphorylation (compare lanes 3 and 4), but no significant change in uninfected (lanes 1 and 2) or trophozoite-stage Pf-RBC phosphorylation (lanes 5 and 6).
Fig. 3.
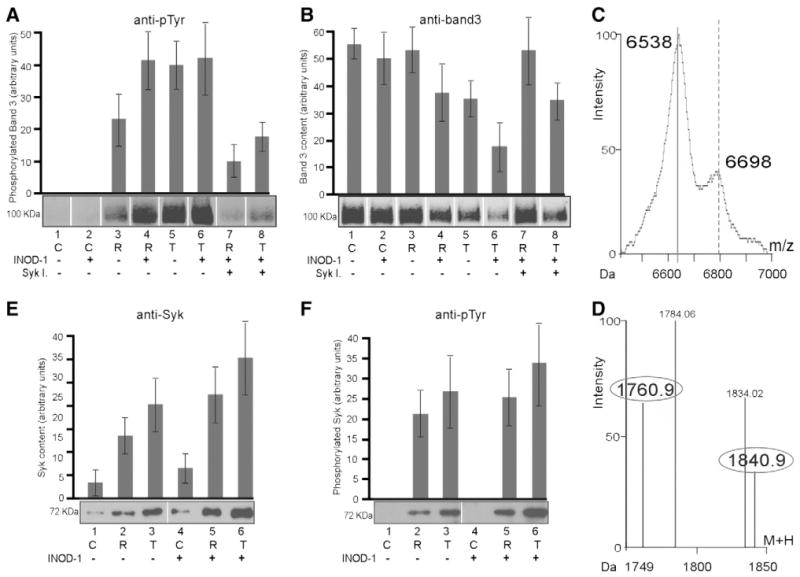
Analysis of band 3 phosphorylation and Syk translocation to the membrane induced by INOD-1. 1-DE maps of membrane proteins were obtained by 8% SDS–PAGE of control RBCs (C), ring-stage RBCs (R), and trophozoite-stage RBCs (T). Gels were analyzed by Western blotting using (A) anti-phosphotyrosine (anti-pTyr) or (B) anti-band 3 antibodies. Control RBCs and ring- and trophozoite-infected RBCs were treated with 100 nM INOD-1 (lanes 2, 4, 6) and with 100 nM INOD-1 in presence of 10 μM Syk inhibitors II and IV (lanes 7, 8). Gel slices corresponding to the band 3 region were subjected to trypsin digestion and analyzed by MALDI-TOF Micro MX (Micromass). The mass spectra revealed band 3 tryptic peptides containing (C) Tyr 8 and 21, 6538 (unphosphorylated) and 6698 Da (double phosphorylated), and (D) Tyr 359, 1760.9 (unphosphorylated) and 1840.9 Da (phosphorylated). Intensity values are presented as the percentage of the highest intensity ion. Membrane proteins were separated by 8% SDS–PAGE and analyzed by Western blotting using (E) anti-Syk and (F) anti-pTyr antibodies. Infected RBCs were treated with (lanes 4–6) or without 300 nM INOD-1 (lanes 1–3). Gels were run under reducing conditions. Images were acquired using a laser IR fluorescence detector (Odyssey, Li-Cor). Quantitative densitometry of band 3 phosphorylation level (A), band 3 content (B), Syk content (E), and Syk phosphorylation level (F) was performed using a laser IR fluorescence detector (Odyssey, Li-Cor) with Odyssey version 3.0 and expressed as arbitrary fluorescence units. Results are averages±SD of five separate experiments.
At the schizont stage, we observed an extensive protein dephosphorylation (data not shown) and a large variability between different parasite preparations. Dephosphorylation and the variability in the patterns observed at schizont stage may be due to the combined effects of variable degrees of proteolysis [26] and to the energetic decay that characterizes the terminal parasite stages [27] (data not shown).
To study the involvement of Syk tyrosine kinase in INOD-1-induced band 3 phosphorylation, we pretreated the Pf-RBCs with Syk kinase inhibitors and examined the effect of INOD-1 on band 3 tyrosine phosphorylation. Fig. 3A, lanes 7 and 8 show that inhibition of Syk markedly decreases both basal and INOD-induced band 3 tyrosine phosphorylation, in both ring- and trophozoite-stage Pf-RBCs (p<0.01).
Anti-band 3 Western blotting has previously shown that band 3 is somewhat depleted in trophozoite-stage Pf-RBCs compared to ring stage and control cells from the same donor [8,24]. More detailed analysis of the gels in Fig. 3B now suggests that INOD-1 treatment further enhances depletion of band 3, in both ring- and trophozoite-stage Pf-RBCs (lanes 3, 4 and 5, 6). Moreover, Syk inhibitors can be seen to suppress the INOD-1-induced band 3 depletion, suggesting that band 3 depletion is influenced by its level of tyrosine phosphorylation (Fig. 3B, lanes 7 and 8).
To further characterize the band 3 phosphorylation process, we mapped the sites of tyrosine phosphorylation by mass spectrometry. This analysis revealed tyrosines 8 and 21 (Fig. 3C) and 359 (Fig. 3D) as the major sites of phosphorylation. Consistent with the inhibition data, phosphorylation of these residues has already been ascribed to the activity of Syk (Tyr 8 and 21) and possibly Lyn (Tyr 359) tyrosine kinases [15].
Activation of Syk kinase frequently involves its own tyrosine phosphorylation and translocation to the membrane[15,16,28]. Therefore, to further explore the involvement of Syk kinase in mediating the effects of INOD treatment in Pf-RBCs, we investigated its phosphorylation along with its translocation to the membrane in response to INOD exposure. Fig. 3E shows that Syk gradually accumulates on the Pf-RBC membrane during parasite maturation (compare lanes 1, 2, and 3) and that INOD-1 addition enhances this accumulation (Fig. 3E, lanes 4, 5, and 6). Moreover, as seen in Fig. 3F, Syk phosphorylation increases in proportion to its accumulation on the membrane, suggesting that the active phosphorylated form of Syk translocates to the membrane. Although Lyn kinase often cooperates with Syk in regulating various processes in hematopoietic cells, no evidence of Lyn translocation to Pf-RBC membranes in either INOD-1-treated or untreated Pf-RBCs could be detected using anti-Lyn antibodies and Western blots (data not shown).
Fig. 4 shows the concentration and time dependence of INOD-induced enhancement of band 3 tyrosine phosphorylation in ring-stage infected and control RBCs. Enhancement of phosphorylation by 100 nM INOD-1 was clearly evident by 60 min of incubation and phosphorylation progressively increased for the 6-h duration of the study. In control RBCs, 100 nM INOD-1 did not cause any measurable enhancement of band 3 tyrosine phosphorylation, even after 6 h of exposure (Fig. 4A). The dose-dependent response to INOD-1 revealed that, in ring-infected RBCs, enhanced tyrosine phosphorylation was evident at 20 nM INOD-1, with maximal activity occurring at 300 nM concentration. In control RBCs, a weak increase in band 3 tyrosine phosphorylation was measurable in the 300–500 nM INOD-1 concentration range, suggesting that very high doses of INOD-1 can activate tyrosine phosphorylation even in uninfected erythrocytes (Fig. 4B).
Fig. 4.
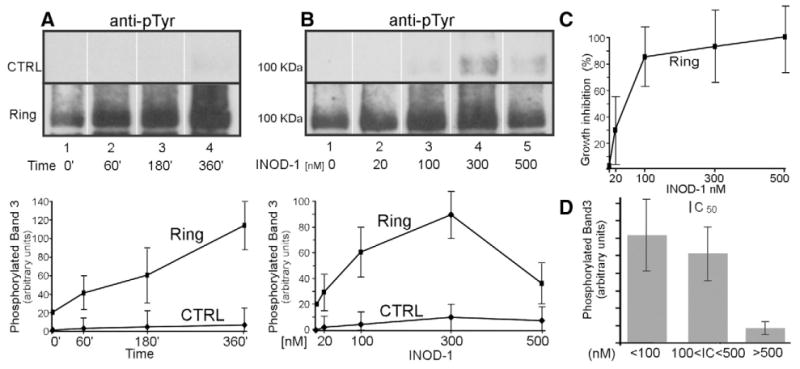
Effect of the antimalarial potency, concentration, and incubation time of INODs on band 3 tyrosine phosphorylation. Proteins were separated by 8% SDS–PAGE and analyzed by Western blotting using an anti-phosphotyrosine (anti-pTyr) antibody. (A) Membrane proteins of control RBCs (CTRL) and ring-infected RBCs (Ring) were treated with 100 nM INOD-1 for various incubation times. (B) Control RBCs and ring-infected RBCs were treated with various concentrations of INOD-1 for 3 h of incubation. Quantitative densitometry of band 3 phosphorylation was performed using a laser IR fluorescence detector (Odyssey, Li-Cor) with Odyssey version 3.0 software and expressed as arbitrary fluorescence units. (C) Percentage of parasite growth inhibition induced by various INOD-1 concentrations at ring stage. (D) Average levels of band 3 tyrosine phosphorylation induced by 64 different INODs are plotted as a function of the IC50 values of the INOD species. For the purpose of simplification, INOD derivatives were grouped into three groups according to their IC50 ranges: 0–100, 100–500, and >500 nM. Data were acquired as described for Figs. 4A and 4B. Results are averages±SD of three separate experiments.
To study the relationship between the antiplasmodial activity of INODs and their abilities to induce band 3 tyrosine phosphorylation, we measured the parasite growth inhibition at various INOD-1 concentrations. As shown in Fig. 4C INOD-1 exerts some antiplasmodial activity in the low-nanomolar range (20 nM), which corresponds to the minimal concentration required to trigger band 3 tyrosine phosphorylation. We do not have direct evidence to explain the decrease in band 3 phosphorylation observed at 500 nM INOD-1 concentration. As this concentration is 6.6-fold higher than the IC50, we hypothesized that it may result from a rapid antiplasmodial action and/or an effect on additional targets. To further establish a causal relationship between INOD antiplasmodial activity and their ability to induce band 3 phosphorylation we tested 66 different INOD compounds displaying widely varying antiplasmodial activities in ring-infected RBCs [2]. Fig. 4D shows the effects of these INODs after grouping them according to their IC50 values for inhibition of parasitemia. In this figure, compounds with IC50 values below 100 nM (compounds 1, 11, 18, 22, 25, 44) were grouped together, as were compounds with IC50 values between 100 and 500 nM (compounds 9, 14, 17, 19, 23, 24, 48) and compounds with IC50 values greater than 500 nM (compounds 5, 16, 60, 62, 43). INODs possessing IC50 values greater than 500 nM induced significantly (p<0.01) less band 3 tyrosine phosphorylation than INODs possessing more potent antiplasmodial activity.
Characterization of the phosphorylated band 3 aggregates
As previously described [8], two major aggregation states of band 3 have been observed during size-exclusion chromatography of detergent-extracted Pf-RBC membrane proteins: a high-molecular-weight fraction (Fig. 5, fraction 1), corresponding to aggregates of band 3 and other proteins, and a lower-molecular-weight fraction (Fig. 5, fraction 2) consisting mainly of band 3 dimers and tetramers. Importantly, INOD-1 (300 nM) treatment of ring-infected RBCs caused a consistent increase in the higher-molecular-weight fraction. Thus, whereas band 3 in uninfected RBCs is invariably found in the lower-molecular-weight fraction (data not shown), a moderate proportion of the band 3 in ring-stage Pf-RBCs segregated into this higher-molecular-weight fraction. Moreover, when these ring-stage Pf-RBCs were treated with INOD-1, almost all of the band 3 shifted into this higher-molecular-weight fraction (compare open and closed circles in Fig. 5A). That this fraction is heavily oxidized can be seen from the facts that: (i) it contains a large amount of denatured hemoglobin products (open and closed squares),and (ii) it can be disaggregated by exposure to dithiothreitol, a disulfide-reducing agent (Fig. 5B, fraction 1). Most significantly, tyrosine-phosphorylated band 3 is seen only in this high-molecular-weight fraction (Fig. 5B, fraction 2, compare lanes 1 and 2), suggesting that the phosphorylation event is somehow linked to band 3 oxidation and aggregation. This hypothesis is, in fact, consistent with the observation that INOD-1 exposure, which also enhances band 3 tyrosine phosphorylation (Figs. 1C and F), similarly increases aggregate formation in Pf-RBCs (Fig. 5A).
Fig. 5.
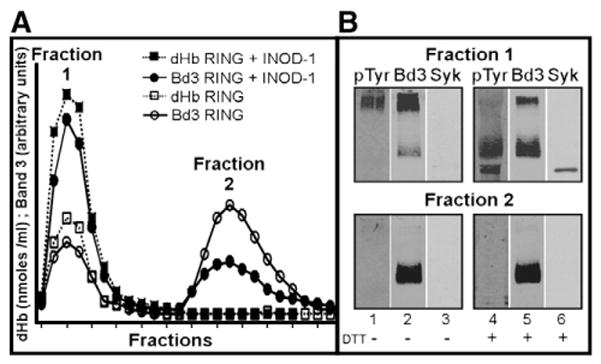
Analysis of the phosphorylation and aggregation states of band 3 complexes in ring-infected RBCs±INOD-1 treatment. Proteins were extracted from membranes of ring-infected RBCs by incubation in 1% Triton X-100 and separated by gel-filtration chromatography on 40×1-cm Sepharose CL-6B. (A) Two fractions were obtained: Fraction 1 comprised higher-molecular-weight components and Fraction 2 comprised lower-molecular-weight components. Eosin maleimide-labeled band 3 fluorescence was measured in each fraction and expressed as arbitrary units. Denatured hemoglobin (dHb) was quantified by visual spectroscopy and expressed as nmol/ml. (B) Fractions 1 and 2 were analyzed by Western blotting with anti-pTyr (lanes 1 and 4), anti-band 3 (Bd3) (lanes 2 and 5), and anti-Syk (lanes 3 and 6) antibodies. Gels were run under nonreducing conditions. Images were acquired using a laser IR fluorescence detector (Odyssey, Li-Cor).
Although an increase in the content of aggregated band 3 and associated denatured hemoglobin products is also eventually seen in the untreated trophozoite-stage Pf-RBCs, no additional effect of INOD-1 administration on either the composition or the quantity of these aggregates could be detected (data not shown). Thus, the aforementioned effects of INOD-1 exposure can be induced prematurely in ring-stage Pf-RBCs, but later occur spontaneously in trophozoite-stage Pf-RBCs.
Effect of INOD-1 on the release of microvesicles in infected RBCs
In Fig. 3B, we observed a decrease in the band 3 content of Pf-RBCs after INOD-1 treatment, suggesting that the band 3 population that is phosphorylated in response to INODs can be lost from the membrane via release of microvesicles [29]. To test this hypothesis, we isolated microvesicles from the supernatants of infected and uninfected RBCs (hematocrit 25%) and measured their band 3 contents. Fig. 6A, lane 4, shows that only a small amount of band 3 is isolated in the microvesicle fraction from untreated ring-stage Pf-RBCs, whereas the major fraction of band 3 is found in the vesicular fraction of trophozoite-stage Pf-RBCs (Fig. 6A, lane 7). Moreover, whereas exposure to 300 nM INOD-1 induces only a small amount of vesiculation in uninfected RBCs (Fig. 6A, lanes 1 and 2), INOD-1 treatment causes dramatic vesiculation in ring-infected Pf-RBCs (Fig. 6A, lanes 4 and 5; p<0.01). In trophozoite-stage cells, in contrast, the effect of INOD-1 addition is not detectable, possibly because of the high level of vesiculation that occurs spontaneously in these Pf-RBCs (Fig. 6A, lanes 7 and 8). Importantly, Syk kinase inhibitors markedly reduced the extent of INOD-1-induced vesiculation in ring-infected RBCs (Fig. 6A, lanes 5 and 6), whereas the same inhibitors had no effect on trophozoite-stage RBCs (Fig. 6A, lane 9).
Fig. 6.
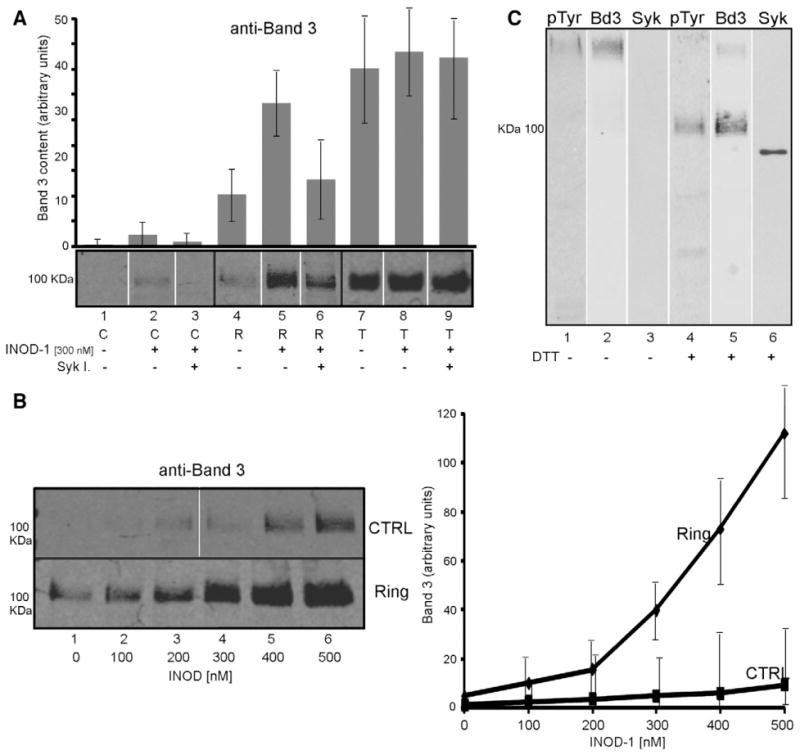
Effect of INOD-1 treatment on the release of microvesicles from control and infected RBCs. Microvesicles were isolated from the supernatants of control (C) and ring (R) and trophozoite (T) infected RBCs, separated by 8% SDS–PAGE, and analyzed by Western blotting using anti-band 3 antibody. (A) Microvesicles released after 3 h treatment with 300 nM INOD-1 in the presence (lanes 3, 6, 9) or absence (lanes 1, 2, 4, 5, 7, 8) of 10 μM Syk inhibitors II and IV. (B) Microvesicles released from control (CTRL) and ring-infected (Ring) RBCs after treatment with increasing concentrations of INOD-1. (C) Microvesicles isolated from ring-infected RBCs visualized with anti-band 3 (Bd3), anti-pTyr, or anti-Syk antibodies under nonreducing and reducing conditions. Images were acquired using Odyssey IR fluorescence detector (Li-Cor). Quantitative densitometry of band 3 (A, B) was performed using a laser IR fluorescence detector (Odyssey; Li-Cor) with Odyssey version 3.0 software and expressed as arbitrary fluorescence units. Results are averages±SD of three separate experiments.
Because the above data suggest a causal relationship between INOD exposure, band 3 tyrosine phosphorylation, and release of band 3-containing vesicles from ring-stage Pf-RBCs, the linkage among these parameters was further examined. As seen in Fig. 6B, as INOD-1 concentration is increased in ring-stage Pf-RBCs, release of band 3-containing vesicles is also increased. Moreover, in Fig. 6C (lane 2), analysis of the released vesicles by SDS–PAGE under nonreducing conditions reveals that all of band 3 is oxidatively cross-linked, as confirmed by its shift to the expected molecular mass of ~100,000 Da upon exposure to dithiothreitol (lane 5). Lanes 1 and 4 further reveal that band 3 in the vesicles is heavily tyrosine phosphorylated and that the band 3 aggregate contains Syk, i.e., the kinase responsible for this phosphorylation (lane 6). And as expected, spectrometric analysis of solubilized vesicle proteins reveals the presence of large amounts of denatured hemoglobin products (~2 nmol/mg protein; data not shown).
Finally, to provide visual evidence for the release of band 3-containing vesicles from Pf-RBCs, ring- and trophozoite-stage Pf-RBCs were stained with an antibody to band 3 and examined by confocal microscopy.
Fig. 7 shows immunofluorescence images of control ring and trophozoite infected red cells treated with 300 nM INOD-1. Parasitized red cells often displayed surface membrane protrusions containing band 3 that may constitute the source of the microvesicles released by Pf-RBC. Interestingly, the same protrusions were also present in untreated infected RBC (data not shown). Because of exceeding differences between cell preparations and cell-to-cell variations, we could not draw quantitative estimates of vesicle release.
Fig. 7.
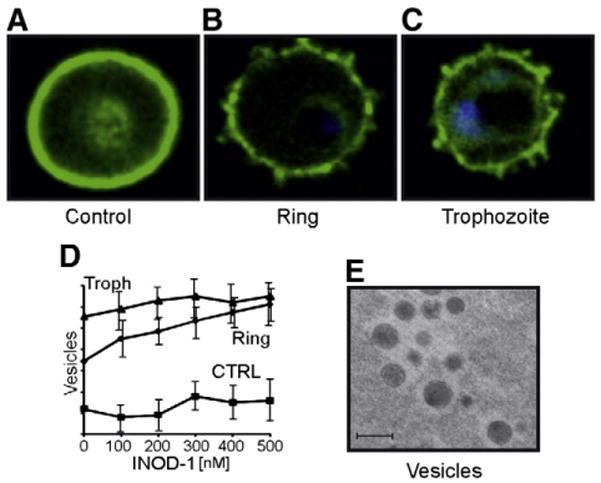
Confocal images of control and infected RBCs after INOD-1 treatment and analysis of released vesicles. (A, B, C) Cells were pretreated with 300 nM INOD-1, isolated, and stained with anti-band 3 antibody (green) and DAPI for DNA (blue). (D) Vesicles released from control and infected RBCs, ring and trophozoite (Troph), after treatment with various concentrations of INOD-1 were quantified by FACS (FACSCalibur cytometer; BD Biosciences) using CellQuest software (BD Biosciences). The number of vesicles was expressed as arbitrary units. Results are averages±SD of three separate experiments. (E) An electron microscopy image of vesicles isolated from infected RBCs. Ultrathin sections were stained with osmium tetraoxide, examined, and photographed using a Zeiss EM 900 transmission electron microscope (Carl Zeiss Jena GmbH, Jena, Germany), operating at 80 kV. Bar, 1 μm.
The number of released vesicles could be readily quantified by flow cytometry, and as seen in Fig. 7D, their abundance increased linearly with INOD-1 concentration. The results shown in Fig. 6 were therefore now confirmed using a different methodology, although the FACS analysis was apparently more sensitive at detecting vesiculation at low INOD-1 concentrations than Western blotting. In contrast, INOD-1 had no added effect on vesicle release from trophozoite-infected RBCs, consistent with the constitutive high rate of vesicle release from this stage of Pf-RBCs. Analysis of the released vesicles by electron microscopy revealed large, electron-dense inclusions (Fig. 7E) that may represent the large membrane aggregates detected in Figs. 5 and 6.
Antiplasmodial effect of INOD-1 at various P. falciparum maturation stages
Because the effects of INOD-1 on the molecular properties of Pf-RBCs were considerably more pronounced during ring than during trophozoite stage, we asked whether INOD inhibition of parasitemia might also be more prominent in ring-stage than in trophozoite-stage cultures. For this purpose, synchronized parasite cultures at ring and trophozoite stages were treated with 100 nM INOD-1 and then examined for parasite maturation. Treatment of ring-infected Pf-RBCs resulted in 85% inhibition of parasite maturation (based on trophozoite numbers 24 h after treatment with INOD-1 versus medium). In contrast, treatment of trophozoite-infected Pf-RBCs resulted in only a 26% decline in the parasite count after 24 h. These data support the involvement of the aforementioned INOD-stimulated changes in ring-stage Pf-RBCs in the therapeutic mechanism of INOD-1. To establish a causal relationship between INODs’ antiplas-modial effect and band 3 hyperphosphorylation we pretreated ring-infected Pf-RBCs with Syk inhibitors (10 μM) before treatment with INOD-1 (100 nM). We found that Syk inhibition caused an ≈ 40% decrease in INOD-1 antiplasmodial activity.
Discussion
Previous work has demonstrated the potent antimalarial activity of INODs in both parasite cultures in vitro and murine models of P. berghei infection in vivo [2]. However, apart from the essential role of their redox-active pharmacophore, little can be speculated about their mechanism of action. Based on the data presented above, we wish to propose the following hypothesis. We hypothesize that P. falciparum requires a stable erythrocyte membrane during early stages of infection for successful parasite maturation. Oxidative stress, a prominent consequence of parasite growth [8,24,30], leads directly to membrane destabilization by: (i) dissociating ankyrin from band 3 via tyrosine phosphorylation of band 3 [21], (ii) weakening the spectrin–protein 4.1–actin junctional complex [31], and (iii) promoting formation of hemichrome-enriched membrane protein aggregates [8]. This membrane destabilization in turn results in membrane vesiculation and a decline in Pf-RBC surface-to-volume ratio. INODs apparently enhance this membrane destabilization by shifting Pf-RBCs, which are already oxidatively stressed [24], toward an even more intense oxidative environment. Within this heightened oxidative milieu, infected erythrocytes become not only less hospitable for parasite maturation, but also less mechanically stable and capable of harboring the developing parasite.
The hypothesized requirement for a stable red cell membrane for completion of the parasite’s life cycle is suggested by the observation that perturbations that compromise the architecture of the membrane, including mutations in critical structural proteins [32–34], defects in hemoglobin stability [35], and deficiencies in important metabolic enzymes [36], confer variable degrees of resistance to malaria or are protective against its more severe complications. Indeed, erythrocytes with naturally unstable membranes (e.g., hereditary spherocytosis, hereditary pyropoikilocytosis, hereditary elliptocytosis, Asian ovalocytosis) do not support Plasmodium proliferation in vitro [32,37–40]. Furthermore, treatment of red cells with morphology-altering drugs has been shown to inhibit entry and maturation of the parasite in cell culture [41]. And most importantly, RESA, a parasite-encoded protein that is critical for Plasmodium proliferation, functions by stabilizing the infected cell membrane during early stages of parasite development; i.e., ring-infected RBCs devoid of RESA undergo membrane vesiculation and loss of parasite viability early in the parasite’s life cycle [42–44].
Data suggesting that oxidative overload creates an inhospitable environment for parasite maturation include the facts that sickle cell disease [45], G6PD deficiency [4,46], α- and β-thalassemias [47], and oxidizing drugs [2,48] all inhibit parasite growth. One might therefore inquire how oxidative overload leads to suppression of parasitemia? Whereas an oxidative milieu can be damaging to almost any protein, detailed studies have shown that specific defects emerge in the red cell membrane in response to oxidation of membrane sulfhydryls. In G6PD-deficient red cells we have recently demonstrated that persistent band 3 oxidation and phosphorylation cause progressive membrane vesiculation and release of hemichromes [16,22]. Interestingly, we have observed that parasites growing in G6PD-deficient red cells exert a marked oxidative effect at early stages of maturation and cause increased vesiculation at this stage (R. Vono et al., unpublished results).
We have demonstrated that band 3 sulfhydryls are oxidatively cross-linked during early stages of parasite maturation [8,30]. The formation of these disulfide bonds between cysteine 201 of one subunit of band 3 and cysteine 317 of the other subunit has been further shown to lead to loss of ankyrin affinity [49]. The resulting dissociation of ankyrin from band 3 due to band 3 oxidation will be further aggravated by oxidation of sulfhydryls in Syk and erythrocyte tyrosine phosphatases, both of which lead to enhancement of band 3 tyrosine phosphorylation [16] and ankyrin dissociation [21]. Oxidation of cysteines in spectrin have also been shown to destabilize the spectrin–protein 4.1–actin junctional complex [31]. Finally, oxidative overload can also promote hemichrome formation, resulting in band 3 clustering, autologous antibody binding, complement deposition, and erythrocyte phagocytosis by macrophages [8,16,30,50]. Not surprisingly, these hemichromes form at an accelerated rate in thalassemic, sickle, and G6PD-deficient erythrocytes [6,8].
The sequelae of oxidative effects on membrane tyrosine phosphate content deserve more detailed elaboration. As noted above, oxidative stress results in a dramatic rise in band 3 tyrosine phosphorylation, which, as shown recently by us [24], triggers complete dissociation of ankyrin from band 3 [21]. Because rupture of the band 3–ankyrin bridge by any mechanism is well documented to lead to membrane vesiculation [21], loss of membrane surface area naturally follows elevation of band 3 tyrosine phosphorylation. Consistent with this possible mechanism, band 3 tyrosine phosphorylation in ring-infected RBCs was found to constitute the major covalent modification induced by INODs. Because this process occurred over the same concentration range as INOD’s antiplasmodial activity, we hypothesized that elevation of band 3 tyrosine phosphorylation and the consequent weakening of membrane interactions play a critical role in the therapeutic action of INODs. INODs showed higher antiplasmodial activity, more band 3 modifications, and membrane vesiculation at the ring stage; this observation has been confirmed by a recent study performed in Cameroon on fresh P. falciparum clinical isolates [51]. A major challenge of the present work was then to establish a causal linkage between phosphorylation of band 3 induced by INODs, increased vesiculation of Pf-RBCs, and antiplasmodial activity. Importantly, several observations were found to support this mechanistic linkage: (i) in ring-infected RBCs, INOD-1 triggered a dose-dependent increase in band 3 phosphorylation and a parallel increase in vesicle release in a concentration range corresponding to its IC50; (ii) Syk kinase inhibitors concomitantly suppressed band 3 phosphorylation, the release of membrane vesicles, and antiplasmodial activity of INODs; (iii) the antiplasmodial activities of a large series of INODs positively correlated with their capability to induce band 3 phosphorylation and vesiculation; (iv) INODs enhanced the phosphorylation of Syk kinase and its consequent translocation to the membrane; (v) INOD treatment promoted the formation of large protein complexes containing Syk kinase, denatured hemoglobin products, and hyperphosphorylated and oxidatively cross-linked band 3; and (vi) vesicles released from INOD-treated Pf-RBCs contained the same protein components seen in the aforementioned high-molecular-weight complexes. Together with our data on oxidative changes in INOD-treated Pf-RBCs, these observations indicate a causal relationship between INOD antiplasmodial activity, band 3 tyrosine phosphorylation, and the consequent red cell membrane destabilization. Together with our data on the selective phosphorylation of the oxidized band 3 molecules [16], we suggest that, similar to other phosphorylation pathways activated by redox signaling [52,53], in erythrocytes band 3 tyrosine phosphorylation may result from the interaction between the oxidative stress exerted by the intracellular parasites and specifically modified free radical compounds.
Acknowledgments
This work was supported by the European Union (Redox Anti-malarial Drug Discovery, Read-Up, FP6-2004-LSH-2004-2.3.0-7, Strep No. 018602). We thank Elena Valente and Daniela Ulliers for technical assistance.
Abbreviations
- Pf-RBC
Plasmodium falciparum red blood cell
- INOD
indolone-N-oxide derivative
- G6PD
glucose-6-phosphate dehydrogenase
References
- 1.Dondorp AM, Yeung S, White L, Nguon C, Day NP, Socheat D, et al. Arte-misinin resistance: current status and scenarios for containment. Nat Rev. 2010;8:272–280. doi: 10.1038/nrmicro2331. [DOI] [PubMed] [Google Scholar]
- 2.Nepveu F, Kim S, Boyer J, Chatriant O, Ibrahim H, Reybier K, et al. Synthesis and antiplasmodial activity of new indolone N-oxide derivatives. J Med Chem. 2010;53:699–714. doi: 10.1021/jm901300d. [DOI] [PubMed] [Google Scholar]
- 3.Ginsburg H, Atamna H. The redox status of malaria-infected erythrocytes: an overview with an emphasis on unresolved problems. Parasite (Paris) 1994;1:5–13. doi: 10.1051/parasite/1994011005. [DOI] [PubMed] [Google Scholar]
- 4.Cappadoro M, Giribaldi G, O’Brien E, Turrini F, Mannu F, Ulliers D, et al. Early phagocytosis of glucose-6-phosphate dehydrogenase (G6PD)-deficient erythrocytes parasitized by Plasmodium falciparum may explain malaria protection in G6PD deficiency. Blood. 1998;92:2527–2534. [PubMed] [Google Scholar]
- 5.Ayi K, Cappadoro M, Branca M, Turrini F, Arese P. Plasmodium falciparum glutathione metabolism and growth are independent of glutathione system of host erythrocyte. FEBS Lett. 1998;424:257–261. doi: 10.1016/s0014-5793(98)00185-9. [DOI] [PubMed] [Google Scholar]
- 6.Ayi K, Turrini F, Piga A, Arese P. Enhanced phagocytosis of ring-parasitized mutant erythrocytes: a common mechanism that may explain protection against falciparum malaria in sickle trait and beta-thalassemia trait. Blood. 2004;104:3364–3371. doi: 10.1182/blood-2003-11-3820. [DOI] [PubMed] [Google Scholar]
- 7.Becker K, Kirk K. Of malaria, metabolism and membrane transport. Trends Parasitol. 2004;20:590–596. doi: 10.1016/j.pt.2004.09.004. [DOI] [PubMed] [Google Scholar]
- 8.Giribaldi G, Ulliers D, Mannu F, Arese P, Turrini F. Growth of Plasmodium falciparum induces stage-dependent haemichrome formation, oxidative aggregation of band 3, membrane deposition of complement and antibodies, and phagocytosis of parasitized erythrocytes. Br J Haematol. 2001;113:492–499. doi: 10.1046/j.1365-2141.2001.02707.x. [DOI] [PubMed] [Google Scholar]
- 9.Akide-Ndunge OB, Tambini E, Giribaldi G, McMillan PJ, Muller S, Arese P, et al. Co-ordinated stage-dependent enhancement of Plasmodium falciparum antioxidant enzymes and heat shock protein expression in parasites growing in oxidatively stressed or G6PD-deficient red blood cells. Malaria J. 2009;8:113. doi: 10.1186/1475-2875-8-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tokumasu F, Fairhurst RM, Ostera GR, Brittain NJ, Hwang J, Wellems TE, et al. Band 3 modifications in Plasmodium falciparum-infected AA and CC erythrocytes assayed by autocorrelation analysis using quantum dots. J Cell Sci. 2005;118(Pt 5):1091–1098. doi: 10.1242/jcs.01662. [DOI] [PubMed] [Google Scholar]
- 11.Dondorp AM, Omodeo-Sale F, Chotivanich K, Taramelli D, White NJ. Oxidative stress and rheology in severe malaria. Redox Rep. 2003;8:292–294. doi: 10.1179/135100003225002934. [DOI] [PubMed] [Google Scholar]
- 12.Omodeo-Sale F, Motti A, Basilico N, Parapini S, Olliaro P, Taramelli D. Accelerated senescence of human erythrocytes cultured with Plasmodium falciparum. Blood. 2003;102:705–711. doi: 10.1182/blood-2002-08-2437. [DOI] [PubMed] [Google Scholar]
- 13.Stocker R, Hunt NH, Buffinton GD, Weidemann MJ, Lewis-Hughes PH, Clark IA. Oxidative stress and protective mechanisms in erythrocytes in relation to Plasmodium vinckei load. Proc Natl Acad Sci U S A. 1985;82:548–551. doi: 10.1073/pnas.82.2.548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Harrison ML, Isaacson CC, Burg DL, Geahlen RL, Low PS. Phosphorylation of human erythrocyte band 3 by endogenous p72syk. J Biol Chem. 1994;269:955–959. [PubMed] [Google Scholar]
- 15.Brunati AM, Bordin L, Clari G, James P, Quadroni M, Baritono E, et al. Sequential phosphorylation of protein band 3 by Syk and Lyn tyrosine kinases in intact human erythrocytes: identification of primary and secondary phosphorylation sites. Blood. 2000;96:1550–1557. [PubMed] [Google Scholar]
- 16.Pantaleo A, Ferru E, Giribaldi G, Mannu F, Carta F, Matte A, et al. Oxidized and poorly glycosylated band 3 is selectively phosphorylated by Syk kinase to form large membrane clusters in normal and G6PD-deficient red blood cells. Biochem J. 2009;418:359–367. doi: 10.1042/BJ20081557. [DOI] [PubMed] [Google Scholar]
- 17.Zipser Y, Piade A, Kosower NS. Erythrocyte thiol status regulates band 3 phosphotyrosine level via oxidation/reduction of band 3-associated phosphotyrosine phosphatase. FEBS Lett. 1997;406:126–130. doi: 10.1016/s0014-5793(97)00263-9. [DOI] [PubMed] [Google Scholar]
- 18.Low PS, Allen DP, Zioncheck TF, Chari P, Willardson BM, Geahlen RL, et al. Tyrosine phosphorylation of band 3 inhibits peripheral protein binding. J Biol Chem. 1987;262:4592–4596. [PubMed] [Google Scholar]
- 19.Campanella ME, Chu H, Low PS. Assembly and regulation of a glycolytic enzyme complex on the human erythrocyte membrane. Proc Natl Acad Sci U S A. 2005;102:2402–2407. doi: 10.1073/pnas.0409741102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Harrison ML, Rathinavelu P, Arese P, Geahlen RL, Low PS. Role of band 3 tyrosine phosphorylation in the regulation of erythrocyte glycolysis. J Biol Chem. 1991;266:4106–4111. [PubMed] [Google Scholar]
- 21.Ferru E, Giger K, Pantaleo A, Campanella E, Grey J, Ritchie K, et al. Regulation of membrane–cytoskeletal interactions by tyrosine phosphorylation of erythrocyte band 3. Blood. 2011;117:5998–6006. doi: 10.1182/blood-2010-11-317024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pantaleo A, Ferru E, Carta F, Mannu F, Simula LF, Khadjavi A, et al. Irreversible AE1 tyrosine phosphorylation leads to membrane vesiculation in G6PD deficient red cells. PLoS One. 2011;6:e15847. doi: 10.1371/journal.pone.0015847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lambros C, Vanderberg JP. Synchronization of Plasmodium falciparum erythrocytic stages in culture. J Parasitol. 1979;65:418–420. [PubMed] [Google Scholar]
- 24.Pantaleo A, Ferru E, Carta F, Mannu F, Giribaldi G, Vono R, et al. Analysis of changes in tyrosine, serine phosphorylation of red cell membrane proteins induced by P, falciparum growth. Proteomics. 2010;10:3469–3479. doi: 10.1002/pmic.201000269. [DOI] [PubMed] [Google Scholar]
- 25.Turrini F, Mannu F, Cappadoro M, Ulliers D, Giribaldi G, Arese P. Binding of naturally occurring antibodies to oxidatively and nonoxidatively modified erythrocyte band 3. Biochim Biophys Acta. 1994;1190:297–303. doi: 10.1016/0005-2736(94)90087-6. [DOI] [PubMed] [Google Scholar]
- 26.Salmon BL, Oksman A, Goldberg DE. Malaria parasite exit from the host erythrocyte: a two-step process requiring extraerythrocytic proteolysis. Proc Natl Acad Sci U S A. 2001;98:271–276. doi: 10.1073/pnas.011413198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kanaani J, Ginsburg H. Compartment analysis of ATP in malaria-infected erythrocytes. Biochem Int. 1988;17:451–459. [PubMed] [Google Scholar]
- 28.Bordin L, Ion-Popa F, Brunati AM, Clari G, Low PS. Effector-induced Syk-mediated phosphorylation in human erythrocytes. Biochim Biophys Acta. 2005;1745:20–28. doi: 10.1016/j.bbamcr.2004.12.010. [DOI] [PubMed] [Google Scholar]
- 29.Wagner GM, Chiu DT, Yee MC, Lubin BH. Red cell vesiculation—a common membrane physiologic event. J Lab Clin Med. 1986;108:315–324. [PubMed] [Google Scholar]
- 30.Turrini F, Giribaldi G, Carta F, Mannu F, Arese P. Mechanisms of band 3 oxidation and clustering in the phagocytosis of Plasmodium falciparum-infected erythrocytes. Redox Rep. 2003;8:300–303. doi: 10.1179/135100003225002943. [DOI] [PubMed] [Google Scholar]
- 31.Chishti AH, Maalouf GJ, Marfatia S, Palek J, Wang W, Fisher D, et al. Phosphorylation of protein 4.1 in Plasmodium falciparum-infected human red blood cells. Blood. 1994;83:3339–3345. [PubMed] [Google Scholar]
- 32.Rank G, Sutton R, Marshall V, Lundie RJ, Caddy J, Romeo T, et al. Novel roles for erythroid Ankyrin-1 revealed through an ENU-induced null mouse mutant. Blood. 2009;113:3352–3362. doi: 10.1182/blood-2008-08-172841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gallagher PG, Forget BG. Hematologically important mutations: band 3 and protein 4.2 variants in hereditary spherocytosis. Blood Cells Mol Dis. 1997;23:417–421. doi: 10.1006/bcmd.1997.0160. [DOI] [PubMed] [Google Scholar]
- 34.Manno S, Takakuwa Y, Mohandas N. Modulation of erythrocyte membrane mechanical function by protein 4.1 phosphorylation. J Biol Chem. 2005;280:7581–7587. doi: 10.1074/jbc.M410650200. [DOI] [PubMed] [Google Scholar]
- 35.Kannan R, Labotka R, Low PS. Isolation and characterization of the hemichrome-stabilized membrane protein aggregates from sickle erythrocytes: major site of autologous antibody binding. J Biol Chem. 1988;263:13766–13773. [PubMed] [Google Scholar]
- 36.Campanella ME, Chu H, Wandersee NJ, Peters LL, Mohandas N, Gilligan DM, et al. Characterization of glycolytic enzyme interactions with murine erythrocyte membranes in wild-type and membrane protein knockout mice. Blood. 2008;112:3900–3906. doi: 10.1182/blood-2008-03-146159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Facer CA. Erythrocytes carrying mutations in spectrin and protein 4.1 show differing sensitivities to invasion by Plasmodium falciparum. Parasitol Res. 1995;81:52–57. doi: 10.1007/BF00932417. [DOI] [PubMed] [Google Scholar]
- 38.Gallagher PG. Hereditary elliptocytosis: spectrin and protein 4.1R. Semin Hematol. 2004;41:142–164. doi: 10.1053/j.seminhematol.2004.01.003. [DOI] [PubMed] [Google Scholar]
- 39.Bunyaratvej A, Butthep P, Kaewkettong P, Yuthavong Y. Malaria protection in hereditary ovalocytosis: relation to red cell deformability, red cell parameters and degree of ovalocytosis. Southeast Asian J Trop Med Public Health. 1997;28(Suppl 3):38–42. [PubMed] [Google Scholar]
- 40.Mohandas N, Lie-Injo LE, Friedman M, Mak JW. Rigid membranes of Malayan ovalocytes: a likely genetic barrier against malaria. Blood. 1984;63:1385–1392. [PubMed] [Google Scholar]
- 41.Ziegler HL, Staerk D, Christensen J, Hviid L, Hagerstrand H, Jaroszewski JW. In vitro Plasmodium falciparum drug sensitivity assay: inhibition of parasite growth by incorporation of stomatocytogenic amphiphiles into the erythrocyte membrane. Antimicrob Agents Chemother. 2002;46:1441–1446. doi: 10.1128/AAC.46.5.1441-1446.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Da Silva E, Foley M, Dluzewski AR, Murray LJ, Anders RF, Tilley L. The Plasmodium falciparum protein RESA interacts with the erythrocyte cytoskeleton and modifies erythrocyte thermal stability. Mol Biochem Parasitol. 1994;66:59–69. doi: 10.1016/0166-6851(94)90036-1. [DOI] [PubMed] [Google Scholar]
- 43.Pei X, Guo X, Coppel R, Bhattacharjee S, Haldar K, Gratzer W, et al. The ring-infected erythrocyte surface antigen (RESA) of Plasmodium falciparum stabilizes spectrin tetramers and suppresses further invasion. Blood. 2007;110:1036–1042. doi: 10.1182/blood-2007-02-076919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mills JP, Diez-Silva M, Quinn DJ, Dao M, Lang MJ, Tan KS, et al. Effect of plasmodial RESA protein on deformability of human red blood cells harboring Plasmodium falciparum. Proc Natl Acad Sci U S A. 2007;104:9213–9217. doi: 10.1073/pnas.0703433104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pantaleo A, Giribaldi G, Mannu F, Arese P, Turrini F. Naturally occurring anti-band 3 antibodies and red blood cell removal under physiological and pathological conditions. Autoimmun Rev. 2008;7:457–462. doi: 10.1016/j.autrev.2008.03.017. [DOI] [PubMed] [Google Scholar]
- 46.Wajcman H, Galacteros F. Glucose 6-phosphate dehydrogenase deficiency: a protection against malaria and a risk for hemolytic accidents. C R Biol. 2004;327:711–720. doi: 10.1016/j.crvi.2004.07.010. [DOI] [PubMed] [Google Scholar]
- 47.Brockelman CR, Wongsattayanont B, Tanariya P, Fucharoen S. Thalassemic erythrocytes inhibit in vitro growth of Plasmodium falciparum. J Clin Microbiol. 1987;25:56–60. doi: 10.1128/jcm.25.1.56-60.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ginsburg H. Redox metabolism in malaria: from genes, through biochemistry and pathology, to drugs. Redox Rep. 2003;8:231–233. doi: 10.1179/135100003225002808. [DOI] [PubMed] [Google Scholar]
- 49.Willardson BM, Thevenin BJ, Harrison ML, Kuster WM, Benson MD, Low PS. Localization of the ankyrin-binding site on erythrocyte membrane protein, band 3. J Biol Chem. 1989;264:15893–15899. [PubMed] [Google Scholar]
- 50.Turrini F, Arese P, Yuan J, Low PS. Clustering of integral membrane proteins of the human erythrocyte membrane stimulates autologous IgG binding, complement deposition, and phagocytosis. J Biol Chem. 1991;266:23611–23617. [PubMed] [Google Scholar]
- 51.Tahar R, Vivas L, Basco L, Thompson E, Ibrahim H, Boyer J, Nepveu F. Indolone-N-oxide derivatives: in vitro activity against fresh clinical isolates of Plasmodium falciparum, stage specificity and in vitro interactions with established antimalarial drugs. J Antimicrob Chemother. 2011;66:2566–2572. doi: 10.1093/jac/dkr320. [DOI] [PubMed] [Google Scholar]
- 52.Winterbourn CC, Hampton MB. Thiol chemistry and specificity in redox signaling. Free Radic Biol Med. 2008;45:549–561. doi: 10.1016/j.freeradbiomed.2008.05.004. [DOI] [PubMed] [Google Scholar]
- 53.Forman HJ. Signal transduction and reactive species. Free Radic Biol Med. 2009;47:1237–1238. doi: 10.1016/j.freeradbiomed.2009.09.002. [DOI] [PubMed] [Google Scholar]
- 54.Bunney JE, Hooper M. Isatogens, Part VII Polarographic reduction of isatogens. J Chem Soc (B) 1970;7:1239–1241. [Google Scholar]
- 55.Winterbourn CC. Reaction of superoxide with hemoglobin. CRC Handbook of Methods for Oxygen Radical Research. 1985:137–141. [Google Scholar]