

Abstract
The myotubularins are a large family of inositol polyphosphate 3-phosphatases that, despite having common substrates, subsume unique functions in cells that are disparate. The myotubularin family consists of 16 different proteins, 9 members of which possess catalytic activity, dephosphorylating phosphatidylinositol 3-phosphate [PtdIns(3)P] and phosphatidylinositol 3,5-bisphosphate [PtdIns(3,5)P2] at the D-3 position. Seven members are inactive because they lack the conserved cysteine residue in the CX5R motif required for activity. We studied a subfamily of homologous myotubularins, including myotubularin-related protein 6 (MTMR6), MTMR7, and MTMR8, all of which dimerize with the catalytically inactive MTMR9. Complex formation between the active myotubularins and MTMR9 increases their catalytic activity and alters their substrate specificity, wherein the MTMR6/R9 complex prefers PtdIns(3,5)P2 as substrate; the MTMR8/R9 complex prefers PtdIns(3)P. MTMR9 increased the enzymatic activity of MTMR6 toward PtdIns(3,5)P2 by over 30-fold, and enhanced the activity toward PtdIns(3)P by only 2-fold. In contrast, MTMR9 increased the activity of MTMR8 by 1.4-fold and 4-fold toward PtdIns(3,5)P2 and PtdIns(3)P, respectively. In cells, the MTMR6/R9 complex significantly increases the cellular levels of PtdIns(5)P, the product of PI(3,5)P2 dephosphorylation, whereas the MTMR8/R9 complex reduces cellular PtdIns(3)P levels. Consequentially, the MTMR6/R9 complex serves to inhibit stress-induced apoptosis and the MTMR8/R9 complex inhibits autophagy.
Inositol lipids play important roles in a variety of intracellular signaling pathways. In response to stimuli, the phosphoinositide profile is regulated by phospholipases, lipid kinases, and phosphatases. Understanding the roles of inositol signaling has expanded during the last decade and a number of these enzymes have been shown to cause diseases when mutated (1). The tumor-suppressor PTEN was discovered through positional cloning as being mutated in several types of cancer (2, 3). PTEN was subsequently shown to be a phosphatase, which dephosphorylates phosphatidylinositol 3,4,5-trisphosphate to generate phosphatidylinositol 4,5-bisphosphate, an activity that is lost in patients with PTEN mutations (4, 5). Mutations in the inositol polyphosphate 5-phosphatase OCRL cause the X-linked disorder Lowe syndrome, which is associated with mental retardation, blindness, and renal failure (6). Mutations in myotubularin cause myotubular myopathy (7), and mutations in myotubularin-related protein 2 (MTMR2) and MTMR13 cause a form of Charcot Marie Tooth disease type 4B, a demyelinating neurodegenerative disorder (8, 9).
The myotubularin family consists of 16 different proteins, 9 members of which possess catalytic activity (10, 11) and 7 members that are inactive. Myotubularin proteins are not redundant and have unique functions within cells by regulating a specific pool of dephosphorylating phosphatidylinositol 3-phosphate [PtdIns(3)P] and phosphatidylinositol 3,5-bisphosphate [PtdIns(3,5)P2] (12–15). Varying tissue expression and subcellular localization play a role in determining the unique function of myotubularin proteins (16–21). One mechanism that regulates the myotubularins is the formation of heterodimers between catalytically active and inactive proteins. The interaction between different myotubularin proteins has a significant effect on enzymatic activity. For example, the association of myotubularin (MTM1) with MTMR12 results in a threefold increase in the 3-phosphatase activity of MTM1, alters the subcellular localization of MTM1 from the plasma membrane to the cytosol, and attenuates the filopodia formation seen with MTM1 overexpression (21, 22). MTMR2 binds to MTMR5 via the coiled-coil domains resulting in a three- to fourfold increase in 3-phosphatase activity and altered subcellular localization of MTMR2 (19). MTMR2 also binds to MTMR13, resulting in a dramatic increase in the catalytic activity of MTMR2 toward both PtdIns(3)P and PtdIns(3,5)P2 (23). In these examples, the binding of a catalytically inactive protein to a catalytically active protein resulted in changes in activity and localization; hence, inactive myotubularin proteins may serve a regulatory role. Mutations in both active and inactive myotubularins are associated with diseases (8, 9), such as myotubular myopathy, Charcot-Marie-Tooth disease, and others, indicating that inactive myotubularin proteins are functionally important. Based on these results, the working hypothesis is that the enzymatically active myotubularin proteins dimerize with enzymatically inactive myotubularin proteins, and the formation of these heteromers can result in altered enzymatic activity and subcellular localization. Myotubularin proteins can be grouped into subfamilies based on homology. Closely related MTMR6, MTMR7, and MTMR8 comprise such a subfamily, and MTMR9 is the sole member of another subfamily. Previous studies have shown that MTMR9 binds to MTMR6 (24) and to MTMR7 (25).
PtdIns(3)P has been proposed to be essential in autophagy, a conserved intracellular process for the degradation of cytoplasmic proteins or organelles. A number of human diseases, including cancer and neurodegenerative disorders, are linked to dysfunctions in autophagy (26). Autophagy has been demonstrated to eliminate aggregated proteins in neurons (27). Previous studies have shown that aggregated proteins have pathological significance with respect to neurodegeneration, as removal of these proteins in mouse models of spinocerebellar ataxia 1 and Huntington disease correlates with reversal of symptoms (28, 29).
Type III PI3K, which generates PtdIns(3)P in mammalian cells, forms a complex with Beclin 1 and controls autophagosome formation (30). Little is known what role the synthesis and degradation of PtdIns(3)P plays in autophagy. It was proposed that when autophagy is suppressed under nutrient-rich conditions, the activity of PtdIns(3)P phosphatases overrides that of type III PI3K (31). Most recently, two members of the MTMR family, Jumpy (MTMR14) and MTMR3, have been shown to be involved in the regulation of autophagy (32, 33). Knockdown of Jumpy enhances autophagy under both nutrient and starvation conditions, whereas a dominant-negative MTMR3 only increases autophagic activity in the presence of nutrients, suggesting that the roles of the two MTMRs in autophagy are different. Down-regulation of both MTMR3 and MTMR14 facilitates initiation as well as the completion of autophagy, indicating that the local PtdIns(3)P level is important for the entire autophagic process. Here, we demonstrate that inactive MTMR9 interacts with active MTMR8. Unlike the MTMR6/R9 complex that regulates PtdIns (3, 5)P2 levels and thereby affects apoptosis, the MTMR8/R9 complex down-regulates the levels of PthIns(3)P and blocks the autophagic process.
Results
Human MTMR9 Binds to MTMR8 and Increases the Stability of MTMR8.
We previously demonstrated that human MTMR6 and MTMR9 directly associate both in vitro and in cells (24). Formation of the MTMR6/R9 complex increased MTMR6’s affinity for phospholipids, catalytic activity, and protein stability. Functionally, the complex inhibited apoptosis (24). To investigate whether the human orthologs of MTMR8 and MTMR9 interact, the human MTMR8 cDNA (GenBank, NM_017677) and human MTMR9 cDNA (GenBank, NM_015458) were cloned, as previously described (24). HA-MTMR8 and FLAG-MTMR9 were coexpressed in HeLa cells and immunoprecipitated using anti-HA and anti-FLAG antibodies. Both MTMR8 and MTMR9 were detected when either MTMR8 or MTMR9 was immunoprecipitated (Fig. 1A).
Fig. 1.
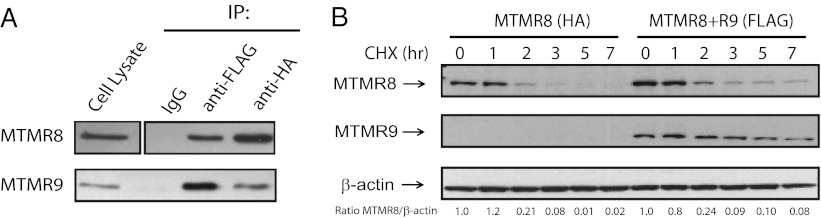
Interaction between MTMR8 and MTMR9. (A) HeLa cells were cotransfected with HA-MTMR8 and MTMR9-FLAG for 24 h, and proteins were immunoprecipitated with anti-HA and anti-FLAG polyclonal antibodies. Rabbit IgG was used as a negative control. Western blotting was done using monoclonal antibodies against HA (Upper) or FLAG (Lower) tags. (B) Stability of MTMR8 is increased by the formation of the complex. HeLa cells were transfected with HA-MTMR8, or HA-MTMR8 plus FLAG-MTMR9 for 24 h. Cycloheximide (150 μg/mL) was added for the times indicated. The levels of MTMR8 and MTMR9 were detected by Western blotting with an anti-HA or anti-FLAG antibody, respectively. β-Actin levels are shown as a loading control. MTMR8 band intensities relative to actin are indicated below each lane.
We next tested whether or not formation of the complex stabilizes the proteins, possibly by decreasing the degradation rate. The levels of MTMR8 were analyzed in cycloheximide-treated HeLa cells in the presence or absence of MTMR9. Higher levels of MTMR8 and slower degradation of MTMR8 are observed in cells cotransfected with both proteins (Fig. 1B).
Catalytic Activity of MTMR6, MTMR6/R9, MTMR8, and MTMR8/R9.
To determine the effect of MTMR9 on the 3-phosphatase activity of both MTMR6 and MTMR8, we determined the catalytic activity for each using radio-labeled PtdIns(3*)P and PtdIns(3*,5)P2, by measuring the release of [32P]-PO4. MTMR9 increased the enzymatic activity of MTMR6 toward PtdIns(3)P only about 2-fold, whereas it enhanced the activity toward PtdIns(3,5)P2 by over 30-fold. By contrast, MTMR9 increased MTMR8 activity 4-fold and 1.4-fold toward PtdIns(3)P and PtdIns(3,5)P2, respectively (Fig. 2 A and B). The cellular level of the product PtdIns(5)P was elevated threefold when both MTMR6 and MTMR9 were coexpressed but no significant increase in the level of PtdIns(5)P was seen by overexpression of MTMR8 plus MTMR9 (Fig. 2C), consistent with the changes in enzymatic activity observed in the in vitro assays. MTMR8 exhibited relatively higher activity toward PtdIns(3)P than MTMR6 or MTMR6 plus MTMR9 in vitro (Fig. 2A); however, no significant decrease was observed in the cellular levels of PtdIns(3)P by overexpression of MTMR8, using an antibody that specifically recognizes PtdIns(3)P. Transfection efficiency was determined in a separate set of plates and found to be greater than 95%. No significant change in PtdIns(3)P levels was observed in cells overexpressing MTMR6 or MTMR6 plus MTMR9 (Fig. 2D). These data are quantified in Fig. 2E, with 50 cells per cover-slip, and three cover-slips counted for each condition. Spots larger than 1 nm were counted as one PI(3)P molecule. Only overexpression of MTMR8 plus MTMR9 altered the cellular levels of PtdIns(3)P significantly, implying that the MTMR6/R9 complex controls PtdIns(3,5)P2, but the MTMR8/R9 complex determines PtdIns(3)P levels, thereby possibly affecting different cellular functions. A moderate decrease was observed in the level of PtdIns(3)P, with overexpression of MTMR9 alone, suggesting that inactive MTMR9 altered these levels through interactions with endogenous MTMR8.
Fig. 2.

Enzymatic activity of MTMR6, MTMR6/R9, MTMR8, and MTMR8/R9 toward PtdIns(3)P (A) or PtdIns(3,5)P2 (B). (C) Measurement of cellular PtdIns(5)P levels. HeLa cells were transfected with vector (control), cotransfected with either MTMR6 plus MTMR9 or MTMR8 plus MTMR9, and PtdIns(5)P levels were measured as previously described (45). Results are presented as relative mass compared with that in vector-transfected cells. (D) Overexpression of MTMR8 plus MTMR9 reduces levels of PtdIns(3)P in COS-7 cells. Cells were stained with anti-PI(3)P antibodies followed by anti-mouse Alexa568-conjugated antibodies. Nuclei are visualized with DAPI. Cotransfection efficiency was determined in a separate set of plates, because of limited available fluorescence channels, and was found to be greater that 95% in all cases. (Magnification: 63×.) (E) Quantification of immunofluorescence shown in D, expressed as an average number of PI(3)P spots per cell normalized to the number seen in MTMR8/R9 expressing cells. (F) Overexpression of MTMR8/R9 has no effect on apoptosis. HeLa cells were treated with the indicated constructs for 36 h, then with 100 µM etoposide to induce apoptosis. After 8 h, cells were treated with APOPercentage dye to selectively stain apoptotic cells with dark spots. Percentage of apoptotic cells are counted as the number of darker cells out of every 100 cells (*P < 0.01, t test).
Role of the MTMR8/R9 Complex in Autophagy.
A number of studies in Caenorhabditis elegans indicate that several myotubularins have nonredundant roles in regulating PtdIns(3)P levels during endocytosis (17, 34). It is likely that MTMR6 and MTMR8 have distinct functions because of different substrate specificities and specific subcellular localization. We have demonstrated that the MTMR6/R9 complex protects cells from etoposide-induced apoptosis (24). However, the antiapoptosis effect was not seen with overexpression of MTMR8 and MTMR9 (Fig. 2F). As our in vitro studies suggested that the MTMR8/R9 complex controls a cellular pool of PtdIns(3)P, we examined the cellular consequences that result from both increasing and decreasing the levels of MTMR8 and MTMR9 in cells.
PtdIns(3)P has been proposed to be essential in autophagy, a conserved intracellular process for the degradation of cytoplasmic proteins or organelles. Overexpression of both MTMR8 and MTMR9 resulted in a significant increase in the level of p62, a protein that is degraded in autophagosomes and is used to monitor autophagy (35) (Fig. 3A). Knockdown of either MTMR8 or MTMR9 alone had no effect on the level of p62 in cells, whereas knockdown of both MTMR8 and MTMR9 significantly reduced the level of p62 in HeLa cells, treated with 100 nM bafilomycin A1 for 3 h to inhibit fusion between autophagosomes and lysosomes (Fig. 3B). The level of MTMR8 following RNAi of MTMR8 was 0.275, when the level of MTMR8 was set at 1.0 in control siRNA, as determined using RT-PCR. Very little effect was seen in the levels of p62 with RNAi of MTMR6 alone or in combination with MTMR9 (Fig. 3C). Thus, inactive MTMR9 regulates its individual binding partners’ discrete functions. Up- or down-regulation of MTMR8 alone had no significant effect on autophagy, as measured by p62 levels, compared with controls (Fig. 3 A and B). Knocking down MTMR9 alone led to a notable effect on autophagy compared with control RNAi or vector (Fig. 3B), implying that there might be other members of this MTMR subfamily involved in the autophagy pathway that are also controlled by MTMR9: for example, MTMR7. HeLa cells transfected with MTMR8 for overexpression, followed by RNAi of MTMR8, show reduced expression of MTMR8, compared with RNAi of vector alone (Fig. 3D). The levels of MTMR8 protein were also further reduced using a combination of RNAi oligonucleotides targeting both MTMR8 and MTMR9 (Fig. 3D, lanes 3 and 6). This finding suggested that the formation of the MTMR8/R9 complex stabilizes the proteins, as we have previously seen with the MTMR6/R9 complex (24). Moreover, when HeLa cells expressing HA-MTMR8 and endogenous MTMR9 were placed in serum-free medium, the complex between these proteins was completely dissociated by 2 h (Fig. 3E).
Fig. 3.

The MTMR8/R9 complex regulates autophagy. (A) HeLa cells were transfected with the indicated constructs. Western blotting was used to detect MTMR8, MTMR9, and p62. A nonspecific band (*) is seen below MTMR9 in the vector and MTMR8 lanes. (B) Western blotting of p62 in HeLa cell extracts transfected with the indicated RNAi constructs. (C) HeLa cells were transfected for 24 h with the indicated RNAis, then treated for 4 h with 100 nM Bafilomycin A1 (BafA1), 10 µg/mL E64d, and 10 µg/mL pepstatin. Cell lysates were immunoblotted with anti-p62 and anti–β-actin, and p62 band intensities relative to actin are indicated below each lane. (D) HeLa cells were transfected with HA-MTMR8 for 24 h, treated with the indicated RNAi constructs for another 24 h, then analyzed by blotting with an antibody against HA tag. (E) The MTMR8 and MTMR9 complex dissociates during starvation induced autophagy. HeLa cells were transfected with HA-MTMR8 for 36 h, followed by serum starvation for the indicated times. Extracts were immunoprecipitated with an antibody against MTMR9 and immunoblotted with an anti-HA antibody to detect MTMR8. The protein levels of MTMR8 and MTMR9 during starvation are shown in input panels.
Another PtdIns(3)P binding autophagy factor, WIPI-1 (WD repeat domain, phosphoinositide interacting 1), is recruited to autophagic membranes in a PtdIns(3)P-dependent fashion (36). The quantification of WIPI-1 protein accumulation can be used to monitor mammalian autophagy (36). HeLa cells were transfected with GFP-WIPI for 24 h, transfected with MTMR6, MTMR8, MTMR6 plus MTMR9, or MTMR8 plus MTMR9 constructs for overexpression or RNAi constructs for knockdowns. The percentage of cells displaying distinct WIPI-I puncta was used to quantify the extent of autophagy. Knockdown of MTMR8 plus MTMR9 significantly induced autophagy, which is not seen in cells treated with RNAis of MTMR6, MTMR8, MTMR9, or the MTMR6/R9 complex (Fig. 4 A and B). Strikingly, overexpression of MTMR8 plus R9 abolished rapamycine-induced autophagy (Fig. 4 C and D).
Fig. 4.

Knockdown of the MTMR8/R9 complex induces autophagy. (A) HeLa cells were transfected with GFP-WIPI-1 for 24 h, then treated with indicated RNAi constructs for 24 h. Autophagy was assessed by measuring WIPI-1 puncta-formation by immunofluorescence. (Magnification: 63×.) (B) Results from a total of 500 cells were counted and the ratios of cells in puncta/nonpuncta status was determined. (*P < 0.01.) (C) Overexpression of the MTMR8/R9 complex suppresses autophagy. HeLa cells were transfected with GFP-WIPI-1 for 24 h, then transfected with the indicated constructs for an additional 24 h. Autophagy was induced by rapamycine for 3 h and then was measured by immunofluorescence. (Magnification: 63×.) (D) The ratios of cells in puncta/nonpuncta status was measured. Cotransfection efficiency was determined to be greater than 95% using a duplicate set of plates. (E) The hypothesized consequential effects of complex formation between MTMR9 and either MTMR8 or MTMR6 are shown in a schematic diagram.
Discussion
We describe here two members of a closely related subfamily of active myotubularins, MTMR6 and MTMR8, both of which partner with the same inactive myotubularin MTMR9. Previous studies have shown an association between MTMR6 and MTMR9 in mouse and C. elegans (24, 34). Complex formation between MTMR6 and MTMR9 increases MTMR6’s affinity for phospholipids, catalytic activity, protein stability, and complex formation inhibits apoptosis (24). We demonstrated that the association of MTMR9 with MTMR6 not only enhances the enzymatic activity of MTMR6, but also determined its substrate preference and thus the levels of its product. We have found that the MTMR6/R9 complex controls PtdIns(3,5)P2 levels and the MTMR8/R9 complex determines PtdIns(3)P levels, thereby affecting different cellular functions. We have shown that the MTMR8/R9 complex functions to reduce autophagy and the MTMR6/R9 complex inhibits apoptosis.
Endocytic membrane trafficking is critically dependent on the local synthesis of PtdInd(3)P and PtdIns(3,5)P2 (37). PtdIns(3)P is generated on both early endosomes and late endosomes by the class III PI3K complex (38). Early endosome antigen 1, a protein essential for endosome fusion, binds to Rab5-GTP and PtdIns(3)P through the FYVE domain on the PIKfyve kinase. PtdIns(3)P is subsequently converted into PtdIns(3,5)P2 by PIKfyve on multivesicular late endosomes, which is required for protein sorting as well as controlling lysosome size (39). Although the mechanisms involved in the synthesis of phosphoinositides at endosomes are well understood, little is known about the lipid phosphatases that degrade endosomal PtdIns(3)P and PtdIns(3,5)P2. It is likely that myotubularin proteins, which dephosphorylate PtdIns(3)P and PtdIns(3,5)P2 at the D3 position, are involved in membrane trafficking. Overexpression of MTM1 leads to enlarged endosomal structures and delayed movement of the epidermal growth factor receptor into the lysosome, similar to the observation made when PtdIns(3)P or PtdIns(3,5)P2 is depleted by mutations in PI3K or PIKfyve (40, 41). A recent study with RNAi depletion of MTM1 also led to accumulation of the epidermal growth factor receptor in distinct endosomes, despite the increased level of PtdIns(3)P on early endosomes (42). Thus, impairment of optimal phosphoinositide levels in endosomes may contribute to the disease phenotype when MTM1 is mutated. We plan to investigate the role of the MTMR6/R9 and the MTMR8/R9 complexes in the endocytosis process, because each complex controls a distinct phosphatidylinositol pool and therefore may regulate different signaling pathways.
MTMR9 seems to play a central role in the regulation of all three active members of this subfamily. Although to date no disease has been associated with mutations in this subfamily, new mutations are being uncovered all the time and the detailed study of this subfamily will lead to a better understanding of the biochemical events underlying human diseases caused by mutations in these proteins. MTMR9 expression has been correlated with obesity in humans, as determined from a large number of gene-based single-nucleotide polymorphisms (43). A replicated association between obesity and a single-nucleotide polymorphism located in the MTMR9 gene was demonstrated in a study comprised of 1,011 obese and 2,171 control individuals, P = 10−7 (43). In this same report, transcription of MTMR9 in the rat hypothalamic region was induced by fasting and reduced by a high-fat diet. Because MTMR9 lacks phosphatase activity, it is likely that it interacts with one of the active phosphatases to cause the obesity phenotype. To define the regulatory role of MTMR9 in vivo, an MTMR9 knockout mouse model is currently being generated (44).
Materials and Methods
All experiments were performed at least three times.
Reagents and Chemicals.
All chemicals and reagents, unless specifically noted, were purchased from Sigma–Aldrich. [γ-32P]-ATP was purchased from MP Biomedicals.
Cloning, Expression, and Purification of Human MTMR6 and MTMR9.
Full-length human MTMR9 was cloned as described previously (24). Constructs for human MTMR6, MTMR8, and MTMR9 were expressed in Sf9 cells and protein was purified, as described previously (24). Briefly, Sf9 cells were transfected with the appropriate construct, and the recombinant flag-tagged proteins were purified using FLAG M1 agarose affinity gel (Sigma-Aldrich) according to the manufacturer’s instructions.
Cell Culture, Transfection, Immunoprecipitation, and Western Blotting.
HeLa cells were maintained in culture using 10% (vol/vol) FBS in DMEM. Unless noted, transfection was conducted by using Lipofectamine 2000 (Invitrogen). RNAi transfections were done using a Nucleofector kit (Amaxa). RNAi duplexes (Ambion) used in transfections are as follows: control RNAi (luciferase) duplex, sense 5′-CUUACGCUGAGUACUUCGAdTdT-3′; antisense, 5′-UCGAAGUACUCAGCGUAAGDTdT-3′; MTMR6 RNAi (R6-1), sense, 5′-GGAAGTCAATGGCACTAATgg-3′; antisense, 5′-TTTAGTGCCATTGACTTCCaa-3′; MTMR8 RNAi (Invitrogen; Stealth RNAi, catalog # HSS124669); MTMR9 RNAi, 5′-CAAAGGAGGTGGCTTTGA Tca-3′ and 5′-TCAAAGCCACCTCCTTTGgc-3′. The specificity and efficacy were determined using quantitative reverse transcription-PCR, with 70%, 70%, and 50% reduction upon the RNAi of MTMR6, MTMR8, and MTMR9, respectively. No cross-reactivity was detected. Immunoprecipitation and Western blotting were done as previously described (45).
Measurement of PtdIns(5)P Mass, 3-Phosphatase Activity, and Protein Stability.
The PtdIns(5)P mass assay was conducted as previously described (45) with minor modifications. HeLa cells were transfected with control vector, MTMR6/R9, and MTMR8/R9 constructs. Total cellular phosphatidylinositol content was purified using a PIP mass purification kit (Echelon Bioscience). For measurement of 3-phosphatase activity, 100 ng of purified, recombinant MTMR protein was added to a reaction mixture containing trace amounts of [32P]PtdIns(3)P and [32P]PtdIns(3,5)P2, prepared as described previously (24). Substrate identity and purity were determined by HPLC analysis. Activity is expressed as a rate constant using the equation [S] = [So]e−kt. Units are min−1, and are graphed as min−1/mg protein. Because of the trace amount of label incorporated into the substrate, a defined specific activity cannot be determined. Protein stability was measured as previously described (24).
Immunofluorescence Microscopy.
Twenty-four hours after transfection, HeLa cells grown on cover-slips were fixed as described previously (45), washed with Tris-buffered saline, and solubilized with 0.5% Triton X-100 in PBS. Antibodies were diluted in PBS, containing 0.1% Triton X-100 and 5% BSA. Cells were incubated for 1 h with primary antibody and for 30 min with secondary antibodies at 37 °C. Cover-slips were washed in PBS, and mounted in Prolong mounting medium (Molecular Probes). Images were taken with an Olympus IX70 inverted microscope and processed with Metamorph software (Molecular Devices).
Acknowledgments
We thank Dr. Nordheim (Tuebingen, Germany) for the gifts of WIPI-1 expression vectors (36); Peter Nicholas and Cecil Buchanan for their helpful assistance; and Dr. Shao-Chun Chang for invaluable advice. This work was supported by National Institutes of Health Grant HL16634 (to P.W.M.) and Children’s Discovery Institute Grant MD-II-2009-174 (to M.P.W.).
Footnotes
The authors declare no conflict of interest.
References
- 1.Majerus PW, York JD. Phosphoinositide phosphatases and disease. J Lipid Res. 2009;50(Suppl):S249–S254. doi: 10.1194/jlr.R800072-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Li J, et al. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science. 1997;275:1943–1947. doi: 10.1126/science.275.5308.1943. [DOI] [PubMed] [Google Scholar]
- 3.Steck PA, et al. Identification of a candidate tumour suppressor gene, MMAC1, at chromosome 10q23.3 that is mutated in multiple advanced cancers. Nat Genet. 1997;15:356–362. doi: 10.1038/ng0497-356. [DOI] [PubMed] [Google Scholar]
- 4.Myers MP, et al. P-TEN, the tumor suppressor from human chromosome 10q23, is a dual-specificity phosphatase. Proc Natl Acad Sci USA. 1997;94:9052–9057. doi: 10.1073/pnas.94.17.9052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Maehama T, Dixon JE. The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. J Biol Chem. 1998;273:13375–13378. doi: 10.1074/jbc.273.22.13375. [DOI] [PubMed] [Google Scholar]
- 6.Attree O, et al. The Lowe’s oculocerebrorenal syndrome gene encodes a protein highly homologous to inositol polyphosphate-5-phosphatase. Nature. 1992;358:239–242. doi: 10.1038/358239a0. [DOI] [PubMed] [Google Scholar]
- 7.Laporte J, et al. A gene mutated in X-linked myotubular myopathy defines a new putative tyrosine phosphatase family conserved in yeast. Nat Genet. 1996;13:175–182. doi: 10.1038/ng0696-175. [DOI] [PubMed] [Google Scholar]
- 8.Bolino A, et al. Charcot-Marie-Tooth type 4B is caused by mutations in the gene encoding myotubularin-related protein-2. Nat Genet. 2000;25:17–19. doi: 10.1038/75542. [DOI] [PubMed] [Google Scholar]
- 9.Senderek J, et al. Mutation of the SBF2 gene, encoding a novel member of the myotubularin family, in Charcot-Marie-Tooth neuropathy type 4B2/11p15. Hum Mol Genet. 2003;12:349–356. doi: 10.1093/hmg/ddg030. [DOI] [PubMed] [Google Scholar]
- 10.Alonso A, et al. Protein tyrosine phosphatases in the human genome. Cell. 2004;117:699–711. doi: 10.1016/j.cell.2004.05.018. [DOI] [PubMed] [Google Scholar]
- 11.Vergne I, Deretic V. The role of PI3P phosphatases in the regulation of autophagy. FEBS Lett. 2010;584:1313–1318. doi: 10.1016/j.febslet.2010.02.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Laporte J, et al. Characterization of the myotubularin dual specificity phosphatase gene family from yeast to human. Hum Mol Genet. 1998;7:1703–1712. doi: 10.1093/hmg/7.11.1703. [DOI] [PubMed] [Google Scholar]
- 13.Clague MJ, Lorenzo O. The myotubularin family of lipid phosphatases. Traffic. 2005;6:1063–1069. doi: 10.1111/j.1600-0854.2005.00338.x. [DOI] [PubMed] [Google Scholar]
- 14.Laporte J, Bedez F, Bolino A, Mandel JL. Myotubularins, a large disease-associated family of cooperating catalytically active and inactive phosphoinositides phosphatases. Hum Mol Genet. 2003;12(Spec No 2):R285–R292. doi: 10.1093/hmg/ddg273. [DOI] [PubMed] [Google Scholar]
- 15.Taylor GS, Dixon JE. PTEN and myotubularins: Families of phosphoinositide phosphatases. Methods Enzymol. 2003;366:43–56. doi: 10.1016/s0076-6879(03)66004-0. [DOI] [PubMed] [Google Scholar]
- 16.Kim SA, Taylor GS, Torgersen KM, Dixon JE. Myotubularin and MTMR2, phosphatidylinositol 3-phosphatases mutated in myotubular myopathy and type 4B Charcot-Marie-Tooth disease. J Biol Chem. 2002;277:4526–4531. doi: 10.1074/jbc.M111087200. [DOI] [PubMed] [Google Scholar]
- 17.Xue Y, et al. Genetic analysis of the myotubularin family of phosphatases in Caenorhabditis elegans. J Biol Chem. 2003;278:34380–34386. doi: 10.1074/jbc.M303259200. [DOI] [PubMed] [Google Scholar]
- 18.Berger P, Schaffitzel C, Berger I, Ban N, Suter U. Membrane association of myotubularin-related protein 2 is mediated by a pleckstrin homology-GRAM domain and a coiled-coil dimerization module. Proc Natl Acad Sci USA. 2003;100:12177–12182. doi: 10.1073/pnas.2132732100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kim SA, Vacratsis PO, Firestein R, Cleary ML, Dixon JE. Regulation of myotubularin-related (MTMR)2 phosphatidylinositol phosphatase by MTMR5, a catalytically inactive phosphatase. Proc Natl Acad Sci USA. 2003;100:4492–4497. doi: 10.1073/pnas.0431052100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lorenzo O, Urbé S, Clague MJ. Analysis of phosphoinositide binding domain properties within the myotubularin-related protein MTMR3. J Cell Sci. 2005;118:2005–2012. doi: 10.1242/jcs.02325. [DOI] [PubMed] [Google Scholar]
- 21.Nandurkar HH, et al. Identification of myotubularin as the lipid phosphatase catalytic subunit associated with the 3-phosphatase adapter protein, 3-PAP. Proc Natl Acad Sci USA. 2003;100:8660–8665. doi: 10.1073/pnas.1033097100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Caldwell KK, Lips DL, Bansal VS, Majerus PW. Isolation and characterization of two 3-phosphatases that hydrolyze both phosphatidylinositol 3-phosphate and inositol 1,3-bisphosphate. J Biol Chem. 1991;266:18378–18386. [PubMed] [Google Scholar]
- 23.Berger P, et al. Multi-level regulation of myotubularin-related protein-2 phosphatase activity by myotubularin-related protein-13/set-binding factor-2. Hum Mol Genet. 2006;15:569–579. doi: 10.1093/hmg/ddi473. [DOI] [PubMed] [Google Scholar]
- 24.Zou J, Chang SC, Marjanovic J, Majerus PW. MTMR9 increases MTMR6 enzyme activity, stability, and role in apoptosis. J Biol Chem. 2009;284:2064–2071. doi: 10.1074/jbc.M804292200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mochizuki Y, Majerus PW. Characterization of myotubularin-related protein 7 and its binding partner, myotubularin-related protein 9. Proc Natl Acad Sci USA. 2003;100:9768–9773. doi: 10.1073/pnas.1333958100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Levine B, Deretic V. Unveiling the roles of autophagy in innate and adaptive immunity. Nat Rev Immunol. 2007;7:767–777. doi: 10.1038/nri2161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yamamoto A, Cremona ML, Rothman JE. Autophagy-mediated clearance of huntingtin aggregates triggered by the insulin-signaling pathway. J Cell Biol. 2006;172:719–731. doi: 10.1083/jcb.200510065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yamamoto A, Lucas JJ, Hen R. Reversal of neuropathology and motor dysfunction in a conditional model of Huntington’s disease. Cell. 2000;101:57–66. doi: 10.1016/S0092-8674(00)80623-6. [DOI] [PubMed] [Google Scholar]
- 29.Zu T, et al. Recovery from polyglutamine-induced neurodegeneration in conditional SCA1 transgenic mice. J Neurosci. 2004;24:8853–8861. doi: 10.1523/JNEUROSCI.2978-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shintani T, Klionsky DJ. Autophagy in health and disease: A double-edged sword. Science. 2004;306:990–995. doi: 10.1126/science.1099993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Noda T, Matsunaga K, Taguchi-Atarashi N, Yoshimori T. Regulation of membrane biogenesis in autophagy via PI3P dynamics. Semin Cell Dev Biol. 2010;21:671–676. doi: 10.1016/j.semcdb.2010.04.002. [DOI] [PubMed] [Google Scholar]
- 32.Vergne I, et al. Control of autophagy initiation by phosphoinositide 3-phosphatase Jumpy. EMBO J. 2009;28:2244–2258. doi: 10.1038/emboj.2009.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Taguchi-Atarashi N, et al. Modulation of local PtdIns3P levels by the PI phosphatase MTMR3 regulates constitutive autophagy. Traffic. 2010;11:468–478. doi: 10.1111/j.1600-0854.2010.01034.x. [DOI] [PubMed] [Google Scholar]
- 34.Dang H, Li Z, Skolnik EY, Fares H. Disease-related myotubularins function in endocytic traffic in Caenorhabditis elegans. Mol Biol Cell. 2004;15:189–196. doi: 10.1091/mbc.E03-08-0605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bjørkøy G, et al. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol. 2005;171:603–614. doi: 10.1083/jcb.200507002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Proikas-Cezanne T, Ruckerbauer S, Stierhof YD, Berg C, Nordheim A. Human WIPI-1 puncta-formation: A novel assay to assess mammalian autophagy. FEBS Lett. 2007;581:3396–3404. doi: 10.1016/j.febslet.2007.06.040. [DOI] [PubMed] [Google Scholar]
- 37.Lindmo K, Stenmark H. Regulation of membrane traffic by phosphoinositide 3-kinases. J Cell Sci. 2006;119:605–614. doi: 10.1242/jcs.02855. [DOI] [PubMed] [Google Scholar]
- 38.Nicot AS, Laporte J. Endosomal phosphoinositides and human diseases. Traffic. 2008;9:1240–1249. doi: 10.1111/j.1600-0854.2008.00754.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shisheva A. PIKfyve: Partners, significance, debates and paradoxes. Cell Biol Int. 2008;32:591–604. doi: 10.1016/j.cellbi.2008.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Robinson FL, Dixon JE. Myotubularin phosphatases: Policing 3-phosphoinositides. Trends Cell Biol. 2006;16:403–412. doi: 10.1016/j.tcb.2006.06.001. [DOI] [PubMed] [Google Scholar]
- 41.Tsujita K, et al. Myotubularin regulates the function of the late endosome through the gram domain-phosphatidylinositol 3,5-bisphosphate interaction. J Biol Chem. 2004;279:13817–13824. doi: 10.1074/jbc.M312294200. [DOI] [PubMed] [Google Scholar]
- 42.Cao C, Backer JM, Laporte J, Bedrick EJ, Wandinger-Ness A. Sequential actions of myotubularin lipid phosphatases regulate endosomal PI(3)P and growth factor receptor trafficking. Mol Biol Cell. 2008;19:3334–3346. doi: 10.1091/mbc.E08-04-0367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yanagiya T, et al. Association of single-nucleotide polymorphisms in MTMR9 gene with obesity. Hum Mol Genet. 2007;16:3017–3026. doi: 10.1093/hmg/ddm260. [DOI] [PubMed] [Google Scholar]
- 44.Zou J, et al. The role of myotubularin-related phosphatases in the control of autophagy and programmed cell death. Adv Enzyme Regul. 2012;52:282–289. doi: 10.1016/j.advenzreg.2011.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zou J, Marjanovic J, Kisseleva MV, Wilson M, Majerus PW. Type I phosphatidylinositol-4,5-bisphosphate 4-phosphatase regulates stress-induced apoptosis. Proc Natl Acad Sci USA. 2007;104:16834–16839. doi: 10.1073/pnas.0708189104. [DOI] [PMC free article] [PubMed] [Google Scholar]