

Abstract
Antifreeze proteins (AFPs) are found in organisms ranging from fish to bacteria, where they serve different functions to facilitate survival of their host. AFPs that protect freeze-intolerant fish and insects from internal ice growth bind to ice using a regular array of well-conserved residues/motifs. Less is known about the role of AFPs in freeze-tolerant species, which might be to beneficially alter the structure of ice in or around the host. Here we report the 0.95-Å high-resolution crystal structure of a 223-residue secreted AFP from the snow mold fungus Typhula ishikariensis. Its main structural element is an irregular β-helix with six loops of 18 or more residues that lies alongside an α-helix. β-Helices have independently evolved as AFPs on several occasions and seem ideally structured to bind to several planes of ice, including the basal plane. A novelty of the β-helical fold is the nonsequential arrangement of loops that places the N- and C termini inside the solenoid of β-helical coils. The ice-binding site (IBS), which could not be predicted from sequence or structure, was located by site-directed mutagenesis to the flattest surface of the protein. It is remarkable for its lack of regularity and its poor conservation in homologs from psychrophilic diatoms and bacteria and other fungi.
Keywords: X-ray crystallography, ice growth inhibition, thermal hysteresis
Various species of fish, insects, plants, fungi, and bacteria that inhabit subzero environments produce antifreeze proteins (AFPs) to modulate the effect of ice on their lives (1–3). In fish and those insects that must avoid freezing, the AFPs adsorb to the surface of seed ice crystals to stop their growth (4). The resulting microcurvature of the ice front around the surface-bound AFPs depresses the nonequilibrium freezing temperature through the Gibbs–Thomson effect (5), and slightly elevates the melting temperature (6). The ability to lower the former temperature below the latter of an ice crystal created in an AFP-containing fluid is defined as thermal hysteresis (TH) and reflects the nature and concentration of the AFP. The thermal hysteresis activity of AFPs in fishes can be sufficient to depress their freezing point to below that of seawater (−1.9 °C). The AFPs in insects are more potent and prevent freezing down to −30 °C with the help of other agents like polyalcohols (2).
The unit structure of an ice crystal is hexagonal (Ih) at one atmosphere pressure, which is defined by three equivalent a-axes (a1–a3) perpendicular to the c-axis (7). Surface adsorption of an AFP is specific for one or more planes of ice, which become crystal facets because ice growth on those surfaces is inhibited (4). The ice crystal morphology produced by moderately active fish AFPs is typically a hexagonal bipyramid. When the freezing hysteresis is exceeded the crystal grows rapidly along the c-axis in a needle shape. The “hyperactive” AFPs produced by insects have higher TH activities because they can also bind to the basal plane of ice to block c-axial growth (8).
Crystal structures have been solved for six different types of AFPs from freeze-avoiding organisms: three from fish (9–11), and three from insects (12–14). They have remarkably diverse protein folds that include an α-helix (9), globular proteins of mixed secondary structure (10, 11), β-solenoids (12, 13), and polyproline type II coils (14). Several of these AFPs have had their ice-binding sites (IBSs) delineated by site-directed mutagenesis (15, 16). Although they come from different structures, the ice-binding sites have key features in common. They tend to be relatively flat and somewhat hydrophobic, and they cover an extensive area on the protein. For the globular type III AFP, small adjoining flat surfaces may combine to form a “compound” ice-binding site (17). The side chains of the ice-binding residues are fixed in position, and it is thought that they organize waters into an ice-like pattern (18, 19) that matches and merges with the quasiliquid layer at the ice water interface such that the AFPs bind by freezing onto the ice (20). A solution NMR study revealed that the organized waters were highly mobile and exchanged with bulk waters on a subnanosecond timescale (21).
In contrast to this wealth of information, very little is known about the structure and function of AFPs from organisms that tolerate freezing. The structure of an “AFP” from an Antarctic bacterium has been recently solved (22). However, this protein makes up one domain of a gigantic protein that appears to be involved in cell adhesion. Also solved recently was the structure of a plant AFP from a freeze-tolerant grass (23). This AFP has weak TH activity but is very effective at inhibiting the recrystallization of ice (24). It has a β-solenoid structure, but unlike those from insects the ice-binding residues are less regular and can take up two or more rotameric conformations. To learn more about this category of AFPs we have solved the structure of one from a cold-adapted fungus.
The snow mold fungus, Typhula ishikariensis, attaches onto dormant plants and secretes its AFP (TisAFP) toward the extracellular space under snow cover (25). Purified TisAFP is a mixture of 223-residue polypeptide isoforms. An isoform denoted TisAFP8 showed high TH activity (2.1 °C) and a dendritic ice growth pattern that suggested it also binds to the basal planes of ice, as do hyperactive AFPs. Here, we have determined the X-ray crystal structure of another isoform, TisAFP6, to a resolution of 0.95 Å. It forms a unique right-handed β-helix with a triangular cross-section where the N- and C-terminal β-helical loops are adjacent to each other within the β-solenoid and the capping structures are formed from internal loops. Mutagenesis experiments suggest that the ice-binding site of TisAFP6 lies on the flattest β-helical surface. Surprisingly, this surface lacks the regular arrays of side-chain OH groups seen on β-helical hyperactive AFPs. Also, this ice-binding site is not well conserved among the microbial AFP homologs in bacteria, algae, and diatoms.
Results and Discussion
TisAFP6 forms a 52-Å-long semipear-shaped structure, whose principle constituent is a six-loop right-handed β-helix having a triangular cross-section (Fig. 1). Surprisingly, the β1 loop or coil originating from near the N terminus, lies adjacent to the β6 loop from the C terminus. In this way, the terminal loops are side by side within the parallel β-helix as illustrated by the spectrum colors showing blue next to red (Fig. 1 A and B). Thus, the six β-helical loops are aligned in an irregular order, β1–β6–β5–β4–β3–β2, which is unusual for a β-helix (26). In addition, TisAFP6 contains a 20-residue α-helix from T76 to A95 that lies alongside and parallel to the β-helix axis.
Fig. 1.

Structure of TisAFP6. (A) Ribbon presentation of TisAFP6 structure in spectral color gradation from blue (N terminus) to red (C terminus). The six loops of the right-handed β-helix are numbered β1 to β6 to emphasize their irregular order, β1–β6–β5–β4–β3–β2, in the helix due to the unique positioning of the C-terminal loop, β6, adjacent to the N-terminal loop, β1. The 22-residue-long α-helix lying parallel to the β-helix axis is colored green. (B) Schematic representation of the structure of TisAFP6 illustrating how each loop comprises three short β-strands twisted at an angle of 60° to form the corners of the β-helix. The structure is colored as in A. (C) Triangular cross-section of a central portion of TisAFP6 comprising loops β1–β6–β5–β4 showing the packing of hydrophobic residues (yellow) in the core and between the α- and β-helices. Amino acid sequence of TisAFP6 is shown in Fig. 5.
The six β-helical loops are of different lengths ranging from 18 to 27 residues, with the number of residues constructing each loop given in parentheses as follows: β1, V15–G32 (18); β6, R206–K223 (18); β5, T188–G205 (18); β4, K167–G187 (21); β3, D140–A166 (27); β2, P117–T139 (23). The minimum length of the loop appears to be 18 residues, as seen in the three consecutive loops (β1, β6, and β5) at the top of the molecule. The bottom loops (β4, β3, and β2) of the β-helix have 3- to 9-residue insertions that extend outward on the same side to form the pear-shaped part of the protein. The three short β-strands in each loop are linked at the corners by residues in a 310-helix arrangement.
The β-helical fold of TisAFP6 is stabilized by the following four elements: (i) The hydrogen-bonding network between peptide CO and NH groups down the length of the β-helix parallel to the helical axis that maintains ∼4.75-Å spacing between the loops. (ii) The internal cap structures that mask the edge effects of the β-strands in the terminal loops β1 and β2 and help cover the hydrophobic core. The capping structures normally reside at the N- and C termini of a β-helical protein, but in TisAFP6 they are formed internally between β1 and the α-helix (S47–S61) and between the α-helix and β2 (D101–T107). (iii) A hydrophobic core that is formed from ∼48 aliphatic and aromatic residues without the support of disulfide bonds (Fig. 1C). (iv) The long α-helix that runs the length of the β-helix and contacts most of the loops on the a-face (Fig. 2A). There are 12 hydrophobic residues at their interface that support the intimate association between these two helices.
Fig. 2.

Mapping the interactions between TisAFP6 and ice. (A) Locations of mutated T82, N178, and V221 in the center of the a–c faces, respectively. (B) Plot of TH vs. protein concentration for the three mutants and wild-type TisAFP6 tagged with GST. (C) FIPA pattern obtained with fluorescently tagged TisAFP6–V221C mutant (without GST) on a single ice-crystal hemisphere mounted with the c-axis along the ice finger. (D) Interpretive illustration of C, indicating the three a-axes relative to the six equivalent prism planes bound by TisAFP6. The basal plane is central underneath the hexagonal pit used to deduce the direction of the a-axes (E) FIPA pattern obtained with TisAFP6 from a different angle. (F) Interpretive illustration of E showing that the six equivalent bound prism planes are distinct from both the primary (1°) and secondary (2°) prism planes.
To our knowledge, the structure of TisAFP6 and that of a recently determined homolog from an Arctic yeast Leucosporidium sp. (27) are the only β-helices that deviate from sequential stacking of β-helical loops. A carbonic anhydrase from Methanosarcina thermophila [Protein Data Bank (PDB) ID code = 1THJ] (28) bears a resemblance in having six 18-residue triangular loops with an α-helix alongside. However, the β-helical core of 1THJ is left-handed, whereas that of TisAFP6 is right-handed. The helical structure of TisAFP6 is surprisingly irregular in comparison with the other β-helical AFPs. The program HHrepID (29) was used to search the amino acid sequence of TisAFP6 for repeats with the propensity to make a helical structure, but without success. This may partly reflect some differences in helix loop length of TisAFP6, but it is also due to the obscurity and variation of the ice-binding motifs. In contrast, TmAFP (12), sbwAFP (13), and MpAFP (22) are built up from definitive 12-, 15-, and 19-residue tandem repeats, respectively, with consensus sequences that contain regularly arrayed TXT or TXN ice-binding motifs. Neither the tandem repeats nor the TXT motif is present, however, in TisAFP6 and the other TisAFP isoforms.
The IBS of TisAFP6 was probed by steric mutations in which a tyrosyl residue replaced a short-chain amino acid on different surfaces of the protein. This same strategy was successfully used to identify the IBS of TmAFP (15), MpAFP (30), and LpAFP (31). The TH activity of mutants T82Y, N178Y, and V221Y, where the substitutions appeared in the middle of the a-, b- and c-faces, respectively (Fig. 2A), was compared with that of wild-type TisAFP6 (Fig. 2B). The N terminus of each recombinant protein was tagged with GST. The mutations have no obvious effect on the protein fold of the samples tagged with GST (SI Text). The GST has no TH activity by itself. The GST tagging lowered the absolute value of TH of the four samples (Fig. 2B) to well below the 2.1 °C value obtained with wild-type TisAFP8 (25), whereas the data clearly showed that the N178Y mutation caused a significant (60%) loss in TH activity. In contrast, T82Y and V221Y lost only 15% of wild-type TH activity. This implicates the b-face in ice binding.
The planes of ice bound by TisAFP6 were determined using fluorescence-based ice plane affinity (FIPA) analysis (17). V221 on the c-face was selected as the attachment point for a fluorescent tag following mutation of the valine to cysteine (V221C). In the absence of a GST tag, fluorescently labeled TisAFP6 illuminated the basal plane and six equivalent prism planes of the ice hemisphere (Fig. 2 C and E). On the basis of the orientation of ice pitting during sublimation, the six equivalent prism planes are neither the primary nor the secondary prism planes (Fig. 2 D and F) as they are not aligned to the a1–a3 axes nor to the a1–a2 vector and its two equivalents. All hyperactive AFPs show binding to the basal plane of ice, but the pattern of prism plane binding shown by TisAFP6 is unique among all of the moderate and hyperactive AFPs tested so far. The FIPA pattern produced by TisAFP6 had many slits and discontinuities in the fluorescence (Fig. 2 C and E), which is another distinction from the smoother, more uniform patterns seen with other AFPs (22).
When TisAFP6 is represented in space-filling format (Fig. 3 A and B) the only extensive flat surface to the protein is the b-face. Flatness is one of the key attributes for an IBS (32). However, the regularity of the residues on that surface is poor in comparison with the hyperactive β-helical AFPs from insects, which have two rigid parallel arrays of Thr residues on their IBS (Fig. 3C). Just recently, the structure of an AFP from grass that is active in ice recrystallization inhibition was solved (23). It has an IBS (Fig. 3 D and F) that is less regular than the insect AFPs and more like the b-face of TisAFP6.
Fig. 3.

Comparison of the TisAFP6 IBS with those of sbwAFP and LpAFP. (A) Front view of the b-face of TisAFP6 showing the putative IBS bound by arrows. Aliphatic side chains, hydroxyl group, and nitrogen atoms are colored yellow, red, and blue, respectively. (B) IBS viewed side on to show its flatness and the pear-shaped other side of TisAFP6. (C) Space-filling view of sbwAFP showing the regularity and flatness of its IBS resulting from the alignment of six TXT ice-binding motifs. Color use is the same as in A. (D) IBS of LpAFP from the grass Lolium perenne. The AFP is a left-handed β-roll with eight 14- to 15-residue coils. (E) Alignment of the β-strand sequences of β1–β6 loops of TisAFP6 that comprise the IBS. Large and small letters indicate the residues having their side chains directed toward the outside and inside, respectively. (F) Alignment of the selected sequences of β1–β8 loops of LpAFP to highlight the residues constructing its putative IBS.
The ice-binding β-sheet of TisAFP6 is assembled from a set of 4- to 5-residue peptides located in the middle of five parallel β-strands; T20-G-V-S-T-V25 (β1), T211-S-V-A-L-Q216 (β6), T193-A-V-T-F-K198 (β5), Q175-A-V-N-T-E180 (β4), and G148-T-L-D-V152 (β3) (Fig. 3E and 4A). Of 29 residues on this face, 22 have hydrophobic side chains. As in spruce budworm AFP (sbwAFP) (Fig. 4B), the backbone conformations of the above segments are identical and superimposable on each other. The first residues of the five hexapeptides (T,T,T,G,G) (Fig. 3E) facilitate the formation of a corner of the triangular molecule. The Thr side chains are directed outside of the β-strands and are stacked with each other in line. The second (G,S,A,A,T), fourth (S,A,T,N,D), and sixth (V,Q,K,E) residues also present their side chains toward the solvent and are stacked to form three straight lines parallel to the helical axis. The third (V,V,V,V,L) and fifth (T,L,F,I,V) residues have their side chains directed toward the inside of the β-helix to form part of the interior hydrophobic core. As a consequence, each β-strand constructs an out-out-in-out-in-out pattern of side chains with respect to the core of the protein, which repeats itself in each loop of the β-helix. This pattern compares with the β-strands of sbwAFP that comprise its IBS (Fig. 4B), whose side chains form an in-out-in-out-out pattern (18).
Fig. 4.

Comparison of IBS and surface waters between TisAFP6 and sbwAFP. (A) N- to C-terminal view down the six parallel β-strands constructing the IBS of TisAFP6 characterized by a repetitive out-out-in-out-in-out side-chain motif. Residue assignments are only indicated for the β1-strand. O atoms are red and N atoms are blue. (B) N- to C-terminal view down the six parallel β-strands constructing the IBS of sbwAFP characterized by a repetitive out-out-in-out-in-out side-chain motif. Residue assignments are only indicated for the β1-strand. (C) Positions of 56 waters contacting the crystal structure of TisAFP6; 18 are located in three troughs created between the linearly aligned first, second, fourth, and sixth residues of the IBS β-strands. (D) Positions of waters determined by MD analysis. Structure shown is that at the start of the 12-ns MD simulation performed at 273 K. A total of 604 frames captured during the 12-ns MD simulation were aligned to the initial structure via the α-carbon of each amino acid residue. The volume map tool of VMD was used to calculate the density for just the oxygen atoms of the water molecules. The resulting water density map is colored green and is contoured at 0.9 σ. (E) Regularly aligned and immobilized waters located in a trough on the IBS of sbwAFP.
The interstrand spacing on the ice-binding β-sheet of TisAFP6 is 4.75 Å on average, similar to most β-helical proteins, which includes several AFPs. This separates the side-chain groups along each first, second, fourth, and sixth line to make a close match to the 4.5-Å repeats of oxygen atoms seen on both the basal and prism planes of ice. The distance between the first, second, fourth, and sixth lines of the TisAFP6 is uniformly 6.7 Å when measured using their Cα atoms, but varies between 4.35 and 7.28 Å when using the side-chain atoms of the outward projecting amino acids. For sbwAFP, the distance between the linearly aligned hydroxyl groups (or methyl groups) of the TXT motif is 7.35 Å, which matches O atom spacing on the (10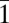 0) prism plane and the 7.8-Å spacing on the (0001) basal plane as well (13). The dimensions of the predicted IBS of TisAFP6 are roughly 17 × 18 Å, with a total solvent accessible area of about 921 Å2, which is comparable to 1,180 Å2 of the IBS-containing face of TmAFP (33). What is especially striking about all of the β-helices/solenoids (26) used as AFPs, including TisAFP6, is the absence of twist around the central axis. This ensures that the ice-binding β-sheet is flat for contacting an ice plane. These comparisons suggest that the TisAFP6 b-face has an ability to bind to two or more ice crystal surfaces, including the basal planes, in a manner similar to other hyperactive AFPs.
0) prism plane and the 7.8-Å spacing on the (0001) basal plane as well (13). The dimensions of the predicted IBS of TisAFP6 are roughly 17 × 18 Å, with a total solvent accessible area of about 921 Å2, which is comparable to 1,180 Å2 of the IBS-containing face of TmAFP (33). What is especially striking about all of the β-helices/solenoids (26) used as AFPs, including TisAFP6, is the absence of twist around the central axis. This ensures that the ice-binding β-sheet is flat for contacting an ice plane. These comparisons suggest that the TisAFP6 b-face has an ability to bind to two or more ice crystal surfaces, including the basal planes, in a manner similar to other hyperactive AFPs.
Along with the repetitive structural motifs of hyperactive AFPs, regularly aligned surface waters have been observed in “trough-like” regions on their ice-binding sites. In sbwAFP for example, aligned surface waters are located in a trough created beside the ranks of threonines of the TXT motif (Fig. 4E), and the spacing between these waters closely matches to waters on both the prism and basal planes of an ice crystal (13). The number and extent of these prominently positioned waters has been limited in insect AFPs because their ice-binding sites, being flat and relatively hydrophobic, form crystal contacts in the unit cell. Fortunately, the hyperactive 34-kDa MpAFP crystallized in such a way as to expose to solvent the IBS of one and a portion of another of the four molecules in the unit cell. On these two surfaces a large number of waters were arranged in an ice-like pattern along the ice-binding side of the β-helix (22). These waters formed cages around hydrophobic groups that were in turn anchored by hydrogen bonding to side-chain and backbone hydrophilic group. The ice-like waters are thought to bind the AFP to ice by an “anchored clathrate mechanism,” which is in line with the hypothesis of Nutt and Smith (20). For TisAFP6, a total of 56 crystallographic waters were found on the predicted IBS, which is completely exposed to solvent within the crystal. Among them are 18 waters located in the three linear troughs made by the residues stacked in the first, second, fourth, and sixth lines (Fig. 4C). In contrast to the insect AFPs, the waters are not perfectly aligned in any of the three troughs, probably due to the structural imperfections resulting from the nonrepetitive nature of the sequence. However, those waters present in the trough between the second and fourth lines seem to be particularly stable, because many of them (cyans in Fig. 4C) are observed in the same positions after molecular dynamics calculation (greens in Fig. 4D). Of the 18 waters, 11 are separated by the interstrand distance (4.75 Å) that matches the distance between oxygen atoms on several ice planes including the (0001) basal plane. The location and abundance of waters on the B-face of TisAFP6 are consistent with it being the IBS and operating by the anchored clathrate mechanism.
To date, amino acid sequences have been determined for at least eight homologs of this ∼23-kDa AFP discovered in fungi (T. ishikariensis (TisAFP6), Lentinula edodes, Flammulina populicola, and Leucosporidium sp AY30); diatoms (Navicula glaciei and Fragilariopsis cylindrus); and bacteria (Colwellia sp. SLW05 and Cytophaga hutchinsonii) (Fig. 5). The present study provides the first tertiary structural information for TisAFP6. Significantly, TisAFP6 and the other seven microbial AFPs share 45–55% sequence identity. An alignment of all homologs shows that some of the most highly conserved residues are glycines and prolines that are involved in turns or the boundaries of secondary structural elements. The next most highly conserved residues are those that form the hydrophobic core. Of 48 hydrophobic residues that contribute to the core of the TisAFP6 structure, 50–60% are identical and 83–95% are similar in the homologs. This analysis suggests that all eight proteins have the same fundamental fold. What is surprising, however, is the amount of variation shown in the residues that make up the ice-binding site (indicated by stars above the alignment in Fig. 5). In previous comparisons of AFP paralogs and orthologs, it has been possible to identify the ice-binding site by its extreme conservation (34). In this example of a fungal AFP structure none of the outer facing surfaces are well conserved and it would not have been possible to identify the IBS of TisAFP6 without using site-directed mutagenesis.
Fig. 5.

Amino acid sequence alignment of TisAFP6 homologs identified in various microorganisms: Tis, Typhula ishikariensis; Led, Lentinula edodes; Fpo, Flammulina populicola; Leu, Leucosporidium sp AY30; Ngl, Navicula glaciei; Fcy, Fragilariopsis cylindrus; Col, Colwellia sp. SLW05; and Chu, Cytophaga hutchinsonii. The positions of α-helices and β-strands in the TisAFP6 structure are indicated below the alignment by cylinders and arrows, respectively. Secondary structure assignments were performed with DSSP (40). Residues conserved between seven or eight species are indicated by a black background, those between five or six species by a blue background, and between four species by an orange background. The asterisks above the alignment show the residues on the putative IBS of TisAFP6. Blue asterisks signify outward pointing residues and orange asterisks signify outward pointing residues. On the Right, numbers indicate the residue number of each polypeptide from the start of the mature protein. Numbers in parentheses indicate the length insertions. Elsewhere, dashes indicate residues missing from one or more homologs.
The appearance of homologs in bacteria, fungi, and diatoms has been attributed to lateral gene transfer (35). The role of these secreted proteins in microorganisms has not yet been established, but it is likely that they modify the texture of ice in the immediate vicinity of their host (36). This is a different function from that of freeze avoidance in fish and insects, and more akin to the role of ice recrystallization inhibition that AFPs are thought to serve in freeze tolerance. The structure of an AFP from a freeze-tolerant grass has just been solved (23). Like TisAFP6, its IBS lacks repetition and regularity. The paradigm of having a highly regular, rigid ice-binding site might not apply to all types and functions of AFPs.
Materials and Methods
Crystallization and X-Ray Structure Analysis of TisAFP6.
Native TisAFP was prepared as described in ref. 37 from the snow mold fungus Typhula ishikariensis strain BRB-1 collected on Spitsbergen Island, Svalbard, Norway. The hanging drop vapor diffusion method (38) was applied to a 10 mg/mL solution of TisAFP. A plate-shaped crystal with dimensions of 0.8 × 0.2 × 0.1 mm was obtained from 100 mM Bis-Tris buffer pH 6.0–6.2, 1.0 M ammonium sulfate, and 100 mM sodium chloride. It belonged to the triclinic space group P1, with unit-cell parameters a = 38.89, b = 41.63, c = 70.25 Å, α = 81.53, β = 83.13, and γ = 62.43 °. X-ray diffraction data were collected to 0.95-Å resolution at −173 °C on BL44B2 at the SPring-8. The electron density map showed one-to-one correspondence with the amino acid sequence of TisAFP6, thereby showing that only TisAFP6 had only been crystallized out of the the mixture of isoforms (25). The crystallographic R factor and free R factor of the final structure were 0.112 and 0.129, respectively. The X-ray data collection and structural refinement of TisAFP6 are summarized in SI Text.
Expression and Site-Directed Mutagenesis of TisAFP6 and Its Mutants.
The cDNAs encoding TisAFP6 and its tyrosine mutants, T82Y, N178Y, and V221Y, were cloned into the expression vector pGEX6P-2 (GE Healthcare) in Escherichia coli BL21 (DE3) Rosetta2 (Novagen) to express TisAFP6 with an N-terminal GST tag. The cultivated cells in Luria–Bertani medium were harvested by centrifugation, washed by resuspension in PBS, and disrupted by sonication. The lysate supernatant was applied into Glutathione Sepharose High-Performance column (GE Healthcare), and the proteins were eluted with 50 mM Tris-HCl pH 8.0 containing 10 mM reduced glutathione.
For preparation of the V221C mutant of TisAFP6, the PCR product was ligated into plasmid pPICZα (Invitrogen) at the corresponding sites immediately downstream of the yeast α-mating factor signal sequence without any affinity tag. The cloned plasmid was linearized with restriction enzyme, transformed into Pichia pastoris X-33 strain, and cultured in a buffered complex glycerol medium at 28 °C. Cells were harvested by centrifugation and resuspended in a buffered complex methanol medium. The supernatant of the culture was dialyzed against 25 mM glycine-HCl buffer pH 3.0, followed by application to an Econo-Pac High S column. The eluted TisAFP6 V221C mutant was then loaded onto an Econo-Pac High Q column, and the flow-through fraction showing TH activities was used for FIPA analysis.
For fluorescence labeling, 1 mL of TisAFP6 V221C mutant solution (1.0 mg/mL) in PBS was incubated with Alexa Fluor 532 C5-maleimide (Invitrogen), which reacts with the SH group of the cysteine. The fluorescence-labeled V221C mutant was dialyzed against distilled water to remove unreacted reagents before the FIPA experiments.
FIPA Analysis and TH Activity Measurements.
FIPA analysis was performed as previously described (17). A single ice crystal hemisphere was immersed and grown in 0.025 mg/mL of fluorescently labeled TisAFP6–V221C. The labeled hemisphere was illuminated under UV light and photographed in a cold room set to −1 °C. TH activity was measured for recombinant samples of TisAFP6 and its three tyrosine mutants (T82Y, N178Y, and V221Y), all of which had a GST tag, using a custom-made photomicroscope system equipped with a temperature-controlled stage and a color-video 3CCD camera (Sony). The activity evaluation was performed using a cooling rate of 0.20 °C/min.
Molecular Dynamics Analysis.
The optimal locations of waters around molecule B of TisAFP6 were determined by molecular dynamics (MD) simulations using the program Gromacs v. 4.5.3 (39). The TisAFP6 crystal structure was solvated in a box containing 7,172 water molecules and two Na+ ions to offset the charge of the protein. Energy minimization and a 50-ps position-restrained MD simulation were initially performed to relax the solvent around the protein before the 12-ns MD simulation. Berendsen temperature and pressure coupling were applied, along with the GROMOS96 43a1 force field and SPC water model.
Supplementary Material
Acknowledgments
The authors thank Dr. Andrew Kajava for performing the program HHrepID to search tandem repeats in the sequence of TisAFP isoforms, Dr. Robert Campbell for molecular dynamics analysis of TisAFP6 and its bound waters, and Nan Xiao for valuable advice. This work was supported by Grant-in-Aid 23310171 and 20570118 for Scientific Research from the Japan Society for the Promotion of Science (JSPS). P.L.D. holds a Canada Research Chair in Protein Engineering and a grant from the Canadian Institutes for Health Research. C.P.G. was supported on an exchange visit to National Institute of Advanced Industrial Science and Technology in Sapporo, Japan by a JSPS summer fellowship.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Data deposition: The atomic coordinates reported in this paper have been deposited in the Research Collaboratory for Structural Bioinformatics Protein Data Bank, http://www.rcsb.org/pdb (RCSB PDB ID code 3VN3).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1121607109/-/DCSupplemental.
References
- 1.Fletcher GL, Hew CL, Davies PL. Antifreeze proteins of teleost fishes. Annu Rev Physiol. 2001;63:359–390. doi: 10.1146/annurev.physiol.63.1.359. [DOI] [PubMed] [Google Scholar]
- 2.Duman JG, Bennett V, Sformo T, Hochstrasser R, Barnes BM. Antifreeze proteins in Alaskan insects and spiders. J Insect Physiol. 2004;50:259–266. doi: 10.1016/j.jinsphys.2003.12.003. [DOI] [PubMed] [Google Scholar]
- 3.Duman JG, Olsen TM. Thermal hysteresis protein activity in bacteria, fungi, and phylogenetically diverse plants. Cryobiology. 1993;30:322–328. [Google Scholar]
- 4.Raymond JA, DeVries AL. Adsorption inhibition as a mechanism of freezing resistance in polar fishes. Proc Natl Acad Sci USA. 1977;74:2589–2593. doi: 10.1073/pnas.74.6.2589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Knight CA, Cheng CC, DeVries AL. Adsorption of α-helical antifreeze peptides on specific ice crystal surface planes. Biophys J. 1991;59:409–418. doi: 10.1016/S0006-3495(91)82234-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Celik Y, et al. Superheating of ice crystals in antifreeze protein solutions. Proc Natl Acad Sci USA. 2010;107:5423–5428. doi: 10.1073/pnas.0909456107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hobbs PV. Ice Physics. London: Oxford Univ Press; 1974. pp. 18–39. [Google Scholar]
- 8.Scotter AJ, et al. The basis for hyperactivity of antifreeze proteins. Cryobiology. 2006;53:229–239. doi: 10.1016/j.cryobiol.2006.06.006. [DOI] [PubMed] [Google Scholar]
- 9.Sicheri F, Yang DSC. Ice-binding structure and mechanism of an antifreeze protein from winter flounder. Nature. 1995;375:427–431. doi: 10.1038/375427a0. [DOI] [PubMed] [Google Scholar]
- 10.Nishimiya Y, et al. Crystal structure and mutational analysis of Ca2+-independent type II antifreeze protein from longsnout poacher, Brachyopsis rostratus. J Mol Biol. 2008;382:734–746. doi: 10.1016/j.jmb.2008.07.042. [DOI] [PubMed] [Google Scholar]
- 11.Antson AA, et al. Understanding the mechanism of ice binding by type III antifreeze proteins. J Mol Biol. 2001;305:875–889. doi: 10.1006/jmbi.2000.4336. [DOI] [PubMed] [Google Scholar]
- 12.Liou YC, Tocilj A, Davies PL, Jia Z. Mimicry of ice structure by surface hydroxyls and water of a β-helix antifreeze protein. Nature. 2000;406:322–324. doi: 10.1038/35018604. [DOI] [PubMed] [Google Scholar]
- 13.Graether SP, et al. β-helix structure and ice-binding properties of a hyperactive antifreeze protein from an insect. Nature. 2000;406:325–328. doi: 10.1038/35018610. [DOI] [PubMed] [Google Scholar]
- 14.Pentelute BL, et al. X-ray structure of snow flea antifreeze protein determined by racemic crystallization of synthetic protein enantiomers. J Am Chem Soc. 2008;130:9695–9701. doi: 10.1021/ja8013538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Marshall CB, Daley ME, Graham LA, Sykes BD, Davies PL. Identification of the ice-binding face of antifreeze protein from Tenebrio molitor. FEBS Lett. 2002;529:261–267. doi: 10.1016/s0014-5793(02)03355-0. [DOI] [PubMed] [Google Scholar]
- 16.DeLuca CI, Davies PL, Ye Q, Jia Z. The effects of steric mutations on the structure of type III antifreeze protein and its interaction with ice. J Mol Biol. 1998;275:515–525. doi: 10.1006/jmbi.1997.1482. [DOI] [PubMed] [Google Scholar]
- 17.Garnham CP, et al. Compound ice-binding site of an antifreeze protein revealed by mutagenesis and fluorescent tagging. Biochemistry. 2010;49:9063–9071. doi: 10.1021/bi100516e. [DOI] [PubMed] [Google Scholar]
- 18.Leinala EK, Davies PL, Jia Z. Crystal structure of β-helical antifreeze protein points to a general ice binding model. Structure. 2002;10:619–627. doi: 10.1016/s0969-2126(02)00745-1. [DOI] [PubMed] [Google Scholar]
- 19.Howard EI, et al. Neutron structure of type-III antifreeze protein allows the reconstruction of AFP-ice interface. J Mol Recognit. 2011;24:724–732. doi: 10.1002/jmr.1130. [DOI] [PubMed] [Google Scholar]
- 20.Nutt DR, Smith JC. Dual function of the hydration layer around an antifreeze protein revealed by atomistic molecular dynamics simulations. J Am Chem Soc. 2008;130:13066–13073. doi: 10.1021/ja8034027. [DOI] [PubMed] [Google Scholar]
- 21.Modig K, Qvist J, Marshall CB, Davies PL, Halle B. High water mobility on the ice-binding surface of a hyperactive antifreeze protein. Phys Chem Chem Phys. 2010;12:10189–10197. doi: 10.1039/c002970j. [DOI] [PubMed] [Google Scholar]
- 22.Garnham CP, Campbell RL, Davies PL. Anchored clathrate waters bind antifreeze proteins to ice. Proc Natl Acad Sci USA. 2011;108:7363–7367. doi: 10.1073/pnas.1100429108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Middleton AJ, et al. Antifreeze protein from freeze-tolerant grass has a beta-roll fold with an irregularly structured ice-binding site. J Mol Biol. 2012;416:713–724. doi: 10.1016/j.jmb.2012.01.032. [DOI] [PubMed] [Google Scholar]
- 24.Knight CA, DeVries AL, Oolman LD. Fish antifreeze protein and the freezing and recrystallization of ice. Nature. 1984;308:295–296. doi: 10.1038/308295a0. [DOI] [PubMed] [Google Scholar]
- 25.Xiao N, et al. Comparison of functional properties of two fungal antifreeze proteins from Antarctomyces psychrotrophicus and Typhula ishikariensis. FEBS J. 2010;277:394–403. doi: 10.1111/j.1742-4658.2009.07490.x. [DOI] [PubMed] [Google Scholar]
- 26.Kajava AV, Steven AC. Beta-rolls, beta-helices, and other beta-solenoid proteins. Adv Protein Chem. 2006;73:55–96. doi: 10.1016/S0065-3233(06)73003-0. [DOI] [PubMed] [Google Scholar]
- 27.Lee JH, et al. Structural basis for the antifreeze activity of an ice-binding protein from an Arctic yeast. J Biol Chem. 2012;287:11460–11468. doi: 10.1074/jbc.M111.331835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kisker C, Schindelin H, Alber BE, Ferry JG, Rees DC. A left-hand beta-helix revealed by the crystal structure of a carbonic anhydrase from the archaeon Methanosarcina thermophila. EMBO J. 1996;15:2323–2330. [PMC free article] [PubMed] [Google Scholar]
- 29.Biegert A, Söding J. HHrepID: De novo protein repeat identification by probabilistic consistency. Bioinformatics. 2008;24:807–814. doi: 10.1093/bioinformatics/btn039. [DOI] [PubMed] [Google Scholar]
- 30.Garnham CP, et al. A Ca2+-dependent bacterial antifreeze protein domain has a novel beta-helical ice-binding fold. Biochem J. 2008;411:171–180. doi: 10.1042/BJ20071372. [DOI] [PubMed] [Google Scholar]
- 31.Middleton AJ, Brown AM, Davies PL, Walker VK. Identification of the ice-binding face of a plant antifreeze protein. FEBS Lett. 2009;583:815–819. doi: 10.1016/j.febslet.2009.01.035. [DOI] [PubMed] [Google Scholar]
- 32.Yang DSC, et al. Identification of the ice-binding surface on a type III antifreeze protein with a “flatness function” algorithm. Biophys J. 1998;74:2142–2151. doi: 10.1016/S0006-3495(98)77923-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Graether SP, Sykes BD. Cold survival in freeze-intolerant insects: The structure and function of β-helical antifreeze proteins. Eur J Biochem. 2004;271:3285–3296. doi: 10.1111/j.1432-1033.2004.04256.x. [DOI] [PubMed] [Google Scholar]
- 34.Baardsnes J, et al. New ice-binding face for type I antifreeze protein. FEBS Lett. 1999;463:87–91. doi: 10.1016/s0014-5793(99)01588-4. [DOI] [PubMed] [Google Scholar]
- 35.Raymond JA, Janech MG. Ice-binding proteins from enoki and shiitake mushrooms. Cryobiology. 2009;58:151–156. doi: 10.1016/j.cryobiol.2008.11.009. [DOI] [PubMed] [Google Scholar]
- 36.Raymond JA. Algal ice-binding proteins change the structure of sea ice. Proc Natl Acad Sci USA. 2011;108:3653–3658. doi: 10.1073/pnas.1106288108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hoshino T, Kiriaki M, Nakajima T. Novel thermal hysteresis proteins from low temperature basidiomycete, Coprinus psychromorbidus. Cryo Lett. 2003;24:135–142. [PubMed] [Google Scholar]
- 38.McPherson A. Current approaches to macromolecular crystallization. Eur J Biochem. 1990;189:1–23. doi: 10.1111/j.1432-1033.1990.tb15454.x. [DOI] [PubMed] [Google Scholar]
- 39.Van Der Spoel D, et al. GROMACS: Fast, flexible, and free. J Comput Chem. 2005;26:1701–1718. doi: 10.1002/jcc.20291. [DOI] [PubMed] [Google Scholar]
- 40.Kabsch W, Sander C. Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers. 1983;22:2577–2637. doi: 10.1002/bip.360221211. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.