

Abstract
Carbon monoxide (CO) is in principle an excellent resource from which to produce industrial hydrocarbon feedstocks as alternatives to crude oil; however, CO has proven remarkably resistant to selective homologation, and the few complexes that can effect this transformation cannot be recycled because liberation of the homologated product destroys the complexes or they are substitutionally inert. Here, we show that under mild conditions a simple triamidoamine uranium(III) complex can reductively homologate CO and be recycled for reuse. Following treatment with organosilyl halides, bis(organosiloxy)acetylenes, which readily convert to furanones, are produced, and this was confirmed by the use of isotopically 13C-labeled CO. The precursor to the triamido uranium(III) complex is formed concomitantly. These findings establish that, under appropriate conditions, uranium(III) can mediate a complete synthetic cycle for the homologation of CO to higher derivatives. This work may prove useful in spurring wider efforts in CO homologation, and the simplicity of this system suggests that catalytic CO functionalization may soon be within reach.
Keywords: reduction, C-C coupling, ethyne diolate, antiferromagnetic exchange coupling
The continued growth and stability of the global economy requires the ready availability of petrochemical feedstocks, but uncertainty in cost and supply, underscored by the energy crisis in the 1970s, has driven the demand for developing alternative sources (1). CO is readily produced by steam reforming reactions and is, thus, an abundant resource that can be used in the production of bulk hydrocarbon feedstocks (2). The concept of directly homologating CO is very appealing, but the triple bond in CO is very strong with a bond energy of ∼257 kcal mol-1, and direct dimerization of CO to O = C = C = O is highly unfavorable with 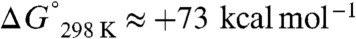 (3). Although direct 1,1-migratory insertion of CO into an organometallic M-R bond to give M-C(O)R is a favorable process for many metals (4) and a key step in industrially important C-C bond formation reactions, e.g., hydroformylation (5), double insertion to give M-C(O)C(O)R is usually energetically unfeasible, but it has been documented (6–8). Thus, processes involving CO can require energy intensive reaction conditions and give varied product distributions that render them economically uncompetitive overall when crude oil remains readily available; however, combining the coupling of CO with reduction results in a thermodynamically feasible process to give a family of oxocarbon dianions
(3). Although direct 1,1-migratory insertion of CO into an organometallic M-R bond to give M-C(O)R is a favorable process for many metals (4) and a key step in industrially important C-C bond formation reactions, e.g., hydroformylation (5), double insertion to give M-C(O)C(O)R is usually energetically unfeasible, but it has been documented (6–8). Thus, processes involving CO can require energy intensive reaction conditions and give varied product distributions that render them economically uncompetitive overall when crude oil remains readily available; however, combining the coupling of CO with reduction results in a thermodynamically feasible process to give a family of oxocarbon dianions 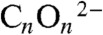 (n = 2–6), including the reductively dimerized ethyne diolate -O-C ≡ C-O-, as building blocks for more complex and value-added organic molecules (9).
(n = 2–6), including the reductively dimerized ethyne diolate -O-C ≡ C-O-, as building blocks for more complex and value-added organic molecules (9).
Molten potassium can effect reductive oligomerization of CO, but the resulting ill-defined salts are thermally unstable and shock-sensitive (10). CO can be electrochemically homologated, but an overpressure of 100–400 bar of CO is required (11). Very few transition metal complexes mediate the reductive coupling of CO (12–17); though, the coupled product could be hydrogenated to free alkene in 60% yield in the presence of hydrogenation cocatalysts such [Rh(PPh3)3Cl] or Pd/C, or under 100 psi of hydrogen, no viable metal complex could be recycled for reuse (14, 15). Conversion of CO to allene has been demonstrated, but five steps are required to close a multicomponent H2/CO/benzyl/Me3SiOTf/Me3SiCl/PhCH2MgCl synthetic cycle (18). The high reduction potentials of low valent f-element complexes suggests a promising approach for the reductive activation of C1-small molecules such as CO (19–22) and CO2 (23); however, only a handful of f-element complexes can affect the reductive homologation of CO under ambient conditions (24, 25). Organometallic uranium complexes can effect reductive homologation of CO to Cn-oligomers (n = 2–4) (26–28), and a small number of uranium triamide and triaryloxide complexes also reductively dimerize CO (29, 30). More recently, an organometallic uranium complex was shown to effect the simultaneous reduction and hydrogenation of CO to give a coordinated methoxide that could be liberated as CH3OSiMe3 (31); however, there have been no reports of successful homologation, functionalization, and removal of Cn-oligomers (n≥2) under mild conditions with retention of a metal complex that may be recycled for reuse in a synthetic cycle as has been demonstrated for CO2 (32, 33), P4 (34), and carbodiimides (35). Thus, the direct homologation of carbon monoxide into oxycarbons in viable, simple, and straightforward synthetic cycles is yet to be accomplished (36).
Here, we show that a simple, preorganized triamidoamine uranium(III) complex can reductively homologate CO to give selectively the ethyne diolate unit. This valuable C2 building block can be functionalized and liberated as bis(organosiloxy)acetylenes following straightforward treatment with organosilylhalides, and these acetylenes readily convert to furanones. A triamidoamine uranium(IV) iodide complex, which is the precursor to the triamido uranium(III) complex, is formed concomitantly, thus, the uranium(III) complex can be recycled. Therefore, using only three steps and four components, all the required transformations to close the synthetic cycle for the homologation of carbon monoxide to higher order derivatives in high yields are now established.
Results and Discussion
Reductive Homologation of CO.
As part of our work examining the chemistry of triamido uranium complexes (37–40), we studied the reaction of [U(TrenDMSB)] [1, ref. 41, TrenDMSB = N(CH2CH2NSiMe2But)3] with excess CO at ambient pressure (1 bar) in pentane. This resulted in a rapid fading of the purple colored solution of 1 to brown to give the dimeric complex [{U(TrenDMSB)}2(μ-η1: η1-OCCO)] (2) in up to 62% isolated yield (Scheme 1). The reaction is quantitative as indicated by 1H NMR spectroscopy, and the yield of 2 reflects its high solubility in hydrocarbons. Crystallization from pentane at -30 °C afforded pale green plates suitable for X-ray diffraction studies, which revealed a reductively homologated CO dimer, bridging two uranium(IV) centers that are related by a crystallographic inversion center.
Scheme 1.
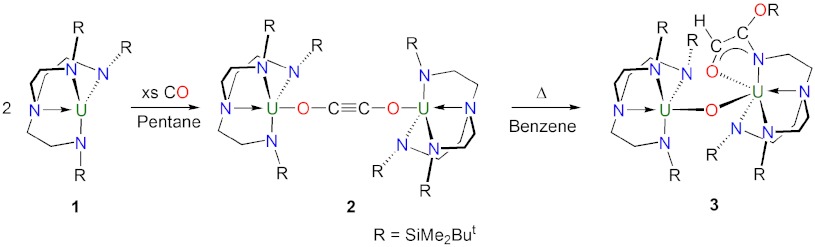
Synthesis of 2 and 3.
The structure of 2 (Fig. 1) shows an almost linear -O-C ≡ C-O- unit that exhibits a O1-C25-C25A angle of 173.8(6)°. The U-Namide and U-Namine bond lengths are unremarkable, but the U-O bond distances of 2.156(3) Å are notably longer than in [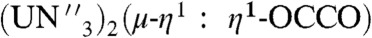 ] [N′′ = N(SiMe3)2, U-O av. = 2.102(2) Å] (29) reflecting the sterically highly congested uranium centers in 2. The C25-C25A bond length of 1.187(8) Å confirms the presence of a C ≡ C triple bond in the ethyne diolate fragment. When the preparation of 2 is repeated with isotope 13C-enriched 13CO, the 13C NMR spectrum of 13C-2 exhibits a resonance at 236 ppm, that corresponds to the ethyne diolate carbon nuclei, and this resonance is not observed in the absence of 13C labeling. The FTIR spectrum of 2 exhibits a band at 1,341 cm-1 assigned as a C-O stretch. This band is shifted to 1,316 cm-1 in 13C-2, which compares well to the calculated isotopomer shift of 1,311 cm-1 from reduced mass considerations. The oxidation of uranium(III) to uranium(IV) during the conversion of 1 to 2 is supported by the UV/visible(vis)/NIR electronic absorption spectrum and variable-temperature superconducting quantum interference device (SQUID) magnetization data for 2. Complex 2 exhibits strong ligand to metal charge transfer absorptions in the region 25,000 - 15,000 cm-1 and weak (ε ∼ 20 M-1 cm-1) bands in the 15,000 - 9,000 cm-1 region that are characteristic of Laporte forbidden f → f transitions for uranium(IV). The magnetic moment of powdered 2 is 4.00 μB at 300 K. This value declines smoothly until ∼50 K after which it decreases more sharply so that at 1.8 K the magnetic moment is 0.69 μB. The SQUID magnetization trace tends to zero, which is consistent with a f2 (
] [N′′ = N(SiMe3)2, U-O av. = 2.102(2) Å] (29) reflecting the sterically highly congested uranium centers in 2. The C25-C25A bond length of 1.187(8) Å confirms the presence of a C ≡ C triple bond in the ethyne diolate fragment. When the preparation of 2 is repeated with isotope 13C-enriched 13CO, the 13C NMR spectrum of 13C-2 exhibits a resonance at 236 ppm, that corresponds to the ethyne diolate carbon nuclei, and this resonance is not observed in the absence of 13C labeling. The FTIR spectrum of 2 exhibits a band at 1,341 cm-1 assigned as a C-O stretch. This band is shifted to 1,316 cm-1 in 13C-2, which compares well to the calculated isotopomer shift of 1,311 cm-1 from reduced mass considerations. The oxidation of uranium(III) to uranium(IV) during the conversion of 1 to 2 is supported by the UV/visible(vis)/NIR electronic absorption spectrum and variable-temperature superconducting quantum interference device (SQUID) magnetization data for 2. Complex 2 exhibits strong ligand to metal charge transfer absorptions in the region 25,000 - 15,000 cm-1 and weak (ε ∼ 20 M-1 cm-1) bands in the 15,000 - 9,000 cm-1 region that are characteristic of Laporte forbidden f → f transitions for uranium(IV). The magnetic moment of powdered 2 is 4.00 μB at 300 K. This value declines smoothly until ∼50 K after which it decreases more sharply so that at 1.8 K the magnetic moment is 0.69 μB. The SQUID magnetization trace tends to zero, which is consistent with a f2 (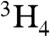 ) electronic configuration that at low temperature has a magnetic singlet state (42).
) electronic configuration that at low temperature has a magnetic singlet state (42).
Fig. 1.

Molecular structure of 2 with displacement ellipsoids at 50% and hydrogen atoms omitted for clarity. Selected bond lengths (Å) and angles (°) for 2:U1-N1, 2.265(4); U1-N2, 2.246(4); U1-N3, 2.263(3); U1-N4, 2.534(3); U1-O1, 2.156(3); O1-C25, 1.302(5); C25-C25A, 1.187(8); U1-O1-C25, 158.8(3); O1-C25-C25A, 173.8(6).
Thermolysis of the Homologated Complex.
Thermolysis of 2 at 80 °C in benzene for 48 h afforded its quantitative conversion to complex 3 as determined by 1H NMR spectroscopy (Scheme 1). Intermediates were not observed under the experimental conditions, and the relatively simple 1H NMR spectrum of 2, which exhibits four resonances, converts to a more complex spectrum containing fourteen resonances. Although the proton, which most likely originates from solvent, on the newly formed alkene unit in 3 was not observed, the complex nature of the 1H NMR spectrum of 3 is consistent with its dinuclear formulation. Complex 3 was isolated in 60% yield as green plates suitable for X-ray diffraction studies, which revealed insertion of the ethyne diolate group into one of the N-Si bonds of the TrenDMSB ligand (Fig. 2). This reaction is accompanied by protonation of the ethyne diolate and insertion of an oxo-bridge, which most likely derives from the glass reaction vessel, between the two uranium(IV) centers. The reduction of the C ≡ C triple bond in the ethyne diolate group is confirmed by the C43-C44 bond length of 1.478(7) Å, and the C-H proton was located in the Fourier difference map. The O2-C43 and N7-C44 bond distances of 1.343(7) and 1.254(6) Å, respectively, are suggestive of significant C = O and N = C double bond character. Taken together with the C43-C44 bond, which is intermediate to single and double C-C bonds, these metrical parameters suggest delocalization around the OCCN fragment. In support of this, the U2-N7 bond length of 2.541(4) Å is closer to the dative U-Namine distances (average 2.636 Å) than the polarized-covalent U-Namide distances (average 2.302 Å) reflecting its dative nature resulting from the strong imino character of N7. Complex 3 has been fully characterized and exhibits a solid state magnetic moment of 4.28 μB at 298 K that decreases to 0.87 μB at 1.8 K consistent with the uranium(IV) formulation. Complex 3 is notable because magnetometric measurements reveal a maximum in a plot of χM vs. T at 3 K (Fig. 3) that suggests weak antiferromagnetic exchange coupling between the two uranium cations through the oxo-bridge. Magnetic exchange coupling between uranium(IV) centers is very rare (43), and, more generally, clear-cut exchange between uranium ions is uncommon because it is often masked by other phenomena (44).
Fig. 2.

Molecular structure of 3 with displacement ellipsoids at 50% and hydrogen atoms omitted for clarity. Selected bond lengths (Å) and angles (°) for 3:U1-N1, 2.300(4); U1-N2, 2.316(4); U1-N3, 2.284(4); U1-N4, 2.580(4); U1-O1, 2.116(3); U2-O1, 2.138(3); U2-O2, 2.186(4); U2-N5, 2.322(4); U2-N6, 2.289(4); U2-N7, 2.541(4); U2-N8, 2.691(4); N7-C44, 1.254(6); C44-O3, 1.372(6); C43-C44, 1.478(7); C43-O2, 1.343(7); U1-O1-U2, 160.87(16); O2-U2-N7, 65.06(14); O2-C43-C44, 109.8(5); C43-C44-N7, 119.6(5).
Fig. 3.

Plot of χM versus T for 3.
The observed insertion that produces 3 contrasts to the only other example of linear ethyne diolate reactivity where a silyl-methyl C-H bond in a uranium amide coordination environment was reported to add across the C ≡ C triple bond (29); however, the straightforward and clean formation of 3 suggested that the ethyne diolate unit in 2 is unusually silicophilic (32, 45, 46) and, therefore, amenable to reaction with organosilyl reagents that could be exploited to close the synthetic cycle.
Liberation Studies and Closing the Synthetic Cycle.
Treatment of a pentane solution of 2 or 13C-2 with Me3SiI or PhMe2SiI quantitatively afforded a mixture of [U(TrenDMSB)I] (4) (47), which is the precursor to 1, and the oils Me3SiOC ≡ COSiMe3 (5a and 13C-5a) or PhMe2SiOC ≡ COSiMe2Ph (5b or 13C-5b) (Scheme 2). In contrast, 3 does not appear to react with Me3SiI or PhMe2SiI. The formation of 4 was confirmed by comparison of its 1H NMR spectrum with that of an authentic, independently prepared sample and also by an X-ray crystallographic unit cell determination. Complex 4 can easily be separated from 5a/5b in essentially quantitative yield and be converted back to 1 in essentially quantitative yield by stirring over a potassium mirror. Compound 1 can be recycled for subsequent reuse over at least four cycles in yields of 80–90% of 4 for each step before separation of 4 from 5 becomes impractical on the scales employed in this study (details are given in the SI Appendix). Addition of Ph2MeSiI or Ph3SiI to 2 or 13C-2 did not liberate the corresponding bis(organosiloxy)acetylenes. Although we cannot rule out electronic effects, these observations suggest that PhMe2SiI represents the optimum steric compromise between the functionalization and liberation reaction proceeding and isolating a well-defined homologated product. The reductive homologation of CO to 5 and 6 affords a route to organic bis(ether)acetylenes and furanones that are precursors to important diols and furans that are produced on industrial scales (8, 48). This method is also a convenient route for incorporating 13C-labeled organic molecules into a synthetic cycle and utilizes organosilyl halides that are industrially produced on a large scale. These findings establish that, under the appropriate conditions, uranium(III) can mediate a complete and straightforward synthetic cycle for the homologation of CO to higher derivatives as illustrated in Scheme 2.
Scheme 2.

The synthetic cycle for the reductive homologation and functionalization of carbon monoxide to 5a/5b and ultimately to 6a/6b.
Postliberation Chemistry of the Bis(organosiloxy)acetylenes.
Following functionalization and liberation of the ethyne diolate group the viscous oils RMe2SiOC ≡ COSiMe2R (R = Me, 5a or 13C-5a; R = Ph, 5b or 13C-5b) are liberated in nonoptimized yields of 90% for 5b/13C-5b and estimated yields of 90% for 5a/13C-5a; however, the stability of these compounds appears to be dependent on the nature of the silyl substituents. Compounds 5b or 13C-5b appear to be more stable than 5a or 13C-5a in neat form possibly due to the extra steric protection afforded by the phenyl substituent. The identities of 5b and 13C-5b (R = Ph) were confirmed by CHN combustion analyses, FTIR spectroscopy, and the presence of the molecular ions [5b - Ph]+ and [13C-5b - Ph]+ at m/z = 249 and 251, respectively, in electrospray ionization (MS-ESI) mass spectra; however, NMR spectra of 5b and 13C-5b could not be obtained because in solution they react further to afford quantitatively the furanones 6b and 13C-6b (Scheme 2) as evidenced by multinuclear NMR and IR spectroscopies and mass spectrometry (see SI Appendix).
For 5a and 13C-5a (R = Me), the resulting bis(trimethylsiloxy)acetylene is unstable and rapidly undergoes subsequent reaction neat or in solution. For example, molecular ions were not observed and instead only the dimerized squarate ions [(Me3SiOCCOSiMe3)2]+ and 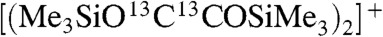 were observed at m/z = 404 and 408, respectively, in MS electron impact ionization (EI) experiments. This is consistent with the literature where ButOCCOBut is the only known stable bis(oxy)acetylene (49). All other derivatives react further at room temperature to give ill-defined mixtures of products (50), and ButOCCOBut can be converted to a squarate derivative by thermolysis (51). This generates a ketene through isobutene elimination and hydrogen migration to give ButOC(H) = C = O that undergoes a [2 + 2]-cycloaddition with the parent ButOCCOBut (52). Although trimethylsilyl is larger than tert-butyl, the Si-O bond is longer than the corresponding C-O bond, which affords less steric protection of the OCCO linkage for the silyl derivative. Thus, although we could record FTIR spectra in Nujol mulls, definitive NMR spectra of 5a or 13C-5a could not be obtained. Whereas the CHN data for 5b or 13C-5b are consistent with the proposed formulations, the CHN data for 5a or 13C-5a consistently deviate from anticipated values and repeatedly return the same elemental composition data showing that subsequent reaction is already and repeatedly occurring at this stage.
were observed at m/z = 404 and 408, respectively, in MS electron impact ionization (EI) experiments. This is consistent with the literature where ButOCCOBut is the only known stable bis(oxy)acetylene (49). All other derivatives react further at room temperature to give ill-defined mixtures of products (50), and ButOCCOBut can be converted to a squarate derivative by thermolysis (51). This generates a ketene through isobutene elimination and hydrogen migration to give ButOC(H) = C = O that undergoes a [2 + 2]-cycloaddition with the parent ButOCCOBut (52). Although trimethylsilyl is larger than tert-butyl, the Si-O bond is longer than the corresponding C-O bond, which affords less steric protection of the OCCO linkage for the silyl derivative. Thus, although we could record FTIR spectra in Nujol mulls, definitive NMR spectra of 5a or 13C-5a could not be obtained. Whereas the CHN data for 5b or 13C-5b are consistent with the proposed formulations, the CHN data for 5a or 13C-5a consistently deviate from anticipated values and repeatedly return the same elemental composition data showing that subsequent reaction is already and repeatedly occurring at this stage.
The FTIR data for 5a and 5b are consistent with the proposed formulations, but we could not unambiguously identify any isotopomer effects as the bands that are anticipated to be affected are in the fingerprint regions that contain overlapping band structures; however, over time the FTIR spectra of neat 5a, 13C-5a, 5b, and 13C-5b change and carbonyl bands are observed to grow in intensity. For 5a and 5b, carbonyl stretches at 1,780 and 1,764 cm-1, respectively, are observed. For 13C-5a and 13C-5b the corresponding stretches are observed at 1,753 and 1,719 cm-1, respectively, which corresponds to isotope shifts of 27 and 45 cm-1, respectively. This compares well to 12C/13C isotopomer shifts expected from reduced mass considerations (usually ∼40 cm-1). Initial 13C NMR spectra (see SI Appendix, Figs. S11–S18) of 5a or 5b show the presence of two organic products. Monitoring over time shows that 5a/5b each convert into a mixture of the corresponding furanones 6a/b with another species tentatively assigned as the tautomeric hydroxyfurans 7a/b. Resonances attributable to 7a/b decay over time to give only 6a/b, which is consistent with the relative acid dissociation constants (pKa) of the C-H and O-H bonds involved in this tautomeric equilibrium. In dry benzene, the conversion of 5b and 13C-5b into 6b or 13C-6b is ∼40% complete after 18 h, and full conversion is achieved within 2 wks, but reaction times can be reduced to within 2 h by the addition of water or heating. The final chemical shifts compare very well to the related furanones 2,3,5,6-tetrakis-O-(trimethylsilyl) ascorbate and 3,4-dimethoxy-5-(hydroxymethyl)-5-methyl-2(5H)-furanone (53, 54).
The proposed mechanism for the conversion of 5a/b to 6a/b is illustrated in Scheme 3. Conversion of bis(oxy)acetylenes to ketenes is well known as are [2 + 2]-cycloadditions of ketenes with alkynes to give squarate derivatives. A π-cycloreversion reaction produces a transient ketene that is converted to a carboxylate. Base-induced elimination of a silyl group opens the way for a subsequent tautomerization that is followed by ring closure. A formal overall sigmatropic rearrangement, which in a concerted mechanism would be symmetry forbidden, can proceed in a stepwise mechanism that may involve ring opening and subsequent reclosure and affords the hydroxyfurans 7a/b that subsequently tautomerize to the furanones 6a/b. This mechanism is supported by the extensive literature precedents for the individual reaction steps (55) and also by the tentative observation of 7a/b as well as 6a/b in NMR spectra recorded promptly after the aqueous washing step, the decay of resonances attributable to 7a/b, and the growth of resonances assigned to 6a/b over time to finally give 6a/b only. The presence of water accelerates these reactions, but it should be noted that trace H+ and hydroxyl groups on the surface of glass flasks can promote this reaction (55).
Scheme 3.

Proposed mechanism for the conversion of the bis(siloxy)acetylenes 5a/5b to the furanones 6a/6b.
Electronic Structure of 2 and Thermodynamic Considerations.
The facile liberation of bis(organosiloxy)acetylenes from 2 and 13C-2 in yields of ca. 90% contrasts with previous reports where coordinated ethyne diolates were reported as impervious to release (29) and forcing conditions resulted in decomposition of the metal complexes (14) that would be highly deleterious to synthetic or catalytic cycles (15). In order to investigate the liberation chemistry of 2, we carried out Density Functional Theory calculations on the whole molecule of 2. The calculated bond lengths and angles are reproduced to within 0.05 Å and 2°, respectively; so, we conclude that the calculations provide a qualitative model of the electronic structure of 2. The HOMO and HOMOs -1, -2, and -3 of 2 are singly occupied and of essentially exclusive 5f character and are delocalized across both uranium centers (Fig. 4). This supports the uranium(IV) formulation suggested by the structural, spectroscopic, and magnetic characterization of 2. HOMO–4 and HOMO–5 describe the principal U-O interactions in the frontier orbital region and represent a quasidegenerate pair of π-donor interactions from oxygen to uranium that approximate to a1u symmetry if the molecule is considered to possess Ci point symmetry. These interactions are polarized towards the oxygen centers as expected for such polar bonds. The calculated spin densities at the uranium, oxygen, amide, and amine centers in 2 are calculated to be 2.33, -0.07, -0.07, and -0.01, respectively, and the U-O, U-Namide, and U-Namine, C-O, and C ≡ C Nalewajski-Mrozek bond indices are 1.25, 1.44, 0.52, 1.49, and 2.54, respectively. These values reflect weak U-O π-donation, stronger, but still relatively modest U-N π-donation, the dative amine donation to uranium, and also the formation of a C ≡ C triple bond.
Fig. 4.

The top six occupied α-spin Kohn–Sham molecular orbitals of 2. Hydrogen atoms are omitted for clarity. (A) HOMO (379a, -2.743 eV); (B) HOMO–1 (378a, -2.745 eV; (C) HOMO–2 (377a, -2.778); (D) HOMO–3 (376a, -2.781); (E) HOMO–4 (375a, -4.399); (F) HOMO–5 (374a, -4.449 eV).
In order to calculate the bond enthalpy change for the reactions
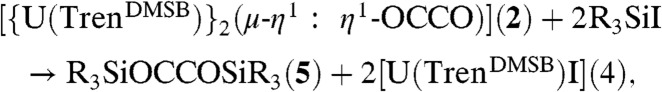 |
[1] |
we calculated the gas phase geometry optimized structures of 2, Me3SiI, PhMe2SiI, Ph2MeSiI, Ph3SiI, Me3SiOCCOSiMe3 (5a), PhMe2SiOCCOSiMe2Ph (5b), Ph2MeSiOCCOSiMePh2, Ph3SiOCCOSiPh3, and 4 to estimate the heats of formation (ΔHfor) for these species. ΔHrxn for the above reaction is
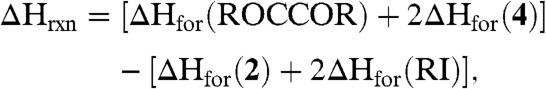 |
[2] |
which reveals that the enthalpy change of the gas phase reaction is exergonic overall at -15.9 kcal mol-1 (R = Me3Si), -15.4 kcal mol-1 (R = PhMe2Si), -13.1 kcal mol-1 (R = Ph2MeSi), and -9.5 kcal mol-1 (R = Ph3Si). Experimentally, the latter two silyl iodides did not affect liberation of the ethyne diolate fragment that we attribute to steric effects that may kinetically block the reaction. Interestingly, the corresponding enthalpies when [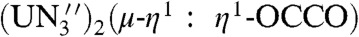 ] (29), which was reported to be inert to liberation using silyl reagents (29), and [
] (29), which was reported to be inert to liberation using silyl reagents (29), and [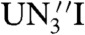 ] are substituted for 2 and 4, respectively, in Eq. 2 are -2.8 kcal mol-1 (R = Me3Si), -2.4 kcal mol-1 (R = PhMe2Si), -0.1 kcal mol-1 (R = Ph2MeSi), and +3.5 kcal mol-1 (R = Ph3Si). The ∼12 kcal mol-1 disparity between these two sets of calculated data may reflect a smaller ligand reorganization energy for preorganized [U(TrenDMSB)] (56) compared to [
] are substituted for 2 and 4, respectively, in Eq. 2 are -2.8 kcal mol-1 (R = Me3Si), -2.4 kcal mol-1 (R = PhMe2Si), -0.1 kcal mol-1 (R = Ph2MeSi), and +3.5 kcal mol-1 (R = Ph3Si). The ∼12 kcal mol-1 disparity between these two sets of calculated data may reflect a smaller ligand reorganization energy for preorganized [U(TrenDMSB)] (56) compared to [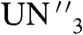 ] that, for the former, is calculated to be 10.8 kcal mol-1 per uranium center. It is interesting to note that the U-O-C angle in 2 is 158.8(3)°; whereas, in [
] that, for the former, is calculated to be 10.8 kcal mol-1 per uranium center. It is interesting to note that the U-O-C angle in 2 is 158.8(3)°; whereas, in [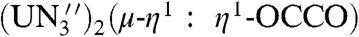 )], the corresponding angle is 177.5°. Although care must be taken when correlating M-O-R bond angles to bond strength (57, 58), it may be that the rigid and steric demands of the TrenDMSB ligand weakens the U-OR linkage in 2 and perhaps opens up the sterically crowded environment to kinetically feasible substitution and product liberation. The solvent and entropy of the reaction are clearly important reaction parameters that are not considered in these gas phase calculations; however it would appear that the steric demands of rigid, preorganized (56) ancillary ligands may play a key role.
)], the corresponding angle is 177.5°. Although care must be taken when correlating M-O-R bond angles to bond strength (57, 58), it may be that the rigid and steric demands of the TrenDMSB ligand weakens the U-OR linkage in 2 and perhaps opens up the sterically crowded environment to kinetically feasible substitution and product liberation. The solvent and entropy of the reaction are clearly important reaction parameters that are not considered in these gas phase calculations; however it would appear that the steric demands of rigid, preorganized (56) ancillary ligands may play a key role.
Summary and Outlook.
To summarize, we have shown that under mild conditions complex 1 can reductively homologate CO to selectively give 2. Following treatment with organosilyl halides, 5 is liberated, and this occurs with concomitant formation of 4, which is the direct precursor to 1. Thus, 1 can be recycled and reused, thereby closing the synthetic cycle with only three steps. We suggest that the unusually facile liberation of 5 may be attributable to the sterically demanding nature of 1 coupled with its rigidity, and this may be an important consideration to apply more generally to the challenge of designing selective catalytic cycles for the reductive homologation and functionalization of CO. The elaboration of 5 into 6 holds promise for the catalytic homologation of CO into more complex and value-added organic molecules. Detailed chemical and engineering studies are currently underway to ameliorate reagent incompatibilities (59), and the simplicity of this system suggests that catalytic CO functionalization may soon be within reach.
Materials and Methods
All operations involving 1–4 were carried out under pure nitrogen or carbon monoxide using Schlenk or glovebox techniques. All reagents and solvents were rigorously dried and degassed prior to use. 1H, 13C, and 29Si NMR spectra were recorded on a Bruker 400 spectrometer operating at 400.2, 100.6, and 79.5 MHz, respectively; chemical shifts are quoted in ppm and are relative to tetramethylsilane. FTIR spectra were recorded on a Bruker Tensor 27 spectrometer. UV/vis/NIR spectra were recorded on a Perkin Elmer Lambda 750 spectrometer. Variable-temperature magnetic moment data were recorded in an applied DC field of 0.1 T on a Quantum Design MPMS XL5 SQUID magnetometer using doubly recrystallized powdered samples. Mass Spectrometry was carried out using Bruker Apex IV Fourier transform ion cyclotron resonance (MS-EI) and Bruker MicroTOF (MS-ESI) instruments. Elemental microanalyses were carried out at the University of Nottingham and by Stephen Boyer at the London Metropolitan University (UK). Density Functional Theory calculations were carried out with Amsterdam Density Functional program version 2010.01 (60). X-ray crystallographic files are available in Datasets 1 and 2 and full details of this and experimental procedures are published in the SI Appendix.
Supplementary Material
Acknowledgments.
We thank Shazad Aslam for the acquisition of NMR data, Dr. Mick Cooper and Graham Coxhill for obtaining MS data, Tong Liu for CHN elemental microanalysis, and Dr. Ross Denton for valuable discussions regarding the conversion of 5 to 6. We are grateful to the European Research Council, the UK Engineering and Physical Sciences Research Council, the University of Nottingham, and the UK National Nuclear Laboratory for generous funding and support and the Royal Society for the award of a University Research Fellowship (S.T.L.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Data deposition: The atomic coordinates have been deposited in the Cambridge Structural Database, Cambridge Crystallographic Data Centre, Cambridge CB2 1EZ, United Kingdom (CCDC 854138 and 854139). These data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1203417109/-/DCSupplemental.
References
- 1.Kerr RA. Bumpy road ahead for world’s oil. Science. 2005;310:1106–1108. doi: 10.1126/science.310.5751.1106. [DOI] [PubMed] [Google Scholar]
- 2.Herrmann WA. Organometallic aspects of the Fischer-Tropsch synthesis. Angew Chem Int Ed. 1982;21:117–130. [Google Scholar]
- 3.Sabzyan H, Noorbala MR. Ab Initio and DFT study of carbon monoxide cyclic oligomers, (CO)2 to (CO)6. J Mol Struct Theochem. 2003;626:143–158. [Google Scholar]
- 4.Marks TJ. Actinide organometallic chemistry. Science. 1982;217:989–997. doi: 10.1126/science.217.4564.989. [DOI] [PubMed] [Google Scholar]
- 5.Hebrard F, Kalck P. Cobalt-catalyzed hydroformylation of alkenes: Generation and recycling of the carbonyl species, and catalytic cycle. Chem Rev. 2009;109:4272–4282. doi: 10.1021/cr8002533. [DOI] [PubMed] [Google Scholar]
- 6.Manriquez JM, Fagan PJ, Marks TJ, Day CS, Day VW. Bis(pentamethylcyclopentadienyl)actinide alkyls: Facile activation of carbon monoxide, carbon-carbon double bond formation, and the production of unusual oxygen-bonded migratory insertion products. J Am Chem Soc. 1978;100:7112–7114. [Google Scholar]
- 7.Fagan PJ, et al. Insertion of carbon monoxide into metal-nitrogen bonds. Synthesis, chemistry, structures, and structural dynamics of bis(pentamethylcyclopentadienyl) organoactinide dialkylamides and η2-carbamoyls. J Am Chem Soc. 1981;103:2206–2220. [Google Scholar]
- 8.Moloy KG, Fagan PJ, Manriquez JM, Marks TJ. A Synthetic and mechanistic study of oxycarbene-like coupling reaction patterns of actinide η2-acyl complexes with carbon monoxide and isocyanides. J Am Chem Soc. 1986;108:56–67. [Google Scholar]
- 9.Wayland B, Fu X. Building molecules with carbon monoxide reductive coupling. Science. 2006;311:790–791. doi: 10.1126/science.1123884. [DOI] [PubMed] [Google Scholar]
- 10.Buchner W. The so-called “alkali carbonyls” III the “alkali carbonyls” as mixtures of an organometallic compound and a metal acetylenediolate. Helv Chim Acta. 1963;46:2111–2120. [Google Scholar]
- 11.Silvestri G, Gambino S, Filardo G, Spadaro G, Palmisano L. The electrochemistry of carbon monoxide reductive cyclotetramerization to squarate anion. Electrochim Acta. 1978;23:413–417. [Google Scholar]
- 12.Bianconi PA, Williams ID, Engeler MP, Lippard SJ. Reductive coupling of two carbon monoxide ligands to form a coordinated alkyne. J Am Chem Soc. 1986;108:311–313. [Google Scholar]
- 13.Vrtis RN, Rao CP, Bott SG, Lippard SJ. Synthesis and stabilization of tantalum-coordinated dihydroxyacetylene from two reductively coupled carbon monoxide ligands. J Am Chem Soc. 1988;110:7564–7566. [Google Scholar]
- 14.Protasiewicz JD, Lippard SJ. Vanadium-promoted reductive coupling of CO and facile hydrogenation to form cis-disiloxyethylenes. J Am Chem Soc. 1991;113:6564–6570. [Google Scholar]
- 15.Carnahan EM, Protasiewicz JD, Lippard SJ. 15 years of reductive coupling: What have we learned? Acc Chem Res. 1993;26:90–97. [Google Scholar]
- 16.Coffin VL, Brennen W, Wayland BB. Thermodynamic studies of competitive adduct formation: Single- and double-insertion reactions of carbon monoxide with rhodium octaethylporpyrin dimer. J Am Chem Soc. 1988;110:6063–6069. doi: 10.1021/ja00226a022. [DOI] [PubMed] [Google Scholar]
- 17.Watanabe T, Ishida Y, Matsuo T, Kawaguchi H. Reductive coupling of six carbon monoxides by a ditantalum hydride complex. J Am Chem Soc. 2009;131:3474–3475. doi: 10.1021/ja9007276. [DOI] [PubMed] [Google Scholar]
- 18.Matsuo T, Kawaguchi H. A synthetic cycle for H2/CO activation and allene synthesis using recyclable zirconium complexes. J Am Chem Soc. 2005;127:17198–17199. doi: 10.1021/ja0567079. [DOI] [PubMed] [Google Scholar]
- 19.Brennan JG, Andersen RA, Robbins JL. Preparation of the first molecular carbon monoxide complex of uranium, (Me3SiC5H4)3UCO. J Am Chem Soc. 1986;108:335–336. [Google Scholar]
- 20.Parry J, Carmona E, Coles S, Hursthouse M. Synthesis and single crystal X-ray diffraction study on the first isolable carbonyl complex of an actinide, (C5Me4H)3UCO. J Am Chem Soc. 1995;117:2649–2650. [Google Scholar]
- 21.Evans WJ, Kozimor SA, Nyce GW, Ziller JW. Comparative reactivity of sterically crowded nf3 (C5Me5)3Nd and (C5Me5)3U complexes with CO: Formation of a nonclassical carbonium ion versus an f element metal carbonyl complex. J Am Chem Soc. 2003;125:13831–13835. doi: 10.1021/ja036631l. [DOI] [PubMed] [Google Scholar]
- 22.Castro-Rodriguez I, Meyer K. Carbon dioxide reduction and carbon monoxide activation employing a reactive uranium(III) complex. J Am Chem Soc. 2005;127:11242–11243. doi: 10.1021/ja053497r. [DOI] [PubMed] [Google Scholar]
- 23.Castro-Rodríguez I, Nakai H, Zakharov LN, Rheingold AL, Meyer K. A linear, O-coordinated η1-CO2 bound to uranium. Science. 2004;305:1757–1759. doi: 10.1126/science.1102602. [DOI] [PubMed] [Google Scholar]
- 24.Evans WJ, Lee DS, Ziller JW, Kaltsoyannis N. Rare earth complexes require 90 pounds per square inch overpressures to homologate carbon monoxide:trivalent [(C5Me5)2(THF)Ln]2(μ-η2: η2-N2) complexes as reducing agents including the reductive homologation of CO to a ketene carboxylate (μ-η4-O2C-C = C = O) J Am Chem Soc. 2006;128:14176–14184. doi: 10.1021/ja0640851. [DOI] [PubMed] [Google Scholar]
- 25.Evans WJ, Grate JW, Hughes LA, Zhang H, Atwood JL. Reductive homologation of CO to a ketenecarboxylate by a low-valent organolanthanide complex: Synthesis and X-ray crystal structure of [(C5Me5)4Sm2(O2CCCO)(THF)]2. J Am Chem Soc. 1985;107:3728–3730. [Google Scholar]
- 26.Summerscales OT, Cloke FGN, Hitchcock PB, Green JC, Hazari N. Reductive cyclotrimerization of carbon monoxide to the deltate dianion by an organometallic uranium complex. Science. 2006;311:829–831. doi: 10.1126/science.1121784. [DOI] [PubMed] [Google Scholar]
- 27.Summerscales OT, Cloke FGN, Hitchcock PB, Green JC, Hazari N. Reductive cyclotetramerization of CO to squarate by a U(III) complex: The X-ray crystal structure of [U(η-C8H6{Sii Pr 3-1,4}2)(η-C5Me4H)]2(μ-η2: η2-C4O4) J Am Chem Soc. 2006;128:9602–9603. doi: 10.1021/ja063222r. [DOI] [PubMed] [Google Scholar]
- 28.Frey ASP, et al. Mechanistic studies on the reductive cyclooligomerisation of CO by U(III) mixed sandwich complexes: The molecular structure of [U(η-C8H6{Sii Pr 3-1,4}2)(η-C5Me4H)]2(μ-η1: η1-C2O2) J Am Chem Soc. 2008;130:13816–13817. doi: 10.1021/ja8059792. [DOI] [PubMed] [Google Scholar]
- 29.Arnold PL, Turner ZR, Bellabarba RM, Tooze RP. Carbon monoxide coupling and functionalisation at a simple uranium coordination complex. Chem Sci. 2011;2:77–79. [Google Scholar]
- 30.Mansell SM, Kaltsoyannis N, Arnold PL. Small molecule activation by uranium Tris(aryloxides): Experimental and computational studies of binding of N2, coupling of CO, and deoxygenation insertion of CO2 under ambient conditions. J Am Chem Soc. 2011;133:9036–9051. doi: 10.1021/ja2019492. [DOI] [PubMed] [Google Scholar]
- 31.Frey ASP, Cloke FGN, Coles MP, Maron L, Davin T. Facile conversion of CO/H2 to methoxide at a uranium(III) Center. Angew Chem Int Ed. 2011;50:6881–6883. doi: 10.1002/anie.201101509. [DOI] [PubMed] [Google Scholar]
- 32.Matson EM, Forrest WP, Fanwick PE, Bart SC. Functionalization of carbon dioxide and carbon disulfide using a stable uranium(III) alkyl complex. J Am Chem Soc. 2011;133:4948–4954. doi: 10.1021/ja110158s. [DOI] [PubMed] [Google Scholar]
- 33.Silvia JS, Cummins CC. Ligand-Based reduction of CO2 to CO mediated by an anionic niobium nitride complex. J Am Chem Soc. 2010;132:2169–2717. doi: 10.1021/ja910445r. [DOI] [PubMed] [Google Scholar]
- 34.Figueroa JS, Cummins CC. Phosphaalkynes from acid chlorides via P for O(Cl) metathesis: A recyclable niobium phosphide (P3-) reagent that effects C-P triple bond formation. J Am Chem Soc. 2004;126:13916–13917. doi: 10.1021/ja0461522. [DOI] [PubMed] [Google Scholar]
- 35.Castro-Rodríguez I, Nakai H, Meyer K. Multiple-Bond metathesis mediated by a sterically pressured uranium complex. Angew Chem Int Ed. 2006;45:2389–2392. doi: 10.1002/anie.200501667. [DOI] [PubMed] [Google Scholar]
- 36.Gladysz JA, et al. Organometallics roundtable 2011. Organometallics. 2012;31:1–18. [Google Scholar]
- 37.Liddle ST, et al. σ and π donation in an unsupported uranium-gallium bond. Angew Chem Int Ed. 2009;48:1077–1080. doi: 10.1002/anie.200805481. [DOI] [PubMed] [Google Scholar]
-
38.Gardner BM, McMaster J, Lewis W, Blake AJ, Liddle ST. A Crystallizable dinuclear tuck-in-tuck-over tuck-over dialkyl tren uranium complex and double dearylation of
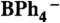 to give the BPh2-Functionalized metallocycle [U{N(CH2CH2NSiMe3)2(CH2CH2NSiMe2CHBPh2)}(THF)] J Am Chem Soc. 2009;131:10388–10389. doi: 10.1021/ja904459q. [DOI] [PubMed] [Google Scholar]
to give the BPh2-Functionalized metallocycle [U{N(CH2CH2NSiMe3)2(CH2CH2NSiMe2CHBPh2)}(THF)] J Am Chem Soc. 2009;131:10388–10389. doi: 10.1021/ja904459q. [DOI] [PubMed] [Google Scholar] - 39.Patel D, et al. A formal high oxidation state inverse-sandwich diuranium complex: A new route to f-block-metal bonds. Angew Chem Int Ed. 2011;50:10388–10392. doi: 10.1002/anie.201104110. [DOI] [PubMed] [Google Scholar]
- 40.Gardner BM, Patel D, Lewis W, Blake AJ, Liddle ST. Photochemically promoted bond-cleavage and -capture in a diazomethane derivative of a triamidoamine uranium(IV) complex. Angew Chem Int Ed. 2011;50:10440–10443. doi: 10.1002/anie.201105098. [DOI] [PubMed] [Google Scholar]
- 41.Roussel P, Scott P. Complex of dinitrogen with trivalent uranium. J Am Chem Soc. 1998;120:1070–1071. [Google Scholar]
- 42.Cooper OJ, et al. Uranium-Carbon multiple bonding: Facile access to the pentavalent uranium carbene [U{C(PPh2NSiMe3)2}(Cl)2(I)] and comparison of UV = C and UIV = C double bonds. Angew Chem Int Ed. 2011;50:2383–2386. doi: 10.1002/anie.201007675. and references therein. [DOI] [PubMed] [Google Scholar]
- 43.Lam OP, Heinemann FW, Meyer K. Activation of elemental S, Se and Te with uranium(III):bridging U-E-U (E = S, Se) and diamond-core complexes U-(E)2-U (E = O, S, Se, Te) Chem Sci. 2011;2:1538–1547. [Google Scholar]
- 44.Mills DP, et al. A delocalised arene-bridged diuranium single molecule magnet. Nat Chem. 2011;3:454–460. doi: 10.1038/nchem.1028. and references therein. [DOI] [PubMed] [Google Scholar]
- 45.Brown JL, Wu G, Hayton TW. Oxo ligand silylation in a uranyl β-ketoiminate complex. J Am Chem Soc. 2010;132:7248–7249. doi: 10.1021/ja1013739. [DOI] [PubMed] [Google Scholar]
- 46.Brown JL, Mokhtarzadeh CC, Lever JM, Wu G, Hayton TW. Facile reduction of a uranyl(VI) β-ketoiminate complex to U(IV) upon oxo silylation. Inorg Chem. 2011;50:5105–5112. doi: 10.1021/ic200387n. [DOI] [PubMed] [Google Scholar]
- 47.Roussel P, et al. Complexes of triamidoamines with the early actinides. synthetic routes to monomeric compounds of tetravalent uranium and thorium containing halide and amide ligands. Inorg Chem. 1999;38:3651–3656. doi: 10.1021/ic990563f. [DOI] [PubMed] [Google Scholar]
- 48.Keay BA, Dibble PW. In: Comprehensive Heterocyclic Chemistry II. Bird CW, editor. Vol 2. New York: Elsevier; 1996. [Google Scholar]
- 49.Pericàs MA, Riera A, Serratosa F. Acetylene diethers. Tetrahedron. 1982;38:1505–1508. [Google Scholar]
- 50.Gleiter R, Werz DB. Alkynes between main group elements: From dumbbells via rods to squares and tubes. Chem Rev. 2010;110:4447–4488. doi: 10.1021/cr9003727. [DOI] [PubMed] [Google Scholar]
- 51.Pericàs MA, Serratosa F. Synthetic applications of di-tert-butoxyethyne:synthesis of deltic and squaric acids. Tetrahedron Lett. 1977;18:4437–4438. [Google Scholar]
- 52.Bou A, Pericàs MA, Serratosa F. Synthetic applications of di-tert-butoxyethyne, ii:new syntheses of squaric, semisquaric, and croconic acids. Tetrahedron Lett. 1982;23:361–364. [Google Scholar]
- 53.Sekine M, Futatsugi T, Hata T, Cramer F. Silyl phosphites 21. A new method for the synthesis of L-ascorbic acid 2-O-phosphate by utilizing phosphoryl rearrangement. J Org Chem. 1982;47:3453–3456. [Google Scholar]
- 54.Kohn H, Abuzar S. Studies on the chemical reactivity of bicyclomycin:acid hydrolysis. J Org Chem. 1988;53:2769–2773. [Google Scholar]
- 55.Tidwell TT. Ketenes II. 2nd Ed. Hoboken, NJ: Wiley-Interscience; 2006. [Google Scholar]
- 56.Morton C, et al. Stabilization of cerium(IV) in the presence of an iodide ligand: Remarkable effects of Lewis acidity on valence state. J Am Chem Soc. 1999;121:11255–11256. [Google Scholar]
- 57.Steffey BD, Fanwick PE, Rothwell IP. Solid state structure of the tantalum bis-aryl compounds Ta(OAr-2,6R2)3(C6H5)2 (R = CH3, Pri; OAr-2,6R2 = 2,6-dialkylphenoxide): Observation of a lack of correlation of M-OR distances and M-O-Ar angles for aryloxide derivatives of niobium(V) and tantalum(V) Polyhedron. 1990;9:963–968. [Google Scholar]
- 58.Rosa Russo M, Kaltsoyannis N, Sella A. Are metal alkoxides linear owing to electrostatic repulsion? Chem Commun. 2002:2458–2459. doi: 10.1039/b207435d. [DOI] [PubMed] [Google Scholar]
- 59.Dmitry VY, Schrock RR. Catalytic reduction of dinitrogen to ammonia at a single molybdenum center. Science. 2003;301:76–78. doi: 10.1126/science.1085326. [DOI] [PubMed] [Google Scholar]
- 60.Te Velde G, et al. Chemistry with ADF. J Comput Chem. 2001;22:931–967. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.