

Abstract
HIV type 1 (HIV-1) persists within resting CD4+ T cells despite antiretroviral therapy (ART). To better understand the kinetics by which resting cell infection (RCI) is established, we developed a mathematical model that accurately predicts (r = 0.65, P = 2.5 × 10−4) the initial frequency of RCI measured about 1 year postinfection, based on the time of ART initiation and the dynamic changes in viremia and CD4+ T cells. In the largest cohort of patients treated during acute seronegative HIV infection (AHI) in whom RCI has been stringently quantified, we found that early ART reduced the generation of latently infected cells. Although RCI declined after the first year of ART in most acutely infected patients, there was a striking absence of decline when initial RCI frequency was less than 0.5 per million. Notably, low-level viremia was observed more frequently as RCI increased. Together these observations suggest that (i) the degree of RCI is directly related to the availability of CD4+ T cells susceptible to HIV, whether viremia is controlled by the immune response and/or ART; and (ii) that two pools of infected resting CD4+ T cells exist, namely, less stable cells, observable in patients in whom viremia is not well controlled in early infection, and extremely stable cells that are established despite early ART. These findings reinforce and extend the concept that new approaches will be needed to eradicate HIV infection, and, in particular, highlight the need to target the extremely small but universal, long-lived latent reservoir.
Keywords: HIV latency, viral kinetics
The persistence of HIV infection despite antiretroviral therapy (ART) is a challenge. Infection quickly establishes viral reservoirs that provide a persistent source of recrudescent viremia following the interruption of ART. HIV reservoirs have been characterized as cells or tissues that restrict virus replication and preserve replication-competent HIV for long periods of time (1). Of these reservoirs, the latent proviral reservoir within resting CD4+ memory T cells is conceptually the most challenging obstacle to viral eradication. The capacity of HIV-1 to establish latent infection within a population of infected but resting CD4+ T cells allows viral persistence despite immune surveillance or ART. The precise measurement of the reservoir of resting CD4+ T-cell infection (RCI) requires a stringent assay that measures replication-competent HIV recovered from these cells, and does not reflect forms of HIV DNA that cannot produce infectious virions: unintegrated linear or circularized molecules, or the predominant population of integrated proviruses that encode dysfunctional genomes (1).
Proviral latency is established in resting CD4+ T cells during early HIV infection (2). It is thought to be established predominantly by the return of infected, cycling cells to the resting state, although direct infection of resting cells may also play a role (3). The frequency of resting CD4+ T-cell infection is thought to represent a balance of the following: (i) the entry of virus into this pool via direct infection of resting cells, or the return to G0 of infected activated cells; and (ii) the loss of cells from this pool by death or activation of infected resting cells, processes that may be influenced by the levels of immune activation and viral replication. The homeostatic proliferation of infected resting CD4+ T cells in the absence of viral expression or infected cell clearance was recently proposed to influence the frequency of RCI (2, 4–6).
Patients treated with ART during acute HIV infection (AHI) present a unique opportunity to study the founding of the reservoir of RCI, as in these cases the duration and extent of viremia is better defined than in patients diagnosed after chronic infection has been established. We studied a cohort of patients identified in AHI via the North Carolina STAT program and the Center for HIV-AIDS Vaccine Immunology (CHAVI) who initiated ART within 45 d of the estimated date of infection (7).
To understand the effect of ART during seronegative HIV infection on the frequency of latently infected cells we used a mathematical model to analyze patient data that includes viral load, CD4+ T-cell counts, and the frequency of latently infected cells in HIV patients receiving ART during acute infection. We found a statistically significant correlation (r = 0.65, P = 2.5 × 10−4) between the measured frequency of latently infected cells and our model prediction showing that the model can describe the data. We show that latently infected cells are largely generated before the initiation of ART during early infection, and that the frequency of latently infected cells often decays during initial antiviral therapy. Our model also suggests that earlier treatment initiation reduces the number of latently infected cells remaining at the time of measurement. These results suggest that the degree of latent infection can be limited by early ART during acute HIV infection.
Methods
Patient Cohort.
We studied a cohort of 27 patients identified in AHI (plasma HIV RNA detected and HIV Western blot negative). One patient (S25) initiated ART 6 mo after the date of infection; all others initiated ART within 45 d of the estimated date of infection. All patients provided written informed consent, and studies were approved by the University of North Carolina Institutional Review Board. Serial measurements of plasma viremia and CD4+ cell count were performed, and when patients were aviremic (<50 HIV RNA copies/mL) on ART for >6 mo, cells were obtained by continuous-flow leukopheresis to measure RCI (8). In 11 of these patients available for study, serial measurements of RCI were performed.
We have previously calculated the SD of our RCI assay performed on different days to be 0.3 log10 or roughly a factor of 2 (8). This value was calculated using a dataset with a large number of assays, and makes the conservative assumption that there is never any real decline of RCI. Therefore, the true variance of our assay is likely to be lower.
Model.
We consider the following variant of a mathematical model (9):
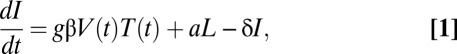 |
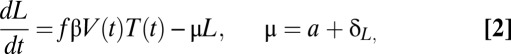 |
in which the target cell level, T(t), and the plasma viral load, V(t), will be specified by the data rather than by explicit equations. Here, I and L represent the density of productively infected cells and latently infected cells, respectively. Target cells become infected at a rate proportional to the product of target cell density and virus concentration with a rate constant β. We assume that a fraction, f, of infection events generates latently infected cells with replication competent genomes, a fraction contains “dead” proviruses detectable by PCR but incapable of reactivation, and the remaining fraction of infection events, g, leads to productively infected cells. Latently infected cells become productively infected at rate a due to stimulation. Productively infected cells and latently infected cells die at rates δ and δL, respectively. During ART, the infection rate β is reduced to the rate (1-ε)β, where ε represents antiviral drug efficacy. A schematic diagram of the model is shown in Fig. S1.
Statistical Analysis.
We approximated the uninfected target cell density, T(t), by the CD4 count. As shown in Fig. 1, the CD4+ T-cell counts measured after the start of ART changed approximately linearly with time. Thus, we performed linear regression to obtain a function approximating T(t) for the time after the initiation of therapy. Moreover, early in HIV-1 infection, a reduction in CD4 count is typically observed (10, 11). Therefore, from the time of infection, t = 0, to a certain time t = tm, we assume that T(t) declines linearly from the initial CD4+ T-cell count, T0 (Fig. 1). Similarly, the viral load V(t) was approximated by a cubic-spline curve fitted to the viral load data (Fig. 1) using the ‘spline.m’ function in MATLAB (MathWorks). For viral load measurements below the detection limit (i.e., 50 HIV-1 RNA copies/mL), we assumed that the viral loads were 25 HIV-1 RNA copies/mL (12). However, we note that choice of a value for undetected viral load does not alter our conclusions as we compute the total area under the viral load curve, which is mainly influenced by the peak viral load. For seven of the 27 patients, we obtained an unrealistic viral load spline curve before the time of ART initiation due to limited data before ART. In those cases, we approximated the early viral kinetics by assuming that the viral load increased exponentially from some initial viral load, V0, to the first viral load measured. Such exponential increases in viral load have been observed in other studies (13). We choose V0 = 0.001 HIV RNA copies/mL as done by Stafford et al. (14) when fitting a model to the kinetics of AHI. Making this value 3 logs lower or 3 logs higher has a negligible influence on our final results. Using T(t) and V(t) approximated from the data, we calculated the latently infected cell density at the final time, tF, i.e., the time at which latently infected cells were first measured by leukopheresis. Solving the model given by Eqs. 1 and 2 one finds
Fig. 1.

Spline curve fit to the patient viral load data and a biphasic linear fit to the patient CD4 count data for four representative patients. Vertical dashed line indicates the time of ART initiation; horizontal dash–dot line represents the assay detection limit. Filled circles denote measured HIV RNA copies/mL; open circles denote values below the limit of quantification. The x axis for each patient extends from the estimated time of infection to the time of first leukopheresis, at which time the RCI frequency was measured.
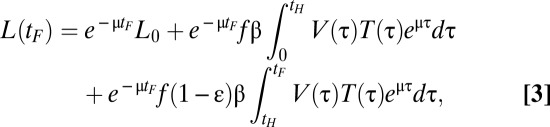 |
where tH is the time of ART initiation, and L0 is the initial latently infected cell density at t = 0, the time of infection. Because it is unlikely that latently infected cells were transmitted at the time of infection we set L0 = 0. Furthermore, the values of the parameters f and β are not known for each patient. Thus, we assumed them to be the same for all patients and use L/fβ, i.e., the number of latently infected cells in arbitrary units, as a measure of the predicted density of latently infected cells in each patient. Assuming that f and β are the same for all patients might have some effect on our results, as patients could differ in infecting viral quasi-species and in the susceptibility of their CD4+ T cells to infection.
We also calculated areas under the V(t), T(t), and V(t)T(t) curves from the time of infection to the time of ART initiation, from the time of ART initiation to the time latently infected cells were measured and from the time of infection to the final time. Using the Statistical Toolbox in MATLAB (MathWorks), we carried out correlation analysis between these areas and the measured frequency of RCI. For our initial computation we used T0 = 1000/μL, μ = 0.0039/d (corresponding to a latently infected cell half-life of 6 mo (15, 16)), ε = 0.75 (17), and tm = tH, the time of ART initiation. However, there are uncertainties about these values; for example, in some studies (18, 19) the half-life of latently infected cells has been estimated as 44 mo, the drug efficacy ε could be higher than 0.75, as observed in Louie et al. (17) for certain drug regimes, and T0, the T-cell count before HIV-1 infection can vary among patients, as can the amount of initial CD4+ T-cell decline after infection (20–22). Therefore, we performed a sensitivity analysis to examine the effects of using different values of T0, tm, μ, and ε.
Results
Model vs. Data.
Viral load and CD4+ T-cell count data, along with the approximating curves V(t) and T(t), are shown in Fig. 1 for representative patients (S19, S31, S237, and S241). We compared the latently infected cell frequency predicted by the model for each subject with the initial RCI frequency measured 6–12 mo after ART initiation. Note that the frequency of latently infected cells predicted by our model is given by L/(T+I+L). However, latently infected cell frequency is typically only a few cells per million, and at the time of leukopheresis there are few infected cells, as most infected cells live only for a short time and ART largely prevents the infection of new cells. Thus, by the time our samples are taken 6 mo or more after ART was initiated, L and I can be ignored in the denominator, and latently infected cell frequency at the time of leukopheresis is approximately given by L(tF)/T(tF). We found a statistically significant correlation between the predicted and measured RCI frequencies (r = 0.65, P = 2.5 × 10−4) (Fig. 2), where the predicted frequency is given in arbitrary units (as in Methods).
Fig. 2.

Correlation between model prediction and the measured frequency of latently infected cells. Predicted infectious units per million resting CD4+ T cells (IUPM) was computed as [L(tF)/(fβ T(tF))] x 106, i.e., the fraction of CD4+ T cells at the time of leukopheresis, tF, that are predicted to be latently infected in 1 million resting CD4+ T cells, and is expressed in arbitrary units of 1014 fβ.
Correlation Analysis of Latently Infected Cells.
We also sought to determine whether the measured frequency of latently infected cells might correlate with simpler viral dynamic features. We analyzed the correlation of the frequency of latently infected cells with the area under the viral load curve, the CD4+ T-cell count curve, and the product of viral load and the CD4+ T-cell count curve [which differs from the model in Eq. 3 in that it does not take into consideration the lifespan of latently infected cells (μ)].
Given that high levels of plasma viremia (23–26) and global activation of CD4+ T cells (27) are seen during primary infection, it has been speculated that viral replication (or accumulated virus) during this period could make a significant contribution to the establishment of latently infected resting CD4+ T cells (28, 29). This period is thus thought to be the most amenable to therapeutic intervention for the purposes of preventing the establishment of latently infected cell reservoirs (28). Supporting this argument, our analysis also shows a significant positive correlation (r = 0.40, P = 0.0401) between the RCI frequency and the area under the pretreatment viral load curve (Fig. S2).
We could not find any statistically significant correlation between the measured RCI frequency and the areas under the T(t) curve or V(t)T(t) curve from the time of infection to the time of ART initiation. However, the RCI frequency showed a significant positive correlation with the areas under the V(t) and V(t)T(t) curves and a significant negative correlation with area under the T(t) curve from the time of ART initiation to the time of leukopheresis (Fig. S2). Moreover, RCI frequency also had significant correlations with the total areas under the V(t), T(t), and V(t)T(t) curves from the time of infection to the time of leukopheresis (Fig. S2).
Dynamics of Latently Infected Cells.
To study RCI over time, we simulated the dynamics of latently infected cells predicted by the model (Fig. 3). The simulations show that RCI is largely generated before ART initiation (Fig. 3). Once ART is initiated, the frequency of latently infected cells declines in most patients, supporting the contention that ART during early infection can reduce RCI. It is noteworthy that after effective ART initiation, few latently infected cells should be generated, and their frequency should decline as existing latently infected cells die or become productively infected cells.
Fig. 3.

Dynamics of the latently infected cell frequency predicted by the model. Vertical dashed line indicates the time of ART initiation; final point represents the time of the first leukopheresis. Predicted IUPM was computed as [L/(fβ T)] x 106, i.e., the fraction of CD4+ T cells that are predicted to be latently infected in 1 million resting CD4+ T cells, and is expressed in arbitrary units of 1014 fβ.
Sensitivity Analysis.
We performed a sensitivity analysis of the correlation between the frequency of latently infected cells predicted by our model and their measured values by changing the values of T0, tm, μ, and ε used in Eq. 3. Changing the initial CD4+ T-cell count, T0, from 600/μL to 1600/μL did not make any change in the P values or r values obtained. We assumed that the time the minimum CD4 count was attained tm = tH, the time of ART initiation for our baseline computation. However, some studies (10) show a decline of CD4+ T cells for the first few weeks of illness only. Thus, we repeated the analysis taking tm = 7, 14, 21, 28, 35, and 42 d, and found that the correlation always remained statistically significant, with P values lying in the range 2.5–3.2 × 10−4 (range of r = 0.64–0.65). On changing μ = loge(2)/180 d−1 [corresponding to 6 mo half-life (t1/2) for latently infected cells, as elsewhere (15, 16)] to μ = loge(2)/1320 d−1 (corresponding to t1/2 = 44 mo) as published (18, 19), the significance of the correlation is preserved with a slight increase in P value from 2.5 × 10−4 to 6.8 × 10−4 and decrease in r from 0.65 to 0.61. Also, the drug efficacy could be lower or higher than the value of 0.75 taken in our base case. Thus, we performed the correlation analysis by taking various values of ε ranging from 0.5 to 0.99. In this range of ε, the correlation always remained statistically significant with the P value varying between 2.2 × 10−4 and 6.9 × 10−4, and r varying between 0.61 and 0.65. The fact that even low values of ε generated statistically significant correlations suggests that most latently infected cells are generated before ART initiation and particularly early in infection, when both viral loads and T-cell counts are high.
Low-Level Viremia Measured by a Single-Copy Assay Is Associated with RCI Frequency.
Approximately 80% of patients on ART with clinically undetectable viremia have stable, persistent, low-level viremia when tested with more sensitive methods (30). In most patients, viral genotypes closely related to those that populate this small plasma pool can be found in resting CD4+ T cells, but this is not always the case (31, 32). We therefore measured low-level viremia using a single-copy assay (SCA) (33) in these patients treated during AHI (Fig. 4A) and in a comparator group of patients treated in chronic HIV infection (CHI) (Fig. 4B), seeking an association between SCA viremia and RCI frequency. In both cohorts, SCA viremia was frequently undetectable (<1 copy/mL) when RCI frequency was <0.3/ million resting CD4+ T cells. Low-level viremia was observed in none of four AHI patients with RCI <0.3 per million, and in four of six patients with RCI > 0.3 per million (Fischer’s two-tailed P = 0.076). A similar trend was observed in 12 CHI patients (one of six and five of six, respectively, P = 0.08), and a significant association (P = 0.04) was seen with the 22 patients evaluated as a group. This observation is consistent with the hypothesis that SCA viremia is the result of expression of a fraction of proviral genomes within the resting cell population.
Fig. 4.

Comparison of low-level viremia (single-copy assay) and RCI frequency in (A) acutely treated and (B) chronically treated HIV-1–infected patients labeled A–L.
RCI Frequency over Time.
The initial evaluation of RCI frequency, which was used to compare with the theoretical analysis above, was performed within the first year of ART suppression. However, 11 patients in this cohort remained on ART (Table S1), and RCI frequency was periodically determined.
Initial RCI in this cohort ranged widely, from more than 12 IUPM to as little as 0.031 IUPM. In three of four patients in whom initial RCI was > 5 IUPM, a precipitous decline of RCI, with mean t1/2 = 5.1 mo, was seen during the initial months of this follow-up period (S257, S368, S416; Table 1 and Fig. 5). In the fourth patient (S25), RCI also declined over time, but with slower and noisier kinetics that resulted in a decay slope that was not significantly different from zero (Table 1 and Fig. 5). The decay of RCI was similar to that observed in several patients reported by Chun et al. (34). However, in patients with an initial RCI frequency of <5 IUPM (S231, S256, S521, S396, and S237; Fig. 5), the decay of RCI was significantly slower, mean t1/2 = 13.7 mo, and in the two patients (S26 and S520) with very low initial RCI (< 0.1 IUPM) there was no decay observed (Table 1 and Fig. S3). Interestingly, in the patients in whom decay was significant, the decay was exponential (Fig. 5) and thus consistent with the mathematical model (Eq. 2).
Table 1.
Characteristics of RCI decay
| Initial RCI | Decay rate | Half-life of RCI | ||
| Patient | (per million) | (per month) | P value* | (mo) |
| Patients with initial RCI >5 IUPM | ||||
| S257 | 11.3 | 0.081 | NA | 8.6 |
| S368 | 10.3 | 0.17 | NA | 4.1 |
| S416 | 5.1 | 0.27 | NA | 2.6 |
| S25 | >5.2 | 0.025 | 0.139 | 27.7 |
| Patients with initial RCI <5 IUPM | ||||
| S231 | 1.24 | 0.028 | 0.016 | 24.8 |
| S256 | 1.2 | 0.055 | 0.034 | 12.6 |
| S521 | 0.98 | 0.09 | NA | 7.7 |
| S396 | 0.92 | 0.12 | 0.11 | 5.8 |
| S237 | 0.31 | 0.039 | 0.033 | 17.8 |
| Patients with initial RCI <0.1 IUPM | ||||
| S26 | 0.064 | −0.032 | 0.034 | NA |
| S520 | 0.095 | −0.041 | 0.557 | NA |
*For patients with only two RCI measurements, a P value cannot be computed for the decay rate being different from zero.
Fig. 5.

Frequency of RCI in 9 of 11 patients on stable ART that exhibited RCI decay. Lines drawn between data points are for ease of viewing and are not meant to predict future trajectories. Dashed line is the best-fit regression line for the data from patient S25.
Discussion
Resting CD4+ T-cell infection, an obstacle to eradication of HIV infection, is established during acute HIV infection (28, 29). We used a mathematical model to analyze the viral load, CD4+ T-cell counts, and the frequency of latently infected cells for HIV patients given ART during acute infection. We found a highly significant correlation (r = 0.65, P = 2.5 × 10−4) between the model prediction of latently infected cell frequency and the experimental measurements, showing that the model can explain the patient data.
Nonetheless, we acknowledge some limitations of our model. As in previous models (9, 35), we considered the experimentally measured peripheral blood CD4+ T-cell count and plasma HIV viral load as measures of target cell and viral density. These quantities may vary in different tissues, and the fraction of CD4+ T cells in blood may change during therapy due to changes in tissue viral burden. Also, as in previous models of latency (6, 9), we did not consider the possibility of productively infected cells surviving and reverting to a resting state, a scenario suggested to be unlikely by Chun et al. (36).
Rapid decay of RCI after ART in acute or early HIV infection has been reported in a study of seven patients (34). In our cohort, in patients with higher initial levels of RCI, a larger, less stable population of cells appears to decay after several years of ART, consistent with an earlier observation in chronically infected patients (37). In patients with early containment of RCI, this smaller pool of persistently infected cells appears to be highly stable, as does a larger pool of resting CD4+ cells measured over time in chronic infection (38). Possible mechanisms for these differences include infection of different populations of memory cells, potent restriction of proviral expression in some cells (e.g., chromatin modifications, or cotranscriptional effects surrounding the proviral integration site), or combinations of these effects. Whether the apparent difference in decay kinetics of RCI defines biological differences in latency is an important area for study as strategies to attack RCI are developed.
Our analysis demonstrates that there is a significant correlation of the RCI frequency measured after suppression of viremia with the area under the viral load curve from the time of infection to the time of ART initiation. This positive correlation between the frequency of latently infected cells and the area under the pretreatment viral load curve suggests that early treatment during primary infection, which reduces the area under the pretreatment viral load curve, reduces the number of latently infected cells. This is especially so in the majority of patients in whom the innate and adaptive immune response do not rapidly and stringently contain HIV replication. In our cohort of all patients with AHI, the median observed peak of viremia is nearly 600,000 copies/mL and can be as high as 84 million copies/mL (7).
It is worth mentioning that Chun et al. (28) did not find a significant correlation between the frequency of latently infected resting CD4+ T cells and the time of initiation of ART after the onset of symptoms of primary HIV-1 infection. Due to significant variations of viral load dynamics among patients from the time of infection to the time of ART initiation, such a correlation analysis cannot account for the viral load dynamics during early infection. On the other hand, the area under the viral load curve considered here takes into account both the viral load that drives the generation of latently infected cells before ART and the time to ART initiation. Our observations are consistent with models in which the generation of latent infection proceeds with similar kinetics during acute and chronic infection.
Because target cells are lost due to infection, we also asked whether there was correlation between the area under the CD4+ T-cell curve and the level of latently infected cells. Viral kinetic theory does not predict any such correlation, and we did not find any significant correlation between the frequency of latently infected cells and the area under the T-cell curve before the initiation of ART. However, the area under the T-cell curve calculated after ART began was negatively correlated with the frequency of latently infected cells. This may be an indirect effect whereby patients with lower CD4+ T-cell counts had higher levels of infection that generated more latently infected cells.
We found that latently infected cells are efficiently generated during primary infection from initiation of infection up to the time of ART. It is this critical period, during which viral loads are high and the immune system may not be in a hyperactivated state, that virions may be more likely to encounter and to infect (albeit inefficiently) CD4 cells that are not maximally activated. Such encounters may be more likely to result in a latent, rather than a productive, infection (3).
Once ART is initiated, there are many fewer infections generating fewer latently infected cells. Therefore, the density of the latently infected cells is predicted to decay during ART. Nonetheless, viral kinetic theory suggests that if there is a constant probability that infection of a target cell will lead to the generation of a latently infected cell, then the total number of latently infected cells should be proportional to the total number of infection events before and after ART. Consistent with this, we found a significant correlation between the frequency of latently infected cells and net infection, as given by Eq. 3, i.e., the area under the V(t)T(t) curve corrected for the effects of latently infected cell loss.
The longitudinal nature of this study allowed examination of the stability of the latently infected cell reservoir in patients in whom ART was initiated during AHI. In patients on ART rapid depletion of RCI was observed, as previously reported (34). However, this process was not uniform across all patients, or across all infected resting CD4+ T cells within a patient. We noted a population of persistently infected resting CD4+ T cells could be observed, usually at a frequency <0.5 per million (Fig. S3 and Table 1). Critically, there was no appreciable decay of infection in this rare population of resting CD4+ T cells. This observation may be related to the apparently stable low level of viremia observed in ART-treated patients after several years of suppression (39).
Of note, in our experience with the RCI assay, the frequency of RCI in patients with chronic HIV infection who have been stably and successfully treated for several years is similar, usually ∼0.5 infected cells per million resting CD4+ T cells (8, 40, 41). One conceptual model that would unify these observations is that “short-lived” latent infections decay over the first 2 y of therapy, as they might represent cells that recognize common antigens that are more likely to be reactivated (37), or proviruses that are less restricted by cellular mechanisms such as histone modification or transcriptional interference and are similarly more likely to be reactivated, or both. Conversely, we hypothesize there exists a population of “long-lived” or deeply latent infections that do not decay, as they represent cells that recognize rare antigens, or contain proviruses that are heavily restricted by cellular mechanisms, or both. It is also possible that this population may be capable of self-renewal.
If this conceptual model is correct, in patients treated in AHI, there is less opportunity for rare events that establish “deep” latent infections, in comparison with patients treated later. Restricting the opportunity for these rare events might be thought of as a vaccine goal—durable, persistent infection may not be established despite early infection and replication events, if the frequency of these events is limited by the immune response to a level that is below that required to generate “deep” latent infections. Although not included in the model, this study may define a very long-lived population, one for which data are not available to allow a mathematical analysis.
In future studies aimed at eradicating persistent infection, patients treated in AHI might be ideal candidates for protocols in which ART interruption is eventually envisioned. Given the report of rapid viral rebound after ART interruption in a single such patient (38), this small enclave of residual infection may be one of great clinical significance, and special efforts to devise an effective therapeutic strategies to prevent “deep” latency during AHI, or target them once established, are essential.
Supplementary Material
Acknowledgments
We thank R. Sackmann, D. Parker, M. Cheema, and N. Dahl for technical support. We also thank Y. Park, A. Afenyi-Annan, and the staff of the University of North Carolina (UNC) Blood Bank, as well as J. Scepanski, A. Crooks, D. Cardona, and A. Sugarbaker for patient follow-up and study coordination, and especially the patients who have participated in these studies. This work was supported by Center for HIV/AIDS Vaccine Immunology (CHAVI) Grant U01-AI067854; National Institutes of Health Grants RR024383 to the North Carolina Translational and Clinical Sciences Institute, AI50410 to the UNC Center for AIDS Research, OD011095, and AI028433 (to A.S.P.); and an equipment grant from the James B. Pendleton Charitable Trust. J.M.C. was a Research Professor of the American Cancer Society, with support from the FM Kirby Foundation.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1120248109/-/DCSupplemental.
References
- 1.Blankson JN, Persaud D, Siliciano RF. The challenge of viral reservoirs in HIV-1 infection. Annu Rev Med. 2002;53:557–593. doi: 10.1146/annurev.med.53.082901.104024. [DOI] [PubMed] [Google Scholar]
- 2.Rong L, Perelson AS. Modeling latently infected cell activation: Viral and latent reservoir persistence, and viral blips in HIV-infected patients on potent therapy. PLOS Comput Biol. 2009;5:e1000533. doi: 10.1371/journal.pcbi.1000533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dai J, et al. Human immunodeficiency virus integrates directly into naive resting CD4+ T cells but enters naive cells less efficiently than memory cells. J Virol. 2009;83:4528–4537. doi: 10.1128/JVI.01910-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chomont N, et al. HIV reservoir size and persistence are driven by T cell survival and homeostatic proliferation. Nat Med. 2009;15:893–900. doi: 10.1038/nm.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kim H, Perelson AS. Viral and latent reservoir persistence in HIV-1-infected patients on therapy. PLOS Comput Biol. 2006;2:e135. doi: 10.1371/journal.pcbi.0020135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rong L, Perelson AS. Modeling HIV persistence, the latent reservoir, and viral blips. J Theor Biol. 2009;260:308–331. doi: 10.1016/j.jtbi.2009.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gay C, et al. Cross-sectional detection of acute HIV infection: Timing of transmission, inflammation and antiretroviral therapy. PLoS ONE. 2011;6:e19617. doi: 10.1371/journal.pone.0019617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Archin NM, et al. Valproic acid without intensified antiviral therapy has limited impact on persistent HIV infection of resting CD4+ T cells. AIDS. 2008;22:1131–1135. doi: 10.1097/QAD.0b013e3282fd6df4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Perelson AS, et al. Decay characteristics of HIV-1-infected compartments during combination therapy. Nature. 1997;387:188–191. doi: 10.1038/387188a0. [DOI] [PubMed] [Google Scholar]
- 10.Little SJ, Perrin L. Management of acute and early HIV-1 infection. AIDS Rev. 2000;2:136–143. [Google Scholar]
- 11.Zaunders J, Carr A, McNally L, Penny R, Cooper DA. Effects of primary HIV-1 infection on subsets of CD4+ and CD8+ T lymphocytes. AIDS. 1995;9:561–566. doi: 10.1097/00002030-199506000-00005. [DOI] [PubMed] [Google Scholar]
- 12.Lynn HS. Maximum likelihood inference for left-censored HIV RNA data. Stat Med. 2001;20:33–45. doi: 10.1002/1097-0258(20010115)20:1<33::aid-sim640>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 13.Ribeiro RM, et al. Estimation of the initial viral growth rate and basic reproductive number during acute HIV-1 infection. J Virol. 2010;84:6096–6102. doi: 10.1128/JVI.00127-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stafford MA, et al. Modeling plasma virus concentration during primary HIV infection. J Theor Biol. 2000;203:285–301. doi: 10.1006/jtbi.2000.1076. [DOI] [PubMed] [Google Scholar]
- 15.Ramratnam B, et al. The decay of the latent reservoir of replication-competent HIV-1 is inversely correlated with the extent of residual viral replication during prolonged anti-retroviral therapy. Nat Med. 2000;6:82–85. doi: 10.1038/71577. [DOI] [PubMed] [Google Scholar]
- 16.Zhang L, et al. Quantifying residual HIV-1 replication in patients receiving combination antiretroviral therapy. N Engl J Med. 1999;340:1605–1613. doi: 10.1056/NEJM199905273402101. [DOI] [PubMed] [Google Scholar]
- 17.Louie M, et al. Determining the relative efficacy of highly active antiretroviral therapy. J Infect Dis. 2003;187:896–900. doi: 10.1086/368164. [DOI] [PubMed] [Google Scholar]
- 18.Finzi D, et al. Latent infection of CD4+ T cells provides a mechanism for lifelong persistence of HIV-1, even in patients on effective combination therapy. Nat Med. 1999;5:512–517. doi: 10.1038/8394. [DOI] [PubMed] [Google Scholar]
- 19.Siliciano JD, et al. Long-term follow-up studies confirm the stability of the latent reservoir for HIV-1 in resting CD4+ T cells. Nat Med. 2003;9:727–728. doi: 10.1038/nm880. [DOI] [PubMed] [Google Scholar]
- 20.Bofill M, et al. Laboratory control values for CD4 and CD8 T lymphocytes. Implications for HIV-1 diagnosis. Clin Exp Immunol. 1992;88:243–252. doi: 10.1111/j.1365-2249.1992.tb03068.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.CASCADE Collaboration Differences in CD4 cell counts at seroconversion and decline among 5739 HIV-1-infected individuals with well-estimated dates of seroconversion. J Acquir Immune Defic Syndr. 2003;34:76–83. doi: 10.1097/00126334-200309010-00012. [DOI] [PubMed] [Google Scholar]
- 22.Reichert T, et al. Lymphocyte subset reference ranges in adult Caucasians. Clin Immunol Immunopathol. 1991;60:190–208. doi: 10.1016/0090-1229(91)90063-g. [DOI] [PubMed] [Google Scholar]
- 23.Clark SJ, et al. High titers of cytopathic virus in plasma of patients with symptomatic primary HIV-1 infection. N Engl J Med. 1991;324:954–960. doi: 10.1056/NEJM199104043241404. [DOI] [PubMed] [Google Scholar]
- 24.Daar ES, Moudgil T, Meyer RD, Ho DD. Transient high levels of viremia in patients with primary human immunodeficiency virus type 1 infection. N Engl J Med. 1991;324:961–964. doi: 10.1056/NEJM199104043241405. [DOI] [PubMed] [Google Scholar]
- 25.Mellors JW, et al. Prognosis in HIV-1 infection predicted by the quantity of virus in plasma. Science. 1996;272:1167–1170. doi: 10.1126/science.272.5265.1167. [DOI] [PubMed] [Google Scholar]
- 26.Piatak M, Jr, et al. High levels of HIV-1 in plasma during all stages of infection determined by competitive PCR. Science. 1993;259:1749–1754. doi: 10.1126/science.8096089. [DOI] [PubMed] [Google Scholar]
- 27.Cossarizza A, et al. Massive activation of immune cells with an intact T cell repertoire in acute human immunodeficiency virus syndrome. J Infect Dis. 1995;172:105–112. doi: 10.1093/infdis/172.1.105. [DOI] [PubMed] [Google Scholar]
- 28.Chun TW, et al. Early establishment of a pool of latently infected, resting CD4(+) T cells during primary HIV-1 infection. Proc Natl Acad Sci USA. 1998;95:8869–8873. doi: 10.1073/pnas.95.15.8869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Noë A, Plum J, Verhofstede C. The latent HIV-1 reservoir in patients undergoing HAART: An archive of pre-HAART drug resistance. J Antimicrob Chemother. 2005;55:410–412. doi: 10.1093/jac/dki038. [DOI] [PubMed] [Google Scholar]
- 30.Maldarelli F, et al. ART suppresses plasma HIV-1 RNA to a stable set point predicted by pretherapy viremia. PLoS Pathog. 2007;3:e46. doi: 10.1371/journal.ppat.0030046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Anderson JA, et al. Clonal sequences recovered from plasma from patients with residual HIV-1 viremia and on intensified antiretroviral therapy are identical to replicating viral RNAs recovered from circulating resting CD4+ T cells. J Virol. 2011;85:5220–5223. doi: 10.1128/JVI.00284-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bailey JR, et al. Residual human immunodeficiency virus type 1 viremia in some patients on antiretroviral therapy is dominated by a small number of invariant clones rarely found in circulating CD4+ T cells. J Virol. 2006;80:6441–6457. doi: 10.1128/JVI.00591-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Palmer S, et al. New real-time reverse transcriptase-initiated PCR assay with single-copy sensitivity for human immunodeficiency virus type 1 RNA in plasma. J Clin Microbiol. 2003;41:4531–4536. doi: 10.1128/JCM.41.10.4531-4536.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chun TW, et al. Decay of the HIV reservoir in patients receiving antiretroviral therapy for extended periods: Implications for eradication of virus. J Infect Dis. 2007;195:1762–1764. doi: 10.1086/518250. [DOI] [PubMed] [Google Scholar]
- 35.Perelson AS, Neumann AU, Markowitz M, Leonard JM, Ho DD. HIV-1 dynamics in vivo: Virion clearance rate, infected cell life-span, and viral generation time. Science. 1996;271:1582–1586. doi: 10.1126/science.271.5255.1582. [DOI] [PubMed] [Google Scholar]
- 36.Chun TW, et al. In vivo fate of HIV-1-infected T cells: Quantitative analysis of the transition to stable latency. Nat Med. 1995;1:1284–1290. doi: 10.1038/nm1295-1284. [DOI] [PubMed] [Google Scholar]
- 37.Strain MC, et al. Heterogeneous clearance rates of long-lived lymphocytes infected with HIV: Intrinsic stability predicts lifelong persistence. Proc Natl Acad Sci USA. 2003;100:4819–4824. doi: 10.1073/pnas.0736332100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chun TW, et al. Rebound of plasma viremia following cessation of antiretroviral therapy despite profoundly low levels of HIV reservoir: Implications for eradication. AIDS. 2010;24:2803–2808. doi: 10.1097/QAD.0b013e328340a239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Palmer S, et al. Low-level viremia persists for at least 7 years in patients on suppressive antiretroviral therapy. Proc Natl Acad Sci USA. 2008;105:3879–3884. doi: 10.1073/pnas.0800050105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Archin NM, et al. Antiretroviral intensification and valproic acid lack sustained effect on residual HIV-1 viremia or resting CD4+ cell infection. PLoS ONE. 2010;5:e9390. doi: 10.1371/journal.pone.0009390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lehrman G, et al. Depletion of latent HIV-1 infection in vivo: A proof-of-concept study. Lancet. 2005;366:549–555. doi: 10.1016/S0140-6736(05)67098-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.