

Abstract
Nicotinic acetylcholine receptors (AChRs) mediate signaling in the central and peripheral nervous systems. The AChR gating conformational change is powered by a low- to high-affinity change for neurotransmitters at two transmitter binding sites. We estimated (from single-channel currents) the components of energy for gating arising from binding site aromatic residues in the α-subunit. All mutations reduced the energy (TyrC1>>TrpB≈TyrC2>TyrA), with TyrC1 providing ∼40% of the total. Considered one at a time, the fractional energy contributions from the aromatic rings were TrpB ∼35%, TyrC1 ∼28%, TyrC2 ∼28%, and TyrA ∼10%. Together, TrpB, TyrC1, and TyrC2 comprise an “aromatic triad” that provides much of the total energy from the transmitter for gating. Analysis of mutant pairs suggests that the energy contributions from some residues are nearly independent. Mutations of TyrC1 cause particularly large energy reductions because they remove two favorable and approximately equal interactions between the aromatic ring and the quaternary amine of the agonist and between the hydroxyl and αLysβ7.
Keywords: allosteric, conformation, ligand-gated
Allosteric proteins receive energy from the environment at sensors and transfer it, via a global conformational change, to an effector site that controls the protein’s functional output. In ion channels, the sensor energy can be chemical, electrical, mechanical, or thermal and the effector is a “gate” that regulates the ionic conductance of the pore. The nicotinic acetylcholine receptor (AChR) is a pentameric ligand-gated ion channel that mediates fast chemical transmission at vertebrate neuromuscular synapses. Agonist molecules, including the neurotransmitter ACh, bind at two sites that can undergo a low- to high-affinity conformational change that increases the probability of the protein adopting an active, ion-permeable shape. In this report, we describe some of the key sources of energy at the transmitter binding sites that serve to promote the AChR gating isomerization.
The adult neuromuscular AChR is composed of four different but homologous subunits (1, 2) (α12βδε; Fig. 1A). Each subunit has a β-barrel and four transmembrane α-helices. The five β-barrels form the extracellular domain, and the five sets of helices comprise the transmembrane domain. The two transmitter binding sites are in the extracellular domain at the α-δ and α-ε subunit interfaces. Each of these sites is ∼55 Å from the gate region in the transmembrane domain.
Fig. 1.

Transmitter binding site. (A) Torpedo AChR (PDB 2bg9) (2). The asterisk indicates a transmitter binding site. (B) Aromatic residues in the Lymnaea AChBP (PDB 1uv6) (32). Loops: A, pink; B, green; and C, tan. Carbamylcholine (CCh), white. (C) Contributions from the side chains. The numbers are energies (kcal/mol) from the corresponding chemical moiety. TrpB, TyrC1, and TyrC2 comprise an “aromatic triad” that contributes most of the total energy. One exit path for energy is TyrC1-Lysβ7.
The extracellular domain of the AChR α1-subunit has 59% of its sequence conserved with the Lymnaea stagnalis acetylcholine binding protein (AChBP). In the X-ray structures of AChBPs, the primary subunit of the ligand binding site (α-subunit in AChRs) has four conserved aromatic residues in three different loops: TyrA, TrpB, TyrC1, and TyrC2 (3) (Fig. 1B). These residues were previously identified in AChRs (4–8), and many studies of the functional consequences of mutations confirm that all four contribute to both the agonist binding and channel gating phases of AChR activation (9–16). In these experiments, the roles of the aromatic residues in AChR activation were assayed by measuring mutation-induced changes in binding affinities, dose–response EC50 values of whole-cell currents, or activation rate constants estimated from single-channel currents. Our goal was to measure the effects of mutations of these four aromatic residues in terms of the energy that they provide toward the liganded gating conformational change.
To estimate these energies, we invoked a cyclical scheme for AChR activation (17–19) (Fig. S1). In this model, an AChR alternatively adopts two global shapes, resting (R) and active (R*), with each having a different affinity for ACh and a different functional output. The R shape has a low affinity and closed channel, and the R* shape has a high affinity and open channel. E2 and E0 are the R ↔ R* “gating” equilibrium constants of diliganded and unliganded AChRs, and Kd and Jd are the equilibrium dissociation constants for ACh in R and R*, respectively. Without any external energy (other than from the agonist) and with two approximately equivalent binding sites (20), E2/E0 = (Kd/Jd)2. By taking the logarithms, this relationship can be expressed as energies: GDL − Gi = 2ΔGB. GDL is the R* vs. R energy difference in diliganded AChRs, Gi is the intrinsic R* vs. R energy difference (with only water at the binding sites), and ΔGB is the binding energy arising from the low- to high-affinity change at each site. ΔGB is the quantity we seek to measure and is related to the experimental equilibrium constants by:
ΔGBACh has been measured previously for adult mouse wild-type (wt) AChRs. E2ACh = 25.4 (21) and E0wt = 7 × 10−7 (20); thus, we calculate ΔGBACh = −5.1 kcal/mol. The increase in affinity at the two transmitter binding sites combined generates −10.2 kcal/mol, which is sufficient to overcome the +8.4-kcal/mol energy difference that separates R and R* (at −100 mV) when the binding sites are unoccupied by agonists to generate a high open-channel probability (22).
To estimate the energy that ACh provides to power gating in mutant AChRs it is necessary to measure both E2ACh and E0 for each construct. Estimating E2ACh in AChRs with binding site mutations is challenging because side chain substitutions here often substantially debilitate both gating (reduce E2ACh) and resting affinity (increase KdACh). Estimating E0, too, is problematic because this constant is small and openings from unliganded AChRs are rare. As described in Methods, we have used a combination of membrane depolarization and background mutations to facilitate the measurement of both E2ACh and E0 in AChRs that have mutations of binding site residues (21).
These engineering techniques, along with single-channel electrophysiology and the cyclical model, have allowed us to estimate ΔGBACh for ∼40 different mutations of the four conserved α-subunit aromatic binding site amino acids. The energy changes caused by a few mutations of these (and other) binding site residues have been reported previously (23–25).
Results
Fig. 1 shows the Torpedo AChR and a ligand binding site of AChBP. The aromatic residues we studied in the AChR are in the primary (α) subunit of the binding site, in loops A (TyrA; αY93 in mouse), B (TrpB; αW149), and C (TyrC1 and TyrC2; αY190 and αY198). These four residues are completely conserved in all AChBPs and mouse AChRs with the exception of α5, which has TyrA-F and TyrC1-D substitutions. Considering other, related pentameric ligand-gated ion channels, the TyrC2 and TrpB positions are also conserved in 5-HT3A, GABAA, and Gly receptors.
For consistency, each aromatic residue was mutated to (at least) F, W/Y, H, S, and A. Both E2ACh and E0 were measured for each construct, from which the low- to high-affinity energy change was calculated by using Eq. 1.
TrpB.
Fig. 2A shows analyses of two TrpB mutants activated by ACh. Openings occurred in clusters that each reflect the activity of an individual AChR. The intracluster effective opening rate increased with increasing [ACh] to reach a plateau at ∼30 mM, signifying that at this concentration, the binding sites were fully occupied by transmitter molecules. From the intracluster opening/closing rate constants, we estimate E2ACh ∼1.1 for TrpB-F and E2ACh ∼0.1 for TrpB-A (Table S1). The wt value is ∼25.4; thus, each of these TrpB mutations caused a substantial decrease in the diliganded gating equilibrium constant.
Fig. 2.

Mutations of TrpB. (A) Diliganded gating. (Upper) Low-time-resolution view of TrpB-F currents (140 mM ACh, +100 mV). Openings (up) occur in clusters, each from one AChR. (Lower) Intracluster opening frequency increases with increasing [ACh] to reach at plateau (arrow, 30 mM ACh). (Inset) Example clusters (TrpB-F). (B) Unliganded gating. (Upper) Low-time-resolution view of unliganded TrpB-F currents (−100 mV). Openings (down) occur in clusters even in the absence of any agonists. (Lower) E0 for each mutation on a log scale. The H and M mutations decreased, and all others increased, E0wt. Traces are examples of unliganded current clusters.
To estimate energy, it is also essential to quantify the effect of a mutation on E0, which is the intrinsic tendency of the protein to adopt the active conformation in the absence of agonists. Fig. 2B shows unliganded gating in TrpB mutants. The F and A substitutions each increased E0 relative to the background by ∼3.3-fold and ∼7.9-fold, respectively (Table S1). For the F and A mutants, we calculate that ΔGBACh was −3.9 kcal/mol and −2.8 kcal/mol, respectively. Both of these energies are smaller (more positive) than that of the wt (−5.1 kcal/mol). To relate energy to physiological impact, note that a +2.0-kcal/mol change in ΔGBACh corresponds to a ∼900-fold lower E2ACh and a decrease in the maximum open probability from 0.97 to 0.03.
We repeated these experiments for an additional eight substitutions at TrpB (Fig. 3 and Table S1). The mutations all decreased E2ACh relative to the wt but could either increase or decrease E0. Considering all side chains, the range in E2ACh was ∼10,200-fold (W-to-M) and that in E0 was ∼370-fold (C-to-H) (Fig. 4). The substantial range in E0 values for the mutants highlights that E2 (or EC50) measurements alone do not generally reflect accurately the energetic consequence of a mutation on binding energy.
Fig. 3.
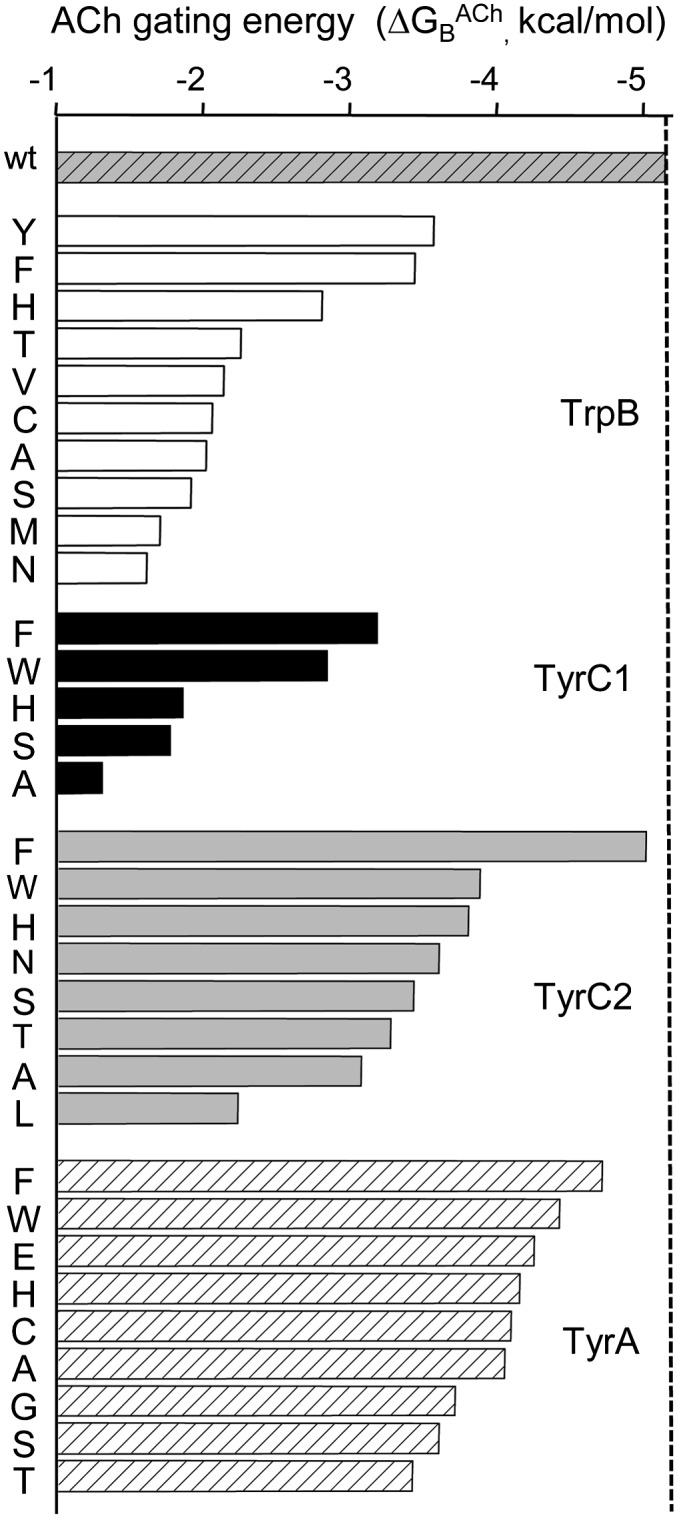
Energies of the aromatic mutants. In wt AChRs, ΔGBACh = −5.1 kcal/mol (topmost bar). All mutations of all the residues reduced ΔGBACh. Mutations of TyrC1 caused the largest reductions, and those of TyrA caused the smallest reductions. The error limits on the ΔGBACh values are given in Table S1.
Fig. 4.

Φ-Analysis. The rate and equilibrium constants were normalized by the wt values. Each symbol is a different mutant (average of ≥3 patches). Filled circles (●) illustrate diliganded ACh gating, and open triangles (△) illustrate gating without any agonists. The slopes (Φ) are somewhat higher with ACh compared with without any agonists.
All the TrpB mutations reduced ΔGBACh in the order (smallest to largest reduction) YFHTAVCSMN. When the aromatic nature was preserved (Y, F, and H), there was a relatively small reduction in ΔGBACh. For the other substitutions, the changes were similar and larger.
TyrC1.
Loop C has two conserved tyrosine residues. Fig. 3 shows the effects of mutating TyrC1. From the opening/closing rate constants at saturation (and after correction for the backgrounds), we estimate E2ACh ∼0.02 in TyrC1-F and only ∼0.00005 in TyrC1-A (Table S1). These effects are greater than those for the same side chain substitutions at TrpB.
Compared with TrpB, the TyrC1 mutations had smaller effects on E0 (Fig. 4). Nonetheless, we were able to measure E2ACh only for a few TyrC1 mutants. Although 10 different side chain substitutions were examined, most of these reduced the resting AChR affinity (increased Kd) to such an extent that the intracluster opening frequency did not reach a plateau even at the highest ACh concentration tested (140 mM). Hence, we were able estimate ΔGBACh only for five TyrC1 substitutions.
The TyrC1-F mutation caused a significant decrease in ΔGBACh. The removal of the hydroxyl group resulted in ∼+2.0 kcal/mol loss of energy for gating from ACh. The order in which TyrC1 mutants reduced ΔGBACh was FWHSA. It is likely that the five side chains for which E2ACh could not be measured caused even larger energy losses (ERTLD).
TyrC2.
The results of mutating the second loop C tyrosine, TyrC2, are also shown in Fig. 3. All substitutions here decreased E2ACh and, like its companion TyrC1, had relatively minor effects on E0 (Fig. 4). Unlike TyrC1, an F substitution at TyrC2 had a small effect on ΔGBACh and reduced this energy relative to the wt by only ∼+0.14 kcal/mol (close to our resolution limit). However, mutation of TyrC2 to A decreased the energy substantially. The other side chain substitutions at TyrC2 caused graded decreases in ΔGBACh, in the order WHNSTAL. The nonpolar, branched Leu weakened the energy by nearly +3 kcal/mol.
TyrA.
Of the four aromatic αTBS residues, mutations of TyrA resulted in the smallest changes to ΔGBACh (Fig. 3). Whether mutated to a negative (E) or small nonpolar (A) side chain, the net energy from ACh decreased only by <∼+1.1 kcal/mol. Here, the order of effect was FWEHCAGST, with the T mutation decreasing the energy by ∼+1.7 kcal/mol. Most TyrA mutations decreased E0, but the largest effect was for the C substitution, which decreased unliganded gating only by ∼11-fold (Fig. 4).
Φ-Analysis.
Single-channel analysis affords the estimation of rate as well as equilibrium constants (Table S2). We plotted (on a log-log scale) the forward, channel-opening rate constant vs. the gating equilibrium constant (normalized by the wt values) for the mutants of all four aromatic residues, for both diliganded and unliganded conditions (Fig. 4). The slope of the linear fit of this rate-equilibrium relationship is called Φ. A Φ-value of 1 (only the opening rate constant changes with mutation) suggests a relatively “early” energy change of the mutated residue in the overall channel-opening process, and a Φ-value of 0 (the opening rate constant does not change) suggests a relatively “late” energy change (26, 27). The Φ-values of all four aromatic positions were high for both the liganded and unliganded conditions, suggesting that these residues in both cases change energy close to the onset of the forward, channel-opening isomerization. However, in most cases, Φ-values were slightly lower for unliganded compared with diliganded gating. The Φ-values for E2ACh were in the range of 0.9–1.0, and those for E0 were in the range of 0.7–0.9 (Table S3). These results suggest that during channel opening, the α-subunit aromatic residues change energy somewhat earlier with vs. without ACh present.
Energetic Interactions Between Aromatic Residues.
We next explored whether or not the energy losses associated with each point mutation are independent of the others. To address this, we made sets of double mutants, either F-F or S-S, for different residue pairs and measured both E2ACh and E0 for each construct. We could not study double S-S mutants using TyrC1 because the huge loss in energy (and resting affinity) caused by even a single mutation here prevented the estimation of E2ACh in the mutant pairs.
The results are given in Table S4. There was only a small amount of energy interaction for the F-F pairs. The largest interaction was between the two loop C tyrosines, but it was only ∼−0.4 kcal/mol. The negative sign indicates that ΔGBACh in the double mutant was more negative (stronger) than expected from independent actions. This result implies that each of these hydroxyls can compensate, to a small extent, for the loss in energy caused by the removal from its companion. With regard to unliganded gating, E0 coupling for the double F-F constructs was also small (Table S5).
Similar results both with and without ACh were found with two tested S-S mutant pairs, between TyrA-TyrC2 and TrpB-TyrC2. Overall, we detected only a small degree of ΔGBACh interactions in eight different double-mutant constructs.
Discussion
Residue Comparison.
All the mutations of the α-subunit binding site aromatic residues decreased ΔGBACh, which is the energy for gating generated by the R → R* affinity increase for ACh at each transmitter binding site. It may be that the wt neuromuscular AChR binding site is optimized to derive the maximum energy from the transmitter molecule to power gating.
The energy changes caused by side chain substitutions common to all four aromatic positions are summarized in Fig. 5A. Regardless of the side chain, the energy loss was always largest at TyrC1. Indeed, considering all substitutions, this residue appears to provide ∼40% of the total energy from the four aromatic residues combined. The energy from the binding site conformational change that powers gating is, to a large extent, derived from TyrC1. The reason for the particularly large energy contribution of TyrC1 is discussed below.
Fig. 5.
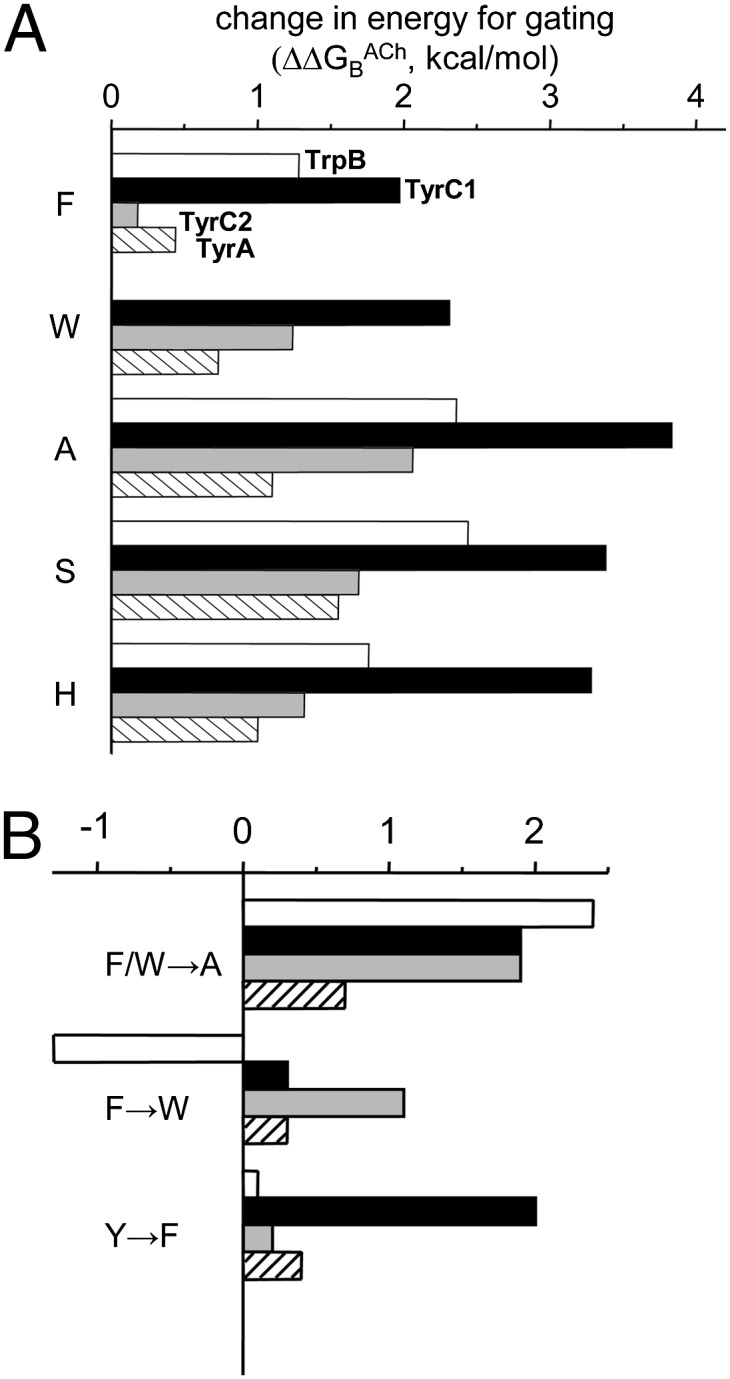
Relative effects of mutations. (A) Change in ΔGBACh for substitutions common to all four aromatic residues. For each substitution, the loss of energy (positive; rightward) was largest for TyrC1 (black) in all cases. The Ala substitution caused the largest energy loss at all positions except TyrA. (B) Energy changes for different substitutions. (Top to Bottom) Removing the aromatic group had the largest effect at TrpB, swapping the aromatic group (F ↔ W) had little effect at TyrC2 and TyrA, and removing the OH group had the largest effect at TyrC1.
The smallest energy changes were for mutations of TyrA. With ACh as the agonist, this residue provides only a small amount of energy. The other two α-subunit binding site aromatics, TyrC2 and TrpB, were of intermediate importance.
Below, we dissect the sources of energy from each aromatic side chain by comparing the consequences of different side chain substitutions. These results are summarized in Fig. 1C. In the following discussion, we will make two assumptions. First, we will assume that removing a chemical moiety from a side chain does not incur new energetic consequences, either at the residue itself or at other structural elements of the binding site. For instance, we will assume that the energy change associated with an F-to-A mutation reflects only the loss of the aromatic ring and not new interactions arising from the A substitution or from changes in the positions of the agonist or other binding site amino acids. Two pieces of evidence support this assumption. The coupling energies we were able to measure were weak (∼0.5 kcal/mol), and A and S substitutions had similar energetic consequences (below). However, we were unable to measure such secondary energetic effects in general (e.g., by more extensive mutant cycle analysis); thus, as a first approximation, we will assume that these were small. Second, in what follows, we will assume that the energetic consequence of each mutation was equivalent at the two transmitter binding sites. Although it has been shown experimentally that in wt AChRs, ΔGBACh is approximately the same at the α-ε and α-δ binding sites (20), we have not tested whether or not the effects of mutations, too, are equal. Hence, the ΔGBACh values we report are the average of the energies at the two transmitter binding sites.
Energy from the Aromatic Group.
For each position, we estimated the energy contribution of just the aromatic moiety by comparing the reduction in ΔGBACh caused by F/W-to-A substitutions (Fig. 5B, Top and Table S6). The reduction was largest at TrpB (+2.3 kcal/mol), somewhat smaller and the same at TyrC1 and TyrC2 (+1.9 kcal/mol), and smallest at TyrA (+0.7 kcal/mol). The pattern of the energy change was similar when we considered F/W-to-S substitutions at each position. The slightly smaller energy losses with S vs. A substitutions at TyrC1 and TyrC2 suggest that the hydroxyl group of a serine in these positions provides a small amount (∼−0.4 kcal/mol) of stabilization energy to ACh.
The ΔGBACh estimates are free energies that incorporate all possible interactions between the side chain and the quaternary amine (QA) of ACh. The primary interaction between the aromatic moieties and the QA is cation-π (16), which is consistent with the observation that differences in ΔGB values between agonists are generated mainly by differences in enthalpy (28). We can compare the above free energy losses consequent to mutations with estimates of the relative strengths, specifically, of cation-π interactions. Fluorination of the aromatic ring, which decreases cation-π forces, increases the EC50 for ACh at TrpB and TyrC2 but not at TyrA (12, 13, 16). That cation-π interactions are large at TrpB and TyrC2 is in agreement with our free energy estimates. No information could be obtained at TyrC1 using the unnatural substitution method because fluorine substitutions eliminated the whole-cell current (12), but our results show that the aromatic-QA free energy provided by TyrC1 is about the same as that provided by TyrC2. In previous studies (16, 29), it was stated that the strength of the TrpB cation-π interaction with ACh is ∼−2 kcal/mol, whereas our free energy estimate is ∼−0.3 kcal/mol greater. This small discrepancy could exist because some of the unnatural mutations changed E0, because not all the interaction is by cation-π forces, or perhaps because the AChRs had different subunit compositions (γ vs. ε). Regardless, our results support the view that most, if not all, of the interaction energies between the aromatic groups and the QA of ACh are cation-π.
From the single mutations, we estimate that the benzene groups of the two loop C tyrosines contribute about equally to the affinity change of the binding site. Considering the mutations one at a time, the fractional contributions of the four aromatic groups were ∼56% for TyrC1 and TyrC2 combined, 35% for TrpB, and only 10% for TyrA. Three amino acids, TyrC1, TyrC2, and TrpB, form an “aromatic triad” that appears to provide much of the energy from the ligand for gating.
The sum of the energy losses for the four F/W-to-A substitutions (+6.9 kcal/mol) is greater than the net energy change for ACh (−5.1 kcal/mol). This suggests that there is some degree of energy coupling between the aromatic groups with regard to their interaction with the QA. We observed a small degree of coupling (−0.4 kcal/mol) between the serine mutations of TyrC2 and TrpB, but we were unable to make similar measurements for TyrC1-TrpB and TyrC1-TyrC2 mutant pairs. This degree of coupling, however, is sufficient to account for the difference between the sum and the net energies. It is possible that the moving parts of the aromatic triad apparatus operate independently to some extent. More measurements of energy coupling may illuminate this issue further.
We can compare the energies of benzene vs. indole rings by comparing the consequence of an F-to-W substitution at each of the four positions (Fig. 5B, Middle). The largest effects on ΔGBACh with this exchange were at TrpB (∼−1.3 kcal/mol, stronger energy with W) and TyrC2 (∼+1.1 kcal/mol, stronger energy with F). The effect of the substitution was negligible at both TyrC1 and TyrA. Hence, the energies provided by these different aromatic groups are different at TrpB and TyrC2 but are almost the same at TyrC1 and TyrA. Note that after F, Y, and W, the next smallest energy loss at TrpB, TyrC1, and TyrC2 was for H, a side chain that has some capacity for cation-π bonding.
Energy from the Hydroxyl Group.
We can estimate the effect of removing the hydroxyl group at the three tyrosines by comparing the energy losses of F mutations (Fig. 5B, Bottom). The effects of OH removal at both TyrC2 and TyrA were small, <+0.5 kcal/mol. (Also, there was little effect of OH removal at TrpB, comparing Y vs. F mutations.) Kearney et al. (12) reported that substituting the hydroxyl group of TyrA and TyrC1 increased the EC50, whereas substitutions at TyrC2 had little effect. In agreement, our energy estimates show a large contribution from the hydroxyl at TyrC1 and almost none at TyrC2. However, we also observed only a small energy change consequent to hydroxyl removal at TyrA, which does not support the conclusion that this moiety is important for ligand gating. It is likely that the large loss in gating observed by Kearney et al. (12) was caused mainly by the substantial reduction in E0 apparent with the TyrA-F mutation (Table S1). Our results indicate that the OH groups of TyrC2 and TyrA contribute little to stabilizing the liganded, high-affinity R* conformation of the transmitter binding site.
There was, however, a large energy loss consequent to the removal of the OH group at TyrC1 (∼+2.0 kcal/mol). For comparison, this amount of energy loss is only marginally smaller than was observed with the complete elimination of TrpB (the W-to-A mutation). Clearly, the TyrC1 hydroxyl group has a major effect on stabilizing the liganded R* binding site.
Why does removing the OH group have such a large effect at TyrC1? We think that the answer has already been provided by Mukhtasimova et al. (30). The hydroxyl group of TyrC1 faces away from the agonist in the X-ray structures of AChBP and does not interact directly with the ligand, but it does approach a conserved lysine in the β7 strand, Lysβ7 (αK145 in mouse). These authors showed that there is a strong energetic interaction (with regard to E2ACh) between the mutations TyrC1-F and Lysβ7-Q. In addition, other studies show that mutations of Lysβ7 can reduce E2ACh substantially, with a Φ-value of ∼1 (31). We hypothesize that the special character of TyrC1 is caused by this side chain making two equivalent energy contributions to the R* transmitter binding region, one between the benzene ring and the QA of the agonist (+1.9 kcal/mol) and the other between the OH group and the ε-amino group of Lysβ7 (+2.0 kcal/mol).
Energy Changes in the Binding Site Region.
The interaction between TyrC1 and Lysβ7 highlights the fact that ΔGB (or EC50) measurements of mutant AChRs do not exclusively reflect interactions between side chains and the agonist molecule. The low-affinity ↔ high-affinity conformational change of the binding site region certainly must involve structural changes that allow energy to exit the binding pocket to launch the global isomerization. In addition, some of an energy loss consequent to a binding site mutation may reflect a change in the shape of the overall pocket that may influence the energy that exits, the energy from interactions of the aromatic triad and the agonist, or both. For example, mutations of a conserved glycine in loop B (GlyB2; αG147 in mouse) reduce ΔGBACh substantially (>+2 kcal/mol), even though this residue is far from the agonist in AChBP structures (25).
Although the TyrC1 energy exit path seems clear, we are unable to identify such exit routes for the energies generated by TrpB, TyrC2, and TyrA. It is possible that the energy changes at these three positions trigger structural changes in backbone atoms or water that move energy out of the immediate vicinity, or that they serve only to support the TyrC1-Lysβ7 exit pathway. It is unclear whether or not there are multiple routes for energy to escape from the binding sites and contribute to the global conformational change of the protein.
We have shown that it is possible to pinpoint, group-by-group, the sources of free energy for the affinity change provided by residues at the AChR binding site. It may be worthwhile to apply these methods to other AChR amino acids and agonists, as well as to other allosteric proteins, to understand better the energy sources from ligands that power protein conformational change.
Methods
The QuikChange site-directed mutagenesis kit (Stratagene) was used to make mutant cDNAs of mouse AChR α-subunits, and their sequences were verified by dideoxy sequencing. Transient transfection of HEK 293 cells was performed using calcium phosphate precipitation. Subunit cDNA (3.5 μg) was added at the ratio 2:1:1:1 (α, β, δ, ε) to each 35-mm culture dish of cells. Cells were incubated for ∼16 h at 37 °C and were then washed with fresh media. Single-channel electrophysiological recordings (cell-attached patches) were performed ∼20 h posttransfection. A detailed description of the methods is given in SI Methods.
Supplementary Material
Acknowledgments
We thank M. Shero, M. Merritt, and M. Teeling for technical assistance. This work was supported by National Institutes of Health Grant NS-064969 and a research fellowship (to I.B.) by the Canadian Institute of Health Research.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1203633109/-/DCSupplemental.
References
- 1.Noda M, et al. Structural homology of Torpedo californica acetylcholine receptor subunits. Nature. 1983;302:528–532. doi: 10.1038/302528a0. [DOI] [PubMed] [Google Scholar]
- 2.Unwin N. Refined structure of the nicotinic acetylcholine receptor at 4A resolution. J Mol Biol. 2005;346:967–989. doi: 10.1016/j.jmb.2004.12.031. [DOI] [PubMed] [Google Scholar]
- 3.Brejc K, et al. Crystal structure of an ACh-binding protein reveals the ligand-binding domain of nicotinic receptors. Nature. 2001;411:269–276. doi: 10.1038/35077011. [DOI] [PubMed] [Google Scholar]
- 4.Cohen JB, Sharp SD, Liu WS. Structure of the agonist-binding site of the nicotinic acetylcholine receptor. [3H]acetylcholine mustard identifies residues in the cation-binding subsite. J Biol Chem. 1991;266:23354–23364. [PubMed] [Google Scholar]
- 5.Dennis M, et al. Amino acids of the Torpedo marmorata acetylcholine receptor alpha subunit labeled by a photoaffinity ligand for the acetylcholine binding site. Biochemistry. 1988;27:2346–2357. doi: 10.1021/bi00407a016. [DOI] [PubMed] [Google Scholar]
- 6.Galzi JL, et al. Identification of a novel amino acid alpha-tyrosine 93 within the cholinergic ligands-binding sites of the acetylcholine receptor by photoaffinity labeling. Additional evidence for a three-loop model of the cholinergic ligands-binding sites. J Biol Chem. 1990;265:10430–10437. [PubMed] [Google Scholar]
- 7.Kao PN, et al. Identification of the alpha subunit half-cystine specifically labeled by an affinity reagent for the acetylcholine receptor binding site. J Biol Chem. 1984;259:11662–11665. [PubMed] [Google Scholar]
- 8.Middleton RE, Cohen JB. Mapping of the acetylcholine binding site of the nicotinic acetylcholine receptor: [3H]nicotine as an agonist photoaffinity label. Biochemistry. 1991;30:6987–6997. doi: 10.1021/bi00242a026. [DOI] [PubMed] [Google Scholar]
- 9.Akk G. Aromatics at the murine nicotinic receptor agonist binding site: Mutational analysis of the alphaY93 and alphaW149 residues. J Physiol. 2001;535:729–740. doi: 10.1111/j.1469-7793.2001.00729.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Aylwin ML, White MM. Ligand-receptor interactions in the nicotinic acetylcholine receptor probed using multiple substitutions at conserved tyrosines on the alpha subunit. FEBS Lett. 1994;349(1):99–103. doi: 10.1016/0014-5793(94)00649-0. [DOI] [PubMed] [Google Scholar]
- 11.Chen J, Zhang Y, Akk G, Sine S, Auerbach A. Activation kinetics of recombinant mouse nicotinic acetylcholine receptors: Mutations of alpha-subunit tyrosine 190 affect both binding and gating. Biophys J. 1995;69:849–859. doi: 10.1016/S0006-3495(95)79959-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kearney PC, et al. Dose-response relations for unnatural amino acids at the agonist binding site of the nicotinic acetylcholine receptor: Tests with novel side chains and with several agonists. Mol Pharmacol. 1996;50:1401–1412. [PubMed] [Google Scholar]
- 13.Nowak MW, et al. Nicotinic receptor binding site probed with unnatural amino acid incorporation in intact cells. Science. 1995;268:439–442. doi: 10.1126/science.7716551. [DOI] [PubMed] [Google Scholar]
- 14.Sine SM, Quiram P, Papanikolaou F, Kreienkamp HJ, Taylor P. Conserved tyrosines in the alpha subunit of the nicotinic acetylcholine receptor stabilize quaternary ammonium groups of agonists and curariform antagonists. J Biol Chem. 1994;269:8808–8816. [PubMed] [Google Scholar]
- 15.Tomaselli GF, McLaughlin JT, Jurman ME, Hawrot E, Yellen G. Mutations affecting agonist sensitivity of the nicotinic acetylcholine receptor. Biophys J. 1991;60:721–727. doi: 10.1016/S0006-3495(91)82102-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhong W, et al. From ab initio quantum mechanics to molecular neurobiology: A cation-π binding site in the nicotinic receptor. Proc Natl Acad Sci USA. 1998;95:12088–12093. doi: 10.1073/pnas.95.21.12088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Auerbach A. The gating isomerization of neuromuscular acetylcholine receptors. J Physiol. 2010;588:573–586. doi: 10.1113/jphysiol.2009.182774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Karlin A. On the application of “a plausible model” of allosteric proteins to the receptor for acetylcholine. J Theor Biol. 1967;16:306–320. doi: 10.1016/0022-5193(67)90011-2. [DOI] [PubMed] [Google Scholar]
- 19.Monod J, Wyman J, Changeux JP. On the nature of allosteric transitions: A plausible model. J Mol Biol. 1965;12:88–118. doi: 10.1016/s0022-2836(65)80285-6. [DOI] [PubMed] [Google Scholar]
- 20.Jha A, Auerbach A. Acetylcholine receptor channels activated by a single agonist molecule. Biophys J. 2010;98:1840–1846. doi: 10.1016/j.bpj.2010.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jadey SV, Purohit P, Bruhova I, Gregg TM, Auerbach A. Design and control of acetylcholine receptor conformational change. Proc Natl Acad Sci USA. 2011;108:4328–4333. doi: 10.1073/pnas.1016617108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nayak TK, et al. The intrinsic energy of the gating isomerization of a neuromuscular acetylcholine receptor channel. J Gen Physiol. 2012;139:349–358. doi: 10.1085/jgp.201110752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bafna PA, Jha A, Auerbach A. Aromatic Residues εTrp-55 and ΔTrp-57 and the Activation of Acetylcholine Receptor Channels. J Biol Chem. 2009;284:8582–8588. doi: 10.1074/jbc.M807152200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Purohit P, Auerbach A. Energetics of gating at the apo-acetylcholine receptor transmitter binding site. J Gen Physiol. 2010;135:321–331. doi: 10.1085/jgp.200910384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Purohit P, Auerbach A. Glycine hinges with opposing actions at the acetylcholine receptor-channel transmitter binding site. Mol Pharmacol. 2011;79:351–359. doi: 10.1124/mol.110.068767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fersht AR. Characterizing transition states in protein folding: An essential step in the puzzle. Curr Opin Struct Biol. 1995;5(1):79–84. doi: 10.1016/0959-440x(95)80012-p. [DOI] [PubMed] [Google Scholar]
- 27.Zhou Y, Pearson JE, Auerbach A. Phi-value analysis of a linear, sequential reaction mechanism: Theory and application to ion channel gating. Biophys J. 2005;89:3680–3685. doi: 10.1529/biophysj.105.067215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gupta S, Auerbach A. Temperature dependence of acetylcholine receptor channels activated by different agonists. Biophys J. 2011;100:895–903. doi: 10.1016/j.bpj.2010.12.3727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Beene DL, et al. Cation-pi interactions in ligand recognition by serotonergic (5-HT3A) and nicotinic acetylcholine receptors: The anomalous binding properties of nicotine. Biochemistry. 2002;41:10262–10269. doi: 10.1021/bi020266d. [DOI] [PubMed] [Google Scholar]
- 30.Mukhtasimova N, Free C, Sine SM. Initial coupling of binding to gating mediated by conserved residues in the muscle nicotinic receptor. J Gen Physiol. 2005;126:23–39. doi: 10.1085/jgp.200509283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Purohit P, Auerbach A. Acetylcholine receptor gating: Movement in the alpha-subunit extracellular domain. J Gen Physiol. 2007;130:569–579. doi: 10.1085/jgp.200709858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Celie PH, et al. Nicotine and carbamylcholine binding to nicotinic acetylcholine receptors as studied in AChBP crystal structures. Neuron. 2004;41:907–914. doi: 10.1016/s0896-6273(04)00115-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.