

Myo9b is a motor–RhoGAP chimera that has been implicated in inflammatory bowel disease. Findings suggest that Myo9b is essential during both collective and individual wound-induced cell migration. It is also important for maintaining tight junction barrier integrity.
Abstract
Polymorphisms in the gene encoding the heavy chain of myosin IXb (Myo9b) have been linked to several forms of inflammatory bowel disease (IBD). Given that Myo9b contains a RhoGTPase-activating protein domain within its tail, it may play key roles in Rho-mediated actin cytoskeletal modifications critical to intestinal barrier function. In wounded monolayers of the intestinal epithelial cell line Caco2BBe (BBe), Myo9b localizes to the extreme leading edge of lamellipodia of migrating cells. BBe cells exhibiting loss of Myo9b expression with RNA interference or Myo9b C-terminal dominant-negative (DN) tail-tip expression lack lamellipodia, fail to migrate into the wound, and form stress fiber–like arrays of actin at the free edges of cells facing the wound. These cells also exhibit disruption of tight junction (TJ) protein localization, including ZO-1, occludin, and claudin-1. Torsional motility and junctional permeability to dextran are greatly increased in cells expressing DN-tail-tip. Of interest, this effect is propagated to neighboring cells. Consistent with a role for Myo9b in regulating levels of active Rho, localization of both RhoGTP and myosin light chain phosphorylation corresponds to Myo9b-knockdown regions of BBe monolayers. These data reveal critical roles for Myo9b during epithelial wound healing and maintenance of TJ integrity—key functions that may be altered in patients with Myo9b-linked IBD.
INTRODUCTION
Class IX myosins are unique among known members of the myosin family of actin-based molecular motors in that their tail domain contains a RhoGTPase-activating protein (RhoGAP) domain (Bahler, 2008). Myosin IXb (Myo9b) is one of two class IX myosins expressed in mammals. The GAP domain of Myo9b specifically targets Rho, not Rac or Cdc42 (Muller et al., 1997; Post et al., 1998). Myo9b is a single-headed, processive motor (Post et al., 2002). Although a baculovirus-expressed, tail-truncated form of Myo9b has been reported to move toward the minus (pointed) end of the actin filament (Inoue et al., 2002), both full-length Myo9b (O'Connell and Mooseker, 2003) and a CFP-tagged, truncated from of Myo9b purified from mammalian cells (O'Connell et al., 2007) are plus (barbed) end–directed motors, as are full-length and tail-truncated forms of Caenorhabditis elegans Myo9 (Liao et al., 2010). Given that most membrane-associated actin filaments are oriented with their plus ends at the membrane, the plus end–directed movement of Myo9b would propel its GAP activity to sites of membrane-bound, active Rho.
Polymorphisms in the gene encoding Myo9b heavy chain have been linked to several forms of inflammatory bowel disease (IBD), including Crohn's disease, celiac disease, and ulcerative colitis (Monsuur et al., 2005; van Bodegraven et al., 2006; Nunez et al., 2007; Cooney et al., 2009). This may be due to defects in Myo9b-dependent immune cell responses, since macrophages purified from Myo9b-knockout mice exhibit defects in chemotactic motility (Hanley et al., 2010). However, IBD is often characterized by increased paracellular permeability of the intestinal epithelium (Turner et al., 1997; Bruewer et al., 2003; Berkes et al., 2003; Clayburgh et al., 2004). Because Myo9b is also expressed in the intestinal epithelial cell (Bement et al., 1994), it could play key roles in Rho-mediated regulation of the mucosal barrier, disruption of which could also contribute to IBD.
In this study, the effects of loss of Myo9b function on epithelial wound healing and tight junction integrity and permeability were examined in the intestinal epithelial cell line Caco2BBe (BBe). There are two major findings in this study with regard to loss of Myo9b function with RNA interference or expression of the C-terminal dominant-negative tail tip of Myo9b (DN-tail-tip). Cells exhibiting Myo9b knockdown or expressing DN-tail-tip fail to migrate in response to wounding, and this nonmigratory phenotype is concurrent with increased accumulations of filamentous actin and associated cytoskeletal machinery. Second, Myo9b loss of function results in a nearly complete disruption of tight junction (TJ) protein localization, resulting in a “leaky” monolayer. These data suggest that in Myo9b-associated IBD, intestinal barrier function may be compromised as a result of these specific cellular disruptions with loss of Myo9b function.
RESULTS
Loss of Myo9b function disrupts wound closure and wound-induced changes in actin cytoskeletal organization in BBe cells
The role of Myo9b in enterocyte function was investigated using the BBe subclone (Peterson and Mooseker, 1992) of the Caco-2 intestinal epithelial cell line (Grasset et al., 1984). BBe cells differentiate into an epithelial monolayer within 2 wk, with features closely mimicking the intestinal epithelium in vivo, including a regulated tight junction–mediated paracellular barrier and enterocytes with a well-ordered apical brush border (BB; Peterson and Mooseker, 1992; Peterson et al., 1993).
Myo9b function in the wound-healing response was examined, due to the underlying requirement for dramatic actin cytoskeletal reorganization upon wound closure. BBe cells were fixed and costained with anti-Myo9b tail antibody and rhodamine–phalloidin to detect filamentous actin (F-actin). Within fully differentiated BBe monolayers, where F-actin localizes primarily to BB microvilli (Figure 1A), Myo9b localization was diffuse within the cytoplasm and was present along lateral membranes (Figure 1B). Immediately upon suction wounding with a Pasteur pipette, this localization was retained (Figure 1, D–F). However, upon wound-induced migration after 3 or 20 h, a striking redistribution occurred, in which lateral Myo9b localization completely diminished as Myo9b shifted to the leading edge, colocalizing with F-actin (Figure 1, G–L). Myo9b expression levels were unchanged in response to wounding, based on immunoblot analysis of BBe monolayers subjected to multiple wounds (unpublished data).
FIGURE 1:

Myo9b localizes to the leading edge of migrating BBe cells. (A–C) Within differentiated BBe monolayers, F-actin accumulates within the apical BB (A) with diffuse Myo9b localization (B). (D–F) On wounding, this localization is retained. (G–I) At 3 h postwounding, Myo9b has already localized to the extreme leading edge of the developing lamellipodia (H), colocalizing with F-actin (G). (J–L) At 20 h postwounding, Myo9b is still localized to the leading edge and also within the lamellar region. Bar, 10 μm.
The transformation of the BBe monolayer during wound closure is best appreciated in representative Z-plane images (Figure 2). F-Actin is clearly present within the apical BB in unwounded monolayers and immediately after suction wounding (Figure 2, A and B). Unlike cell migration in fibroblasts, which immediately results in the formation of forward protrusions, BBe cells must overcome the “cliff-like” height of the wound (Figure 2A), dedifferentiating and then projecting a very thin lamellipodia, where Myo9b is at the extreme leading edge (Figure 2C). At the 20-h time point, Myo9b is not only at the leading edge, but is also within the entire lamellar space (Figure 2D). We also examined the expression profile in response to wounding of two other RhoGAPs expressed in BBe cells, Myo9a (Abouhamed et al., 2009) and p190RhoGAP (Foster et al., 1994). Both exhibited diffuse cytoplasmic staining in unwounded cells, where Myo9a did not redistribute to the leading edge and p190RhoGAP was present at low levels (Supplemental Figure S1). Therefore it was of particular interest to determine the potential role of Myo9b in wound-induced migration with this redistribution to the extreme leading edge of lamellipodia.
FIGURE 2:

Myo9b localizes early to the leading edge of migrating BBe cells. (A) F-Actin (red) specifically localizes to the apical BB and lateral membranes, whereas Myo9b (green) is punctate within the cell body. (B) On wounding, cells have to overcome the cliff-like height to move into the wound space. Arrow indicates the wound margin. (C) Myo9b and F-actin colocalize at the extreme leading edge (arrow) of the developing lamellipodia 3 h postwounding. (D) F-Actin and Myo9b fill the lamellar space while still localizing to the extreme leading edge (arrow) of the large lamellipodia after 20 h of wound-induced migration. (E) Migration is disrupted with Myo9b siRNA transfection, with increased F-actin density at the wound edge. Bar, 5 μm.
To assess Myo9b loss of function, RNA interference (RNAi) was used, with three oligonucleotide target sets to Myo9b. BBe cells underwent two rounds of transfection with nontargeting control oligonucleotide (NTControl) or a pool of three Myo9b-targeting oligonucleotides (Myo9bKD). Cells were permitted to differentiate for at least 1 wk and were wounded to assess cell migration and actin cytoskeletal changes. This transfection method was determined to be effective to obtain 70–80% knockdown of Myo9b expression when probed by Western blot (Supplemental Figure S1). Confirmation of Myo9b loss of expression is displayed biochemically by a representative Western blot, where Myo9b-targeting small interfering RNA (siRNA) oligonucleotides were transfected individually and in combination (Supplemental Figure S1A). Knockdown in BBe cells did not exceed ∼80% due to the mosaic nature of BBe cell culture, where cells initially grow in islands, fuse to confluency, and finally fully differentiate (Peterson and Mooseker, 1992). These islands are somewhat heterogeneous (Supplemental Figure S1B) and therefore resulted in a mixture of Myo9bKD (∼100%) and “resistant” cell populations within a single coverslip (Peterson and Mooseker, 1992; Tyska and Mooseker, 2004). Therefore regions of Myo9b knockdown were determined by immunodetection of anti–Myo9b tail costained with rhodamine–phalloidin. Western blot densitometry measurements over three separate experiments were quantified (Supplemental Figure S1C), indicating that using the mix of three Myo9b-targeting oligonucleotides (3mix; Myo9bKD) consistently resulted in ∼80% knockdown compared with controls. NTControl and untransfected cells exhibited equivalent levels of Myo9b expression.
Wounded regions of NTControl cells exhibited broad lamellipodial formation, with Myo9b localization at the leading edge after 18 h, similar to untransfected cells (Figure 3, A–C). Of interest, when wound closure was permitted in cell populations in which Myo9b expression was knocked down, cell migration was stunted, with F-actin accumulating at the wound edge (Figures 2E and 3, D–F). The Z-plane image captures the high density of F-actin at the wound edge after 20 h, which appears to be a barrier to forward migration (Figure 2E). A representative Western blot of Myo9b protein levels in lysates from NTControl (NT) and Myo9bKD (3mix) BBe cells is displayed as an inset (Figure 3F).
FIGURE 3:

Cell migration is disrupted with loss of Myo9b function together with actomyosin accumulation. (A–C) NTControl cells, expressing nontargeting siRNA oligonucleotide, exhibit normal lamellipodia formation upon wounding. (D–F) Myo9bKD cells expressing Myo9b-targeting siRNA oligonucleotides fail to migrate into the wound space, with F-actin accumulation at the wound edge (D). Representative Western blot of Myo9b knockdown with a mix of three targeting siRNA oligonucleotides (3mix) is displayed as an inset. (G) NTControl cells exhibit a band of Myo2a (green) within the lamellipodia. (H) Myo9bKD cells fail to migrate, corresponding with bands of F-actin (red) and Myo2a at the wound edge. (I) Quantification of migration speed from live imaging of NTControl and Myo9bKD cells indicates a significant (p < 0.002) disruption in wound-induced migration. Bar, 10 μm.
The strikingly different cytoskeletal phenotypes between NTControl and Myo9bKD cell populations were examined by detergent extracting cells before fixation to fully expose the cytoskeleton, which were immunostained with anti–myosin IIa (Myo2a; Figure 3, G and H). Parallel cell populations were probed for Myo9b expression within the same experiment to ensure successful Myo9b knockdown, as both antibodies were rabbit polyclonal and could not be used together (unpublished data). In NTControl cells, Myo2a localized within a discrete band (Figure 3G) behind the extreme leading edge where Myo9b and F-actin were colocalized (Figure 3C). However, within Myo9bKD populations, a dense band of Myo2a colocalized with F-actin (Figure 3H), suggesting a contractile actomyosin phenotype in the absence of Myo9b expression. To quantify this disruption in cell migration, we performed live imaging of NTControl and Myo9bKD wound-induced migration over 45 min (imaging acquisition 60 min postwounding to permit BBe monolayers to recuperate from suction wounding) and quantified migration speed (μm/min; Figure 3I). A statistically significant (p ≤ 0.002) decrease in migration was seen in Myo9bKD cells, which were nearly static, corresponding to the lack of migration into the wound space seen after 18 h (Figures 2E and 3, D–F).
Another way to interfere with Myo9b function involved expression of the C-terminal tip of the tail domain of Myo9b fused to green fluorescent protein (GFP). Numerous studies have used expression of various myosin tail fragments to inhibit specific downstream effects of these motors (Wu et al., 1998; Reck-Peterson et al., 1999; Tyska and Mooseker, 2004; Krendel et al., 2007). To determine whether expression of the C-terminal tail-tip region of Myo9b (amino acids 1954–2126) would result in such inhibition, BBe cells were transfected with either GFP alone (pEGFP-C2) as a negative control or GFP-tagged Myo9b tail tip (DN-tail-tip). Cells were permitted to differentiate and were then suction wounded to induce directed cell migration. As with Myo9b KD, expression of the DN-tail-tip disrupted cell migration (Figure 4). GFP-alone control cells migrated normally, with large lamellae formation, F-actin at the extreme leading edge, and diffuse GFP throughout the migrating cell (Figure 4, A–C). Of interest, both DN-tail-tip cells and neighboring nonexpressing cells failed to migrate into the wound and exhibited F-actin striation similar to that of Myo9bKD cell populations, confirming the results in Figure 3 using RNA interference (Figure 4, D–F). Therefore both methods of disrupting Myo9b were used in subsequent experiments. Live imaging of migrating BBe cells expressing GFP alone or DN-tail-tip is included within Supplemental Data to demonstrate the phenotypic differences between the two conditions and to also display the localization of DN-tail-tip, which is both nuclear and cytoplasmic (Supplemental Movies S1 and S2). Migration speed of GFP- and DN-tail-tip–expressing cells was quantified from four live-imaging experiments. Similar to Myo9bKD cells, there was a statistically significant (p ≤ 0.001) decrease in DN-tail-tip cell migration compared with GFP control cells (Figure 3G; DN-TT).
FIGURE 4:

The C-terminal tail-tip region of Myo9b inhibits collective wound closure in BBe cells. (A–C) BBe cell monolayers expressing control pEGFP-C2 migrate normally into the wound space. (D–F) BBe cells expressing GFP-tagged DN-tail-tip exhibited diminished wound closure and dense actin fiber formation (D), where neighboring cells also failed to migrate. (G) Quantification of wound-induced cell migration of GFP control and GFP-DN-tail-tip–expressing cells indicates a significant (p < 0.001) disruption in migration. Bar, 10 μm.
Because the cell migration defect was quite striking, it did not appear as though another RhoGAP was compensating for loss of Myo9b function. To determine whether there was any indication of this, we immunostained Myo9bKD cells with antibodies against p190RhoGAP and Myo9a (Supplemental Figure S1). Of interest, neither p190RhoGAP (Supplemental Figure S1F) nor Myo9a (Supplemental Figure S1I) redistributed to the wound edge in these cells, suggesting that Myo9b is the predominant RhoGAP during wound-induced migration in BBe cells. Protein levels of both p190RhoGAP and Myo9a were also unchanged in Myo9bKD cells (blot inset; NTControl/left and Myo9bKD/right).
Loss of Myo9b function leaves Rho unregulated
To examine Rho activity, differentiated BBe monolayers that had either undergone two rounds of Myo9b siRNA oligo transfection or one round of pEGFP-C2 (alone or containing DN-tail-tip) transfection were wounded and permitted to migrate for 20 h. GTP-bound Rho (RhoGTP) and phosphorylated myosin light chain of Myo2 (MLC-P) levels were examined, which required live detergent extraction, followed by fixation to expose these epitopes (Figure 5). Large lamella in NTControl cells contained discrete RhoGTP staining, which also localized to the extreme leading edge (Figure 5A; green in Z-plane inset). GTP-bound Rho accumulated at the unmoving wound edge of Myo9bKD BBe cells, colocalizing with F-actin bands (Figure 5B, arrows; yellow in Z-plane inset). Overall levels of RhoGTP were comparable when detected by Western blot (not shown), but the difference in localization corresponded to the phenotypic differences in migratory status between NTControl and Myo9bKD cells, suggesting localized defects with loss of Myo9b function. In parallel DN-tail-tip experiments, RhoGTP similarly accumulated at the unmoving wound edge of DN-tail-tip–expressing cells when compared with control cells (unpublished data).
FIGURE 5:

Loss of Myo9b function results in hyperactivation of Rho signaling. (A) GTP-Rho (green in inset) localizes throughout the lamellipodia in NTControl cells. (B) In unmoving Myo9bKD cells, GTP-Rho accumulates at the unmoving wound edge (arrows), colocalizing with F-actin (yellow; Z-plane inset). (C) Downstream of Rho and ROCK, phosphorylated MLC (MLC-P) localizes to the extreme leading edge of NTControl lamellipodia. (D) MLC-P accumulates in the lateral membranes (green in inset) of the cells behind the wound edge of Myo9bKD cells. (E) DN-tail tip expression induces hypertorsional movements of lateral membranes in expressing and nonexpressing neighbor BBe cells (see Supplemental Movies S4 and S5), which decreases as the distance from expressing cells increases. Tracings (left) from every 7 min of 45-min time lapse or every 30 s (right). Bar, 10 μm.
Downstream of Rho activity, a similar trend was seen when examining levels of the phosphorylated form of the regulatory light chain of Myo2 (MLC-P). Activation of the Rho effector ROCK I results in MLC phosphorylation either through direct phosphorylation at Ser-19 or through inactivation of myosin light chain phosphatase (Kimura et al., 1996; Amano et al., 1996). ROCK I is the likely isoform affected, based on reported tissue expression (Riento and Ridley, 2003). Similar to visualizing GTP-bound Rho, overall levels of MLC-P were only slightly enhanced in Myo9bKD cells over that of NTControl cells. Instead, the differences were seen in the pattern of MLC-P localization, where at the nonmigratory wound edge of both Myo9bKD (Figure 5D) and DN-tail-tip (unpublished data) cells, MLC-P intensity at cell–cell contacts was enhanced (green in Z-plane inset). Overall, MLC-P levels were comparable when NTControl and Myo9bKD cell lysates were probed by Western blot (unpublished data), again suggesting that the defective signaling was localized to regions where Myo9b was disrupted at the wound margin. Of interest, when observing monolayers of GFP control (Supplemental Movie S3) and DN-tail-tip–expressing (Supplemental Movie S4) cells via live differential interference contrast (DIC) imaging, there was a detectable difference in torsional motility of the lateral margins of DN-tail-tip monolayers. DIC traces of the lateral margin of DN-tail-tip–expressing cells over a 45-min time course indicate the extent of increased torsional movement over time compared with GFP control cells (Figure 5E). Traces were taken at 7-min intervals to assess the persistence of this effect (light to dark blue = early to late time points); however, traces every 30 s (Figure 5E; right) highlight the frequency of torsional movements in DN-tail-tip–expressing cells. Neighboring non-DN-tail-tip–expressing cells exhibited increased torsional motility similar to the propagated inhibition of migration in neighboring cells at the wound edge (Figure 4F). Of interest, this effect was “diluted” as distance increased from the expressing cell (Figure 5E).
Consistent with elevated levels of active Rho (and downstream hyperactivation of ROCK I and MLC-P) in cells with loss of Myo9b function, inhibition of Myo2 with blebbistatin (Sigma-Aldrich) reverses the effect of Myo9b DN-tail-tip expression on wound closure. In the absence of drug, DN-tail-tip cells are unable to form a lamellipodia and therefore do not migrate into the wound space (Supplemental Movie S2). However, when 20 μM blebbistatin is added to the culture dish, within minutes, DN-tail-tip cells are able to generate lamellipodia, thus rescuing a normal migratory phenotype (Supplemental Movie S5).
Tight junction proteins fail to localize when Myo9b function is disrupted
Although the experiments thus far focused on the role of Myo9b in wound closure, torsional motility of DN-tail-tip cells and MLC-P accumulation at cell–cell contacts in both Myo9b KD and DN-tail-tip cells suggested that there may be perturbations at the junctional level. Inflammatory bowel diseases are characterized by barrier defects, which are sometimes termed “leaky intestine.” Such defects in the intestinal paracellular barrier give rise to leukocyte and pathogen infiltration through the mucosa, resulting in inflammation. Therefore the effects of loss of Myo9b function on tight junction (TJ) integrity were assessed through localization of the TJ component ZO-1.
As BBe cells differentiate in culture, F-actin localization changes significantly and accumulates in the BB and also in rootlets, which is disrupted upon wound closure (Figure 6A). However, within nonconfluent islands, fully differentiated BBe monolayers, and wounded regions, ZO-1 exhibits discrete junctional localization, perfectly outlining each cell (Figure 6C). Within wounded regions of the BBe monolayer where Myo9b expression was knocked down (Figure 6F), a corresponding loss of TJ protein localization was observed (Figure 6G). This loss of TJ protein localization was not restricted to the wound edge or to ZO-1. Within differentiated BBe monolayers, the junctional patterning of ZO-1 (Figure 7A), occludin (Figure 7C), and claudin-1 (Figure 7E) that was seen within NTControl samples was nearly absent in Myo9bKD regions (Figure 7, B, D, and F). This loss of TJ protein localization was specific to regions of the monolayer where Myo9b was completely knocked down. Neighboring areas where Myo9b expression was normal also had normal TJ protein localization. Overall, TJ protein levels were unchanged with Myo9bKD (Western blot insets: NTControl/left and Myo9bKD/right), suggesting instead a loss of localization to TJs or endocytosis of TJ components that may have been lost during fixation and staining. Of interest, this defect was specific to TJs; when examining adherens junction proteins, we found that E-cadherin localization was comparable between NTControl and Myo9bKD cells, and afadin was slightly decreased in Myo9bKD cells (unpublished data).
FIGURE 6:
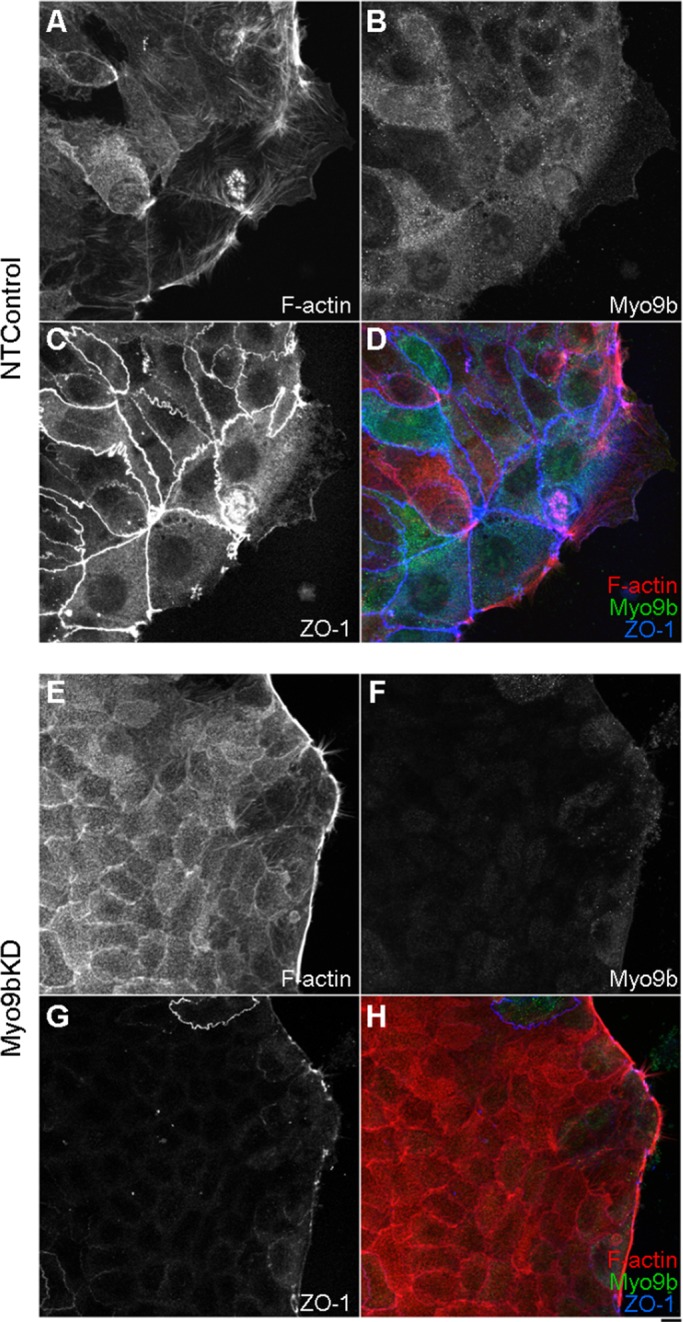
Loss of Myo9b function disrupts ZO-1 junctional localization. (A–D) NTControl cells permitted to migrate after wounding retain apical TJ localization of ZO-1 (C). (E–H) Myo9bKD cells not only fail to migrate into the wound space, but they also exhibit greatly diminished ZO-1 localization (G) in regions where Myo9b is knocked down (F). Bar, 10 μm.
FIGURE 7:

Loss of Myo9b function results in mislocalization of multiple components of the TJ. ZO-1 (A), occludin (C), and claudin (E) outline cell–cell junctions in NTControl cells. This localization is greatly disrupted in regions where Myo9b expression is knocked down (B, D, F). F-Actin localization is normal in these same regions. Western blots of ZO-1, occludin, and claudin (insets) indicate total protein levels are unchanged in NTControl (left) and Myo9bKD (right) cell lysates. Bar, 10 μm.
When examining TJs in DN-tail-tip–expressing cells, we observed a dose-dependent effect, in which increased GFP DN-tail-tip intensity correlated with decreased ZO-1 localization (Figure 8A; left to right, increasing DN-tail-tip expression). Just as was observed for inhibition of cell migration of cells neighboring DN-tail-tip–expressing cells at the wound margin (Figure 4F) and increased torsional motility of DN-tail-tip neighbors (Figure 5E), loss of ZO-1 from junctions was also observed in nonexpressing cells surrounding DN-tail-tip–expressing cells (Figure 8A). Of interest, Abouhamed et al. (2009) observed that knockdown of Myo9a results in similar disruption of TJ protein localization in Caco-2 cells. This suggests that both of these class IX myosins are collectively required for TJ integrity, and that loss of either is sufficient to impact TJ structure.
FIGURE 8:

Loss of Myo9b function results in a compromised “barrier.” (A) ZO-1 TJ localization (red) decreases with increased Myo9b DN-tail-tip expression (green). Z-Plane images are displayed below. (B) Live imaging of small-molecule TRITC-dextran flowing through the midsection of BBe monolayers transfected with pEGFP-C2, where dextran does not permeate the monolayer. (D) In high-expressing DN-tail-tip cells, TRITC-dextran readily passes through the monolayer. Note that permeability of neighboring cells is also affected. Images to the right (C, E) are three-dimensional projections of GFP images taken after the live time course to indicate transfected cells with Z-plane images (inset) after 45 min of flowthrough. (F) Change in TRITC-dextran flowthrough fluorescence intensity over 45 min was quantified and was significantly (p < 0.00001) increased in DN-tail-tip–expressing monolayers. Bar, 10 μm.
Similar to the RhoGTP perturbation that gave rise to a more contractile/adhesive phenotype in wound-healing experiments presented earlier, it was possible that increased GTP-bound Rho perturbed TJ regulation. To determine whether increased Rho activity would disrupt junctions, we incubated differentiated untransfected BBe monolayers with EDTA to disrupt junctions. Cells were permitted to reestablish junctions in the absence or presence of Rho activator (Cytoskeleton). Control cell populations were able to reestablish TJ protein localization within 3 h after addition of media with dimethyl sulfoxide (DMSO; Supplemental Data and Supplemental Figure S2A). However, incubation with Rho activator (50 μg/ml) disrupted ZO-1 localization similar to loss of Myo9b (Supplemental Figure S2B). To determine whether the DN-tail-tip effect on TJ localization could be rescued by targeting Myo2 function, we incubated DN-tail-tip–expressing cells with 20 μM blebbistatin, which rescued ZO-1 localization in these cells when compared with DMSO controls (Supplemental Figure S2C). GFP control cells incubated with blebbistatin exhibited slightly perturbed ZO-1 localization (Supplemental Figure S2C, bottom).
To test the effect of disruption of Myo9b on tight junction permeability, we observed the paracellular movement of tetramethylrhodamine isothiocyanate (TRITC)–labeled dextran (3 kDa) through BBe monolayers expressing GFP alone or GFP-DN-tail-tip. Dextran was added to the medium of BBe monolayers grown on glass-bottom dishes, which have a monolayer height of ∼20 μm. Time-lapse images were acquired for 45 min at a focal plane 10 μm below the apex of the monolayer. There was a striking difference between TRITC-dextran flowthrough in control and DN-tail-tip–expressing cells. TRITC-dextran was already present in the paracellular spaces between cells by 5 min postaddition in DN-tail-tip monolayers and continued to move between cells (Supplemental Movie S7 and Figure 8D), whereas this was barely detectable in control GFP cells (Supplemental Movie S6 and Figure 8B). A three-dimensional projection of GFP and DN-tail-tip expression displays expressing cells (Figure 8, C and E). The Z-plane images postflowthrough are displayed as insets. Note that many cells surrounding the DN-tail-tip–expressing cells also exhibit increased junctional permeability. Fluorescence intensity measurements from the Z-plane of image stacks taken before and after 45-min time courses of TRITC-dextran flowthrough were used to quantify the change in dextran influx from three separate experiments (Figure 8F). The difference between DN-tail-tip and GFP control monolayer dextran influx was highly significant (p ≤ 0.00001), considering that TRITC-dextran was already present between cells upon initiating live image acquisition in DN-tail-tip experiments (Figure 8D).
Loss of Myo9b function disrupts both collective and individual cell migration
The response of an epithelial monolayer to wounding involves a coordinated, collective response of cells at the margin, which retain their junctional connections during migration (Figure 6C; Friedl and Gilmour, 2009). To test whether Myo9b also plays a role in “cell-autonomous” wound induced migration, we examined the effects of Myo9b knockdown on wound healing in HeLa cells. HeLa cells express high levels of Myo9b, and this cell line has been extensively used for assessment of changes in cytoskeletal organization and cell adhesion in response to wounding.
HeLa cells also underwent two rounds of transfection with NT and Myo9b-targeting siRNA oligonucleotides and, upon reaching confluency, were scratch wounded and permitted to migrate for 3 h. Similar to the findings in BBe cells (Figure 3), HeLa NTControl cells migrated into the wound space (Figure 9A), whereas Myo9bKD cells exhibited marked inhibition of wound-induced rearrangements of the actin cytoskeleton at the wound margin (Figure 9B). Low-magnification images of scratch wound closure were used to quantify the ability of HeLa monolayers to close wounds. Images of NTControl and Myo9bKD wounds were taken immediately after wounding and after 6 h of wound closure, at which measurements were taken to calculate the average width of wounds. Myo9bKD regions were significantly (p ≤ 0.001) stunted in migration compared with NTControl regions (Figure 9C; representative of triplicate wounds from two experiments). Knockdown of Myo9b expression was verified by immunoblot analysis (Figure 9D). When examining the cytoskeletal dynamics of Myo9bKD cells, we found stress fiber–like arrays of actin to be present at the free edges of cells facing the wound, similar to that seen in BBe cells (Figure 9B). Focal adhesion proteins paxillin and focal adhesion kinase (FAK), which localized within focal contacts at the leading edge in NTControl cells (Figure 9E), were instead present within static focal adhesions (Figure 9F). This was also true for detection of total Rho levels (unpublished data). These data corresponded to a distinctly adherent and contractile phenotype within Myo9bKD cells, suggesting that Myo9b RhoGAP disruption resulted in unregulated pools of active GTP-bound Rho at the wound edge of individual cells.
FIGURE 9:

Loss of Myo9b function disrupts both collective and individual cell migration. (A) Myo9b (green) localizes to the leading edge of fibroblast-like HeLa cells. (B) HeLa cells expressing Myo9b-targeting siRNA oligonucleotides (Myo9bKD) fail to migrate into the wound space and have corresponding bands of F-actin (red) at the wound edge. (C) Quantification of scratch wound closure confirms this disruption in Myo9bKD HeLa cells. Average change in wound width over 6 h was quantified and significantly decreased (p < 0.001) in Myo9bKD cells. (D) Representative blot showing Myo9b knockdown with both single and combination (3mix) of siRNA oligonucleotide transfection. (E) NTControl HeLa cells exhibit focal contact localization of paxillin and FAK in migrating cells with RhoGTP and MLC-P at the leading edge of lamellipodia. (F) Loss of Myo9b results in accumulation of paxillin, FAK, and RhoGTP in dense focal adhesions at the unmoving wound edge and increased MLC-P localization. (G) Expression of the DN-tail tip of Myo9b disrupts migration only in cells where it is expressed, without affecting neighboring cells (arrow). Bar, 10 μm.
To examine Rho activity and downstream effectors critical to changes in actin dynamics, we used antibodies against RhoGTP and MLC-P to examine the effects of loss of Myo9b function. On wound closure in NTControl HeLa cells, RhoGTP was present within filopodia-like projections at the leading edge of migrating cells (Figure 9E). However, contractile Myo9bKD cells exhibited dense RhoGTP staining at the wound edge, where cells did not progress into the wound space (Figure 9F). Overall levels of MLC-P were also increased in Myo9bKD cells (Figure 9F). Similar to experiments in BBe cells, overall levels of GTP-Rho and MLC-P were comparable, as assayed by immunoblot analysis (unpublished data), again suggesting localized defects in Myo9b function within cells at the wound margin and specific areas of the monolayer (unpublished data) as opposed to global defects with loss of Myo9b function.
Expression of the GFP-tagged DN-tail-tip of Myo9b was also used in HeLa cell experiments (Figure 9G). Again, migration was stunted in DN-tail-tip–expressing cells over cells expressing GFP alone. However, in contrast to BBe cell experiments, in which cells neighboring DN-tail-tip–expressing cells were unable to protrude into the wound margin, the cell migration defect in HeLa cells was restricted to the individual DN-tail-tip–expressing cells. This result is consistent with the previously demonstrated role for Myo9b in the “professionally” motile and autonomous macrophage (Hanley et al., 2010). Thus Myo9b plays critical roles in both epithelial cell collective and cell-autonomous wound-induced migration.
DISCUSSION
In this study, there are two major conclusions with respect to the role of Myo9b in the BBe model of the intestinal epithelium. First, loss of Myo9b function with either RNA interference or expression of the DN-tail-tip results in the inability of wounded monolayers to “heal,” thus implicating Myo9b in directed cell migration. Second, loss of Myo9b function, similar to that reported for Myo9a (Abouhamed et al., 2009), results in the disruption of the TJ complex, in which TJ proteins are mislocalized and the “barrier” is compromised, permitting TRITC-dextran to readily pass through monolayers where Myo9b function has been altered. Of interest, our data suggest that loss of Myo9b function alters RhoGTPase regulation, which can have a number of specific cellular consequences, in this case resulting in two major defects in epithelial cell function. It is also interesting to note that both of these phenotypes directly correlate with the clinical manifestations, in which Myo9b has been implicated as a target of genetic polymorphisms in IBD.
To examine whether Myo9b was required for cell migration, expression was either knocked down (RNAi) or disrupted (DN-tail-tip) in BBe and HeLa cells, and wound-induced migration was examined. Myo9bKD and DN-tail-tip–expressing cells not only failed to migrate, but they also exhibited bands of F-actin and Myo2a at the wound edge. Presumably it is the loss of Myo9b RhoGAP activity that is the basis for the underlying defects. Rho family GTPases are essential to migratory and nonmigratory states, where layers of regulation of the GTP-bound state can dictate specific downstream cytoskeletal changes (Hall, 1998). Rho-mediated cytoskeletal changes are necessary for enterocyte migration during mucosal injury that is essential to maintaining tissue integrity; disruption of Rho regulation could have significant consequences at the tissue level (Santos et al., 1997; Cetin et al., 2004). Therefore one would expect another RhoGAP to compensate for the loss of Myo9b. p190RhoGAP has been implicated in fibroblast migration and in breast cancer metastasis and invasive capacity (Nakahara et al., 1998; Jiang et al., 2008). Myo9a-knockout mice are hydrocephalic, and siRNA knockdown of Myo9a in Caco-2 cells has been shown to disrupt tight junction assembly (Abouhamed et al., 2009). Recently it was also reported that Myo9a is critical to collective wound migration in bronchial epithelial cells (Omelchenko and Hall, 2012). However, expression and localization of both p190RhoGAP and Myo9a were unchanged when Myo9b function was disrupted (Supplemental Figure S1). Therefore there may be cell type–specific roles for class IX myosins, in which one does not compensate for the other, even when expressed. With loss of Myo9b function and without compensation of another RhoGAP, it is likely that GTP-bound Rho is accumulating in Myo9bKD regions, which was observed in both BBe (Figure 5B) and HeLa cells (Figure 9F). Loss of an essential RhoGAP would likely keep downstream targets of GTP-bound Rho, namely ROCK I and its major effector Myo2, in a constant “on” state (Kimura et al., 1996; Amano et al., 1996; Fujisawa et al., 1996).
When the light chains of Myo2 are phosphorylated by upstream targets, the folded, autoinhibited state is released, leaving Myo2 open and able to form filaments and exhibit actin-activated ATPase activity (Scholey et al., 1980). The coordination of Myo2 and actin is essential to the propelling lamellipodia and requires a delicate balance of adhesion sites and arrays of actin (Giannone et al., 2007). It is likely that both isoforms of Myo2 would be hyperphosphorylated downstream of unregulated Rho. However, our observations focused on Myo2a, since it has been implicated in both lamellar dynamics (Cai et al., 2006) and apical junction dynamics downstream of ROCK (Smutny et al., 2010). Loss of Myo9b function and concurrent Myo2a hyperactivation likely result in the recruitment and sequestration of actin into actomyosin arrays, thus inhibiting lamellar actin assembly.
The role of Myo9b in wound-induced migration was examined in both BBe and HeLa cells, and the mode of migration in these two cell types was completely different, yet Myo9b function was essential to both. Under control conditions, HeLa cell migration was amoeboid-like, whereas BBe cells exhibited collective migration. BBe cells near the wound margin dedifferentiated before protrusion, retaining TJs and propelling lamellipodia that were nearly 10 times larger than those generated by HeLa cells. Myo9b localized to the leading edge of both of these cell types during wound-induced migration, and Myo9bKD cells in both systems failed to migrate. The difference between the two modes of migration was highlighted with DN-tail-tip expression, in which nonexpressing neighbors in BBe experiments also failed to migrate, suggesting the importance of the entire “tissue” to migrate into the wound space together. This was not the case in HeLa cells, for which only the DN-tail-tip–expressing cells were affected. The defects in HeLa cells could be paralleled by defective directed migration seen in macrophages isolated from Myo9b-knockout mice (Hanley et al., 2010). Collective migration is often used during development and morphogenesis, and the loss of this ability is usually attributed to the epithelial-to-mesenchymal transition that occurs during metastasis (Hidalgo-Carcedo et al., 2011). Loss of ROCK-mediated Myo2a contractility is essential to both modes of migration, supporting our observations of increased Myo2a accumulation and MLC-P in cells with disrupted Myo9b function (Hanley et al., 2010; Hidalgo-Carcedo et al., 2011). In DN-tail-tip–expressing BBe cells at the wound edge, those cells neighboring the expressing cells were also unable to migrate into the wound space. Could this be attributed to communication between cells through gap junctions? Rho signaling has been implicated in the regulation and gating function of gap junctions (Anderson et al., 2002; Derangeon et al., 2008). However, the increased MLC phosphorylation and corresponding F-actin accumulation at the unmoving wound edge of DN-tail-tip and neighboring cells suggest that mechanical coupling is likely disrupted. Similarly, DIC imaging of DN-tail-tip–expressing cells within unwounded monolayers displays the hypertorsional lateral movements of both expressing and nonexpressing neighbors. Actomyosin filament accumulation could be exerting enough tension to propagate to neighboring cells. Therefore, in contrast to Myo9bKD, the DN-tail-tip is acting as a “gain-of-function” mutation. For example, if the tail tip interacts with an as-yet-unidentified binding partner that is critical for proper junctional and lamellar organization/contractility, DN-tail tip might act as a sequestration sink for this protein, whereas in Myo9bKD cells, the localization/function of this binding partner would not be inhibited.
This propagation of a phenotype to cells neighboring a DN-tail-tip–expressing cell was also seen in TRITC-dextran flowthrough experiments (Figure 8). Although there are major differences between the two functions examined in this study, it is likely that disruption of wound-induced migration and epithelial barrier function are associated with Myo2a hyperactivation. Balanced Rho and downstream ROCK activity is essential to the assembly and function of TJs in intestinal epithelial cells (Walsh et al., 2001). Within T84 epithelial cell monolayers, constitutive Rho family GTPase activation by the Escherichia coli toxin CNF-1 results in disruption of both the gate and fence functions of the TJ, and ROCK inhibition alters actin organization and increases TJ permeability (Walsh et al., 2001). Again, signaling downstream of Rho/ROCK affects Myo2 activity, which has been shown to be critical to developing and maintaining intestinal barrier function. Regulation of MLC phosphorylation with respect to TJs is a delicate balance, in which Myo2 mediates contraction of the perijunctional actomyosin ring (Turner et al., 1997, 1999). When MLC phosphorylation is selectively induced in polarized Caco-2 cells, ZO-1, occludin, and F-actin are redistributed, resulting in defects in permeability (Shen et al., 2006). MLCK inhibition prevents these changes, not only suggesting the importance of MLC phosphorylation, but also emphasizing its exclusivity in TJ regulation (Shen et al., 2006). ROCK and Myo2 hyperactivation has been attributed to intestinal barrier dysfunction, which often results from the internalization of TJ components (Utech et al., 2005, 2010; Samarin et al., 2007). Therefore down-regulation of Rho-dependent pathways within the intestinal epithelium is critical to maintaining junctional integrity. Loss of a major RhoGAP would likely shift the equilibrium, raising havoc with both TJ assembly and integrity of the corresponding barrier. Although F-actin accumulated at the unmoving wound edge of Myo9bKD and DN-tail-tip cells, TJ-associated F-actin localization was comparable to controls, although it was difficult to differentiate from brush-border microvillar F-actin (Figure 2). However, if there is increased perijunctional contractility/tension on junctional elements such as ZO-1, such effects could lead to dramatic effects on junctional integrity without any change in the total amounts of junction-associated F-actin.
The fact that neither p190RhoGAP nor Myo9a compensates for loss of Myo9b function suggests that Myo9b is the predominant RhoGAP in this system. Why would this be the case? Models for class IX myosins suggest that these myosins use their plus-end motor activity to carry their own RhoGAP cargo to the plasma membrane and thus do not need additional binding protein interactions to effect GAP function (Bahler, 2008). The observation that expression of the DN-tail-tip phenocopies Myo9b knockdown suggests that there is (are) Myo9b tail interacting protein(s). The Myo9b tail tip domain shares little homology with that of Myo9a (Chieregatti et al., 1998), which does not redistribute to the lamellipodia of wounded cells (Supplemental Figure S1). This putative, yet-to-be-identified Myo9b tail–binding protein may be essential to Myo9b-dependent regulation of actin dynamics at the leading edge and Myo2-dependent regulation of TJs. This would also help to explain the localized as opposed to global changes in RhoGTP and MLC-P levels downstream of Myo9b disruption.
An argument against Myo9b playing the predominant role in regulating wound-induced cell migration and regulation of TJs is that, as noted, knockdown of Myo9a also disrupts TJs in Caco-2 cells (Abouhamed et al., 2009). In this regard, it will be of interest to determine whether Myo9a knockdown similarly inhibits the wound-healing response and whether the redistribution of Myo9b to the lamellipodia is inhibited. Such future studies would indicate whether the GAP functions of these two myosins are shared in junctional regulation but not wound-induced cell migration.
With respect to the role of Myo9b in inflammatory bowel diseases, dysfunction at the cellular level has often been attributed to problems with cells of the submucosal immune system (Franchimont et al., 2004; Kamada et al., 2005; Niess et al., 2005). Neutrophils derived from Myo9b KO mice exhibit defective cell migration (Hanley et al., 2010). Intraepithelial infiltration of immune cells exhibiting defective Myo9b and therefore Rho signaling would eventually result in inflammation. However, if the intestinal epithelial barrier is also defective due to Myo9b defects, the combinatorial effect of a leaky intestinal barrier and unregulated immune cell migration would be extremely detrimental at the tissue level.
MATERIALS AND METHODS
Cell culture
Caco2BBe (BBe) and HeLa cells were cultured as previously described (Peterson and Mooseker, 1992; Tyska and Mooseker, 2004; Krendel et al., 2007). BBe and HeLa cells were maintained in DMEM (Life Technologies, Carlsbad, CA) supplemented with 10% fetal bovine serum (FBS; Hyclone, Logan, UT) and 1× penicillin–streptomycin–Fungizone (Life Technologies) at 37°C and 5% CO2. Cells were grown on sterilized glass coverslips, WillCo Wells glass-bottom dishes (Warner Instruments, Hamden, CT), or tissue culture–treated plates (Corning Life Sciences, Tewksbury, MA) to confluency, with BBe cells maintained for 2 wk to ensure differentiation of confluent monolayers. To examine directed cell migration, we scratch wounded confluent HeLa monolayers with a pipette tip, whereas differentiated BBe monolayers were suction wounded with a Pasteur pipette.
Materials and antibodies
The following antibodies were used at 20 μg/ml for immunostaining and 1 μg/ml for Western blotting: anti–Myo9b tail (Post et al., 1998); anti–ZO-1, anti-occludin and anti–claudin-1 (Zymed/Invitrogen, San Francisco, CA); anti-Myo2 (Biomedical Technologies, Stoughton, MA); anti–MLC phospho-Ser-19 (Cell Signaling Technology, Beverly, MA); anti-Rho and anti-RhoGTP (NewEast Biosciences, King of Prussia, PA); anti-p190RhoGAP (BD Biosciences, San Diego, CA); and anti-Myo9a (courtesy of Martin Bähler). Alexa 488, 568, and 633 secondary antibodies (Molecular Probes/Invitrogen, Carlsbad, CA) were diluted 1:700 for immunostaining and horseradish peroxidase antibodies (Pierce, Rockford, IL) at 1:5000 for Western blotting. Rhodamine–phalloidin and TRITC-dextran 3000 molecular weight (lysine fixable) were purchased from Molecular Probes/Invitrogen and Rho activator from Cytoskeleton (Denver, CO). Unless noted, other reagents were purchased from Sigma-Aldrich (St. Louis, MO).
RNA interference and transfection
RNA interfering oligonucleotides designed to target Myo9b expression were purchased from Ambion/Applied Biosystems (Foster City, CA). Specifically, Myo9b oligonucleotides s715, s716, and s718 were used both individually and in combination. Silencer Select Negative Control #1 siRNA was used as a nontargeting control (Ambion). The C-terminal tail-tip domain of Myo9b (amino acids 1954–2126) was cloned into pEGFP-C2 (Molecular Probes/Invitrogen). For cDNA transfection, 2 μg (HeLa) or 5 μg (BBe) of pEGFP-C2 (with or without the Myo9b DN-tail-tip) was transfected with Lipofectamine 2000 per 35-mm glass-bottom dish or well of a six-well plate. For RNAi, both HeLa and BBe cells underwent two rounds of siRNA transfection 24 h apart. The first round was done when the cells were 60–70% confluent, with Lipofectamine 2000–mediated transfection and 2 (HeLa) or 5 μl (BBe) oligonucleotide (10 μM). The second round included a gentle trypsinization step (to round the cells without liftoff) before adding the Lipofectamine/oligo mix, in order to increase the surface area of uptake. This transfection method was determined to be effective to obtain ∼70–80% knockdown of Myo9b expression when probed by Western blot. In BBe cells especially, knockdown did not exceed this due to the mosaic nature of BBe cell culture. Cells initially grow in islands, with islands fusing to confluency. These islands are somewhat heterogeneous (Peterson and Mooseker, 1992) and therefore resulted in a mixture of Myo9bKD (where knockdown was ∼100%) and “resistant” cell populations within the same well of a six-well plate. Gel samples were prepared from HeLa and BBe cell lysates to probe for Myo9b expression as previously described (Peterson and Mooseker, 1992). Western blots probed with anti-GFP indicated lysates from GFP control and GFP-DN-tail-tip BBe and HeLa lysates exhibited equivalent expression levels. ImageJ (National Institutes of Health, Bethesda, MD) and Excel (Microsoft, Redmond, WA) software were used for Western blot densitometry analysis.
Immunofluorescence staining
HeLa and BBe cells were plated on sterile glass coverslips. Confluent HeLa monolayers were wounded with a 200-μl pipette tip, whereas differentiated BBe cells were suction wounded with a sterile Pasteur pipette and were permitted to migrate for the indicated time after replacing media. BBe cells were fixed by using a previously described pH shift fixation method (Tyska and Mooseker, 2004). Briefly, cells were washed twice with 1× phosphate-buffered saline (PBS), followed by incubation with 4% paraformaldehyde (PFA) in K–1,4-piperazinediethanesulfonic acid (PIPES; pH 6.5) for 5 min. After removal of PIPES-PFA, cells were incubated with 4% PFA in sodium borate solution (pH 11.0) for 10 min. After two washes with 1× PBS, cells were twice incubated with 1 mg/ml sodium borohydride in PBS for 15 min. Cells were washed twice with 1× PBS, permeabilized in 0.1% Triton X-100 for 3 min, and blocked in 5% bovine serum albumin (BSA) in PBS for 30 min at 37°C. HeLa cells were fixed with 4% PFA diluted in 1× PBS for 40 min, followed by permeabilization for 2 min in 0.1% Triton X-100 and 5% BSA blocking for 30 min at 37°C. For both cell types, primary antibodies were diluted in 1% BSA, and coverslips were incubated in a humidified chamber for 45 min. After two washes each in 1% BSA and 1× PBS, secondary antibodies were diluted in 1% BSA with rhodamine–phalloidin (0.3 μM), and cells were incubated at 37°C for 30 min. Coverslips were washed twice with 1% BSA and three times with 1× PBS and were mounted with Prolong Gold (Invitrogen).
Confocal microscopy
Fluorescence was visualized on a laser scanning confocal microscope (model LSM510; Carl Zeiss Microimaging, Jena, Germany) equipped with 488/568/633 lasers using 20×/1.0 or 63×/1.4 Plan Apochromat objectives. Vertical sections (Z) from the level of the coverslip to the brush border (based on F-actin staining in fixed sections) were acquired at a low scanning rate with averaging of two to four scans to ensure high resolution. Two- or three-color images were acquired with sequential scanning; 568- and 633-nm wavelengths were separated with Zeiss META detection. Images were acquired and pseudocolored using ZEN (Zeiss) with three-dimensional reconstruction and assembly of figures in ImageJ. Cell migration and wound closure analysis was performed using ImageJ and Excel software.
TRITC-dextran experiment
BBe cells grown on glass-bottom culture dishes were transfected with pEGFP-C2 (with or without the Myo9b tail tip) when subconfluent and were permitted to differentiate for at least 10 d. On cell polarization and differentiation, cells were used for live-imaging experiments to examine the passive flowthrough of small–molecular weight, fluorescently tagged dextran. Cells were rinsed with warm PBS, and clear DMEM media (10% FBS) was added containing 150 μg/ml TRITC-dextran 3000 molecular weight. Imaging began within 5 min after addition of TRITC-dextran, which included a Z-stack at time 0, followed by a live time course at a Z-plane exactly halfway through the monolayer for 90 cycles every 30 s (45 min total), followed by a post-live Z-stack. This permitted analysis of live imaging as TRITC-dextran was passing through (or not) the monolayer, along with postexperiment Z-plane analysis of TRITC-dextran levels between individual cells. ImageJ software was used to extrapolate fluorescence intensity across Z-plane images of stacks taken before and after live TRITC-dextran flowthrough imaging.
Supplementary Material
Acknowledgments
We thank Emily Osterweil for making the GFP-DN-tail-tip construct and Martin Bähler for the Myo9a antibody. We thank members of the Mooseker laboratory for numerous discussions regarding the data presented here. This work was supported by grants from the National Institutes of Health (GM073823 and DK025387; MSM) and the Crohn's and Colitis Foundation of America and by a postdoctoral fellowship from the American Cancer Society, New England Division (118001-PF-09-260-01-CSM; S.K.C.).
Abbreviations used:
- BB
brush border
- DN
dominant negative
- GAP
GTPase-activating protein
- IBD
inflammatory bowel disease
- Myo9b
myosin IXb
- TJ
tight junction
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E11-09-0803) on May 9, 2012.
REFERENCES
- Abouhamed M, Grobe K, San IV, Thelen S, Honnert U, Balda MS, Matter K, Bahler M. Myosin IXa regulates epithelial differentiation and its deficiency results in hydrocephalus. Mol Biol Cell. 2009;20:5074–5085. doi: 10.1091/mbc.E09-04-0291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amano M, Ito M, Kimura K, Fukata Y, Chihara K, Nakano T, Matsuura Y, Kaibuchi K. Phosphorylation and activation of myosin by Rho-associated kinase (Rho-kinase) J Biol Chem. 1996;271:20246–20249. doi: 10.1074/jbc.271.34.20246. [DOI] [PubMed] [Google Scholar]
- Anderson SC, Stone C, Tkach L, SundarRaj N. Rho and Rho-kinase (ROCK) signaling in adherens and gap junction assembly in corneal epithelium. Invest Ophthalmol Vis Sci. 2002;43:978–986. [PubMed] [Google Scholar]
- Bahler M. Class IX myosins. In: Coluccio LM, editor. Myosins: A Superfamily of Molecular Motors. Dordrecht, Netherlands: Springer; 20082008. pp. 391–401. [Google Scholar]
- Bement WM, Hasson T, Wirth JA, Cheney RE, Mooseker MS. Identification and overlapping expression of multiple unconventional myosin genes in vertebrate cell types. Proc Natl Acad Sci USA. 1994;91:6549–6553. doi: 10.1073/pnas.91.14.6549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berkes J, Viswanathan VK, Savkovic SD, Hecht G. Intestinal epithelial responses to enteric pathogens: effects on the tight junction barrier, ion transport, and inflammation. Gut. 2003;52:439–451. doi: 10.1136/gut.52.3.439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruewer M, Luegering A, Kucharzik T, Parkos CA, Madara JL, Hopkins AM, Nusrat A. Proinflammatory cytokines disrupt epithelial barrier function by apoptosis-independent mechanisms. J Immunol. 2003;171:6164–6172. doi: 10.4049/jimmunol.171.11.6164. [DOI] [PubMed] [Google Scholar]
- Cai Y, et al. Nonmuscle myosin IIA-dependent force inhibits cell spreading and drives F-actin flow. Biophys J. 2006;91:3907–3920. doi: 10.1529/biophysj.106.084806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cetin S, Ford HR, Sysko LR, Agarwal C, Wang J, Neal MD, Baty C, Apodaca G, Hackam DJ. Endotoxin inhibits intestinal epithelial restitution through activation of Rho-GTPase and increased focal adhesions. J Biol Chem. 2004;279:24592–24600. doi: 10.1074/jbc.M313620200. [DOI] [PubMed] [Google Scholar]
- Chieregatti E, Gartner A, Stoffler HE, Bahler M. Myr 7 is a novel myosin IX-RhoGAP expressed in rat brain. J Cell Sci. 1998;111:3597–3608. doi: 10.1242/jcs.111.24.3597. [DOI] [PubMed] [Google Scholar]
- Clayburgh DR, Shen L, Turner JR. A porous defense: the leaky epithelial barrier in intestinal disease. Lab Invest. 2004;84:282–291. doi: 10.1038/labinvest.3700050. [DOI] [PubMed] [Google Scholar]
- Cooney R, Cummings JR, Pathan S, Beckly J, Geremia A, Hancock L, Guo C, Morris A, Jewell DP. Association between genetic variants in myosin IXB and Crohn's disease. Inflamm Bowel Dis. 2009;15:1014–1021. doi: 10.1002/ibd.20885. [DOI] [PubMed] [Google Scholar]
- Derangeon M, Bourmeyster N, Plaisance I, Pinet-Charvet C, Chen Q, Duthe F, Popoff MR, Sarrouilhe D, Herve JC. RhoA GTPase and F-actin dynamically regulate the permeability of Cx43-made channels in rat cardiac myocytes. J Biol Chem. 2008;283:30754–30765. doi: 10.1074/jbc.M801556200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster R, Hu KQ, Shaywitz DA, Settleman J. p190 RhoGAP, the major RasGAP-associated protein, binds GTP directly. Mol Cell Biol. 1994;14:7173–7181. doi: 10.1128/mcb.14.11.7173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franchimont D, et al. Deficient host-bacteria interactions in inflammatory bowel disease? The toll-like receptor (TLR)-4 Asp299gly polymorphism is associated with Crohn's disease and ulcerative colitis. Gut. 2004;53:987–992. doi: 10.1136/gut.2003.030205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedl P, Gilmour D. Collective cell migration in morphogenesis, regeneration and cancer. Nat Rev Mol Cell Biol. 2009;10:445–457. doi: 10.1038/nrm2720. [DOI] [PubMed] [Google Scholar]
- Fujisawa K, Fujita A, Ishizaki T, Saito Y, Narumiya S. Identification of the Rho-binding domain of p160ROCK, a Rho-associated coiled-coil containing protein kinase. J Biol Chem. 1996;271:23022–23028. doi: 10.1074/jbc.271.38.23022. [DOI] [PubMed] [Google Scholar]
- Giannone G, et al. Lamellipodial actin mechanically links myosin activity with adhesion-site formation. Cell. 2007;128:561–575. doi: 10.1016/j.cell.2006.12.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grasset E, Pinto M, Dussaulx E, Zweibaum A, Desjeux JF. Epithelial properties of human colonic carcinoma cell line Caco-2: electrical parameters. Am J Physiol. 1984;247:C260–C267. doi: 10.1152/ajpcell.1984.247.3.C260. [DOI] [PubMed] [Google Scholar]
- Hall A. Rho GTPases and the actin cytoskeleton. Science. 1998;279:509–514. doi: 10.1126/science.279.5350.509. [DOI] [PubMed] [Google Scholar]
- Hanley PJ, Xu Y, Kronlage M, Grobe K, Schon P, Song J, Sorokin L, Schwab A, Bahler M. Motorized RhoGAP myosin IXb (Myo9b) controls cell shape and motility. Proc Natl Acad Sci USA. 2010;107:12145–12150. doi: 10.1073/pnas.0911986107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hidalgo-Carcedo C, Hooper S, Chaudhry SI, Williamson P, Harrington K, Leitinger B, Sahai E. Collective cell migration requires suppression of actomyosin at cell-cell contacts mediated by DDR1 and the cell polarity regulators Par3 and Par6. Nat Cell Biol. 2011;13:49–58. doi: 10.1038/ncb2133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue A, Saito J, Ikebe R, Ikebe M. Myosin IXb is a single-headed minus-end-directed processive motor. Nat Cell Biol. 2002;4:302–306. doi: 10.1038/ncb774. [DOI] [PubMed] [Google Scholar]
- Jiang W, Betson M, Mulloy R, Foster R, Levay M, Ligeti E, Settleman J. p190A RhoGAP is a glycogen synthase kinase-3-beta substrate required for polarized cell migration. J Biol Chem. 2008;283:20978–20988. doi: 10.1074/jbc.M802588200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamada N, et al. Abnormally differentiated subsets of intestinal macrophage play a key role in Th1-dominant chronic colitis through excess production of IL-12 and IL-23 in response to bacteria. J Immunol. 2005;175:6900–6908. doi: 10.4049/jimmunol.175.10.6900. [DOI] [PubMed] [Google Scholar]
- Kimura K, et al. Regulation of myosin phosphatase by Rho and Rho-associated kinase (Rho-kinase) Science. 1996;273:245–248. doi: 10.1126/science.273.5272.245. [DOI] [PubMed] [Google Scholar]
- Krendel M, Osterweil EK, Mooseker MS. Myosin 1E interacts with synaptojanin-1 and dynamin and is involved in endocytosis. FEBS Lett. 2007;581:644–650. doi: 10.1016/j.febslet.2007.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao W, Elfrink K, Bahler M. Head of myosin IX binds calmodulin and moves processively toward the plus-end of actin filaments. J Biol Chem. 2010;285:24933–24942. doi: 10.1074/jbc.M110.101105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monsuur AJ, et al. Myosin IXB variant increases the risk of celiac disease and points toward a primary intestinal barrier defect. Nat Genet. 2005;37:1341–1344. doi: 10.1038/ng1680. [DOI] [PubMed] [Google Scholar]
- Muller RT, Honnert U, Reinhard J, Bahler M. The rat myosin myr 5 is a GTPase-activating protein for Rho in vivo: essential role of arginine 1695. Mol Biol Cell. 1997;8:2039–2053. doi: 10.1091/mbc.8.10.2039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakahara H, Mueller SC, Nomizu M, Yamada Y, Yeh Y, Chen WT. Activation of beta1 integrin signaling stimulates tyrosine phosphorylation of p190RhoGAP and membrane-protrusive activities at invadopodia. J Biol Chem. 1998;273:9–12. doi: 10.1074/jbc.273.1.9. [DOI] [PubMed] [Google Scholar]
- Niess JH, et al. CX3CR1-mediated dendritic cell access to the intestinal lumen and bacterial clearance. Science. 2005;307:254–258. doi: 10.1126/science.1102901. [DOI] [PubMed] [Google Scholar]
- Nunez C, et al. MYO9B polymorphisms in patients with inflammatory bowel disease. Gut. 2007;56:1321–1322. doi: 10.1136/gut.2007.121905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Connell CB, Mooseker MS. Native myosin-IXb is a plus-, not a minus-end-directed motor. Nat Cell Biol. 2003;5:171–172. doi: 10.1038/ncb924. [DOI] [PubMed] [Google Scholar]
- O'Connell CB, Tyska MJ, Mooseker MS. Myosin at work: motor adaptations for a variety of cellular functions. Biochim Biophys. Acta. 2007;1773:615–630. doi: 10.1016/j.bbamcr.2006.06.012. [DOI] [PubMed] [Google Scholar]
- Omelchenko T, Hall A. Myosin-IXA regulates collective epithelial cell migration by targeting RhoGAP activity to cell-cell junctions. Curr Biol. 2012;22:278–288. doi: 10.1016/j.cub.2012.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson MD, Bement WM, Mooseker MS. An in vitro model for the analysis of intestinal brush border assembly. II. Changes in expression and localization of brush border proteins during cell contact-induced brush border assembly in Caco-2BBe cells. J Cell Sci. 1993;105:461–472. doi: 10.1242/jcs.105.2.461. [DOI] [PubMed] [Google Scholar]
- Peterson MD, Mooseker MS. Characterization of the enterocyte-like brush border cytoskeleton of the C2BBe clones of the human intestinal cell line, Caco-2. J Cell Sci. 1992;102:581–600. doi: 10.1242/jcs.102.3.581. [DOI] [PubMed] [Google Scholar]
- Post PL, Bokoch GM, Mooseker MS. Human myosin-IXb is a mechanochemically active motor and a GAP for rho. J Cell Sci. 1998;111:941–950. doi: 10.1242/jcs.111.7.941. [DOI] [PubMed] [Google Scholar]
- Post PL, Tyska MJ, O'Connell CB, Johung K, Hayward A, Mooseker MS. Myosin-IXb is a single-headed and processive motor. J Biol Chem. 2002;277:11679–11683. doi: 10.1074/jbc.M111173200. [DOI] [PubMed] [Google Scholar]
- Reck-Peterson SL, Novick PJ, Mooseker MS. The tail of a yeast class V myosin, myo2p, functions as a localization domain. Mol Biol Cell. 1999;10:1001–1017. doi: 10.1091/mbc.10.4.1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riento K, Ridley AJ. Rocks: multifunctional kinases in cell behaviour. Nat Rev Mol Cell Biol. 2003;4:446–456. doi: 10.1038/nrm1128. [DOI] [PubMed] [Google Scholar]
- Samarin SN, Ivanov AI, Flatau G, Parkos CA, Nusrat A. Rho/Rho-associated kinase-II signaling mediates disassembly of epithelial apical junctions. Mol Biol Cell. 2007;18:3429–3439. doi: 10.1091/mbc.E07-04-0315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos MF, McCormack SA, Guo Z, Okolicany J, Zheng Y, Johnson LR, Tigyi G. Rho proteins play a critical role in cell migration during the early phase of mucosal restitution. J Clin Invest. 1997;100:216–225. doi: 10.1172/JCI119515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholey JM, Taylor KA, Kendrick-Jones J. Regulation of non-muscle myosin assembly by calmodulin-dependent light chain kinase. Nature. 1980;287:233–235. doi: 10.1038/287233a0. [DOI] [PubMed] [Google Scholar]
- Shen L, Black ED, Witkowski ED, Lencer WI, Guerriero V, Schneeberger EE, Turner JR. Myosin light chain phosphorylation regulates barrier function by remodeling tight junction structure. J Cell Sci. 2006;119:2095–2106. doi: 10.1242/jcs.02915. [DOI] [PubMed] [Google Scholar]
- Smutny M, Cox HL, Leerberg JM, Kovacs EM, Conti MA, Ferguson C, Hamilton NA, Parton RG, Adelstein RS, Yap AS. Myosin II isoforms identify distinct functional modules that support integrity of the epithelial zonula adherens. Nat Cell Biol. 2010;12:696–702. doi: 10.1038/ncb2072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner JR, Angle JM, Black ED, Joyal JL, Sacks DB, Madara JL. PKC-dependent regulation of transepithelial resistance: roles of MLC and MLC kinase. Am J Physiol. 1999;277:C554–C562. doi: 10.1152/ajpcell.1999.277.3.C554. [DOI] [PubMed] [Google Scholar]
- Turner JR, Rill BK, Carlson SL, Carnes D, Kerner R, Mrsny RJ, Madara JL. Physiological regulation of epithelial tight junctions is associated with myosin light-chain phosphorylation. Am J Physiol. 1997;273:C1378–C1385. doi: 10.1152/ajpcell.1997.273.4.C1378. [DOI] [PubMed] [Google Scholar]
- Tyska MJ, Mooseker MS. A role for myosin-1A in the localization of a brush border disaccharidase. J Cell Biol. 2004;165:395–405. doi: 10.1083/jcb.200310031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Utech M, Ivanov AI, Samarin SN, Bruewer M, Turner JR, Mrsny RJ, Parkos CA, Nusrat A. Mechanism of IFN-gamma -induced endocytosis of tight junction proteins: myosin II-dependent vacuolarization of the apical plasma membrane. Mol Biol Cell. 2005;16:5040–5052. doi: 10.1091/mbc.E05-03-0193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Utech M, Mennigen R, Bruewer M. Endocytosis and recycling of tight junction proteins in inflammation. J Biomed Biotechnol. 2010;2010:484987. doi: 10.1155/2010/484987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Bodegraven AA, et al. Genetic variation in myosin IXB is associated with ulcerative colitis. Gastroenterology. 2006;131:1768–1774. doi: 10.1053/j.gastro.2006.09.011. [DOI] [PubMed] [Google Scholar]
- Walsh SV, Hopkins AM, Chen J, Narumiya S, Parkos CA, Nusrat A. Rho kinase regulates tight junction function and is necessary for tight junction assembly in polarized intestinal epithelia. Gastroenterology. 2001;121:566–579. doi: 10.1053/gast.2001.27060. [DOI] [PubMed] [Google Scholar]
- Wu X, Bowers B, Rao K, Wei Q, Hammer JA 3rd. Visualization of melanosome dynamics within wild-type and dilute melanocytes suggests a paradigm for myosin V function in vivo. J Cell Biol. 1998;143:1899–1918. doi: 10.1083/jcb.143.7.1899. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.