

The mechanisms of progesterone inhibition of milk protein gene expression in mammary epithelial cells involve progesterone receptor mediated changes in chromatin at the promoter and enhancer.
Abstract
Differentiated HC-11 cells ectopically expressing progesterone receptor (PR) were used to explore the molecular mechanisms by which progesterone suppresses β-casein gene transcription induced by prolactin (PRL) and glucocorticoids in the mammary gland. As detected by chromatin immunoprecipitation assays, treatment of cells with the progestin agonist R5020 induced a rapid recruitment (5 min) of PR to the proximal promoter (−235 bp) and distal enhancer (−6 kb upstream of transcription start site) of β-casein. PR remained bound for 4 h and was dissociated by 24 h after treatment. Despite efficient binding, the hormone agonist-occupied PR did not stimulate transcription of the β-casein gene. Recruitment of signal transducer and activator of transcription 5a, glucocorticoid receptor, and the CCAAT enhancer binding protein β to the enhancer and proximal promoter of β-casein induced by PRL and glucocorticoids was blocked by progestin cotreatment, whereas PR binding was induced under these conditions. PRL/glucocorticoid-induced histone acetylation and the recruitment of the coactivator p300 and RNA polymerase II required for gene activation were also inhibited by progestin. In addition, progestin prevented dissociation of the corepressors Yin and Yang 1 and histone deacetylase 3 from the promoter, and demethylation of lysine 9 of histone 3 induced by PRL and glucocorticoids. These studies are consistent with the conclusion that progesterone interferes with PRL/glucocorticoid induction of β-casein transcription by a physical interaction of PR with the promoter and enhancer that blocks assembly of a transcriptional activation complex and dissociation of corepressors and promotes repressive chromatin modifications. These studies define a novel mechanism of steroid receptor-mediated transcriptional repression of a physiologically important gene in mammary gland development and differentiation.
Culture systems of differentiated mammary epithelial cells, such as HC-11, have been instrumental in defining mechanisms of lactogenic hormone [prolactin (PRL) and glucocorticoid] regulation of milk protein genes (1). PRL activation of β-casein is mediated by the PRL receptor/Janus kinase (Jak)-2/signal transducer and activator of transcription (Stat)5 signaling pathway. Stat5 resides in the cytoplasm in an inactive form, becomes tyrosine phosphorylated by Jak2 in response to PRL binding to its receptor, and it interacts with specific response elements in the promoter and enhancer of the β-casein gene (2, 3). Stat5a contains a C-terminal transcriptional activation domain that binds coactivators such as p300/cAMP response element-binding protein-binding protein that are essential for mediating transcription (4, 5). Of the two Stat5 isotypes, Stat5a is more important for PRL-dependent mammary gland development and lactation, whereas Stat5b is more involved in GH signaling (6, 7). Glucocorticoids alone have little to no ability to induce expression of β-casein. However, they synergize with PRL through positive cooperative interactions between the glucocorticoid receptor (GR) and Stat5a (1, 8–11). More recently, the essential nature of Stat5 for PRL/glucocorticoid induction of β-casein gene expression was shown by small hairpin RNA knockdown of Stat5 in primary mouse mammary epithelial cell cultures (12).
Mammary gland-specific expression of milk protein genes does not involve a tissue- specific transcription factor but rather the unique combinatorial interactions of ubiquitous Stat5, GR, and other transcription factors. The β-casein promoter (−230 bp from the transcription start site) contains binding sites for Stat5, CCAAT/enhancer-binding protein β (C/EBPβ), the transcriptional repressor Yin and Yang (YY)1 and multiple glucocorticoid response element (GRE) half-sites (3). An evolutionarily conserved distal enhancer with multiple binding sites for Stat5, C/EBPβ, and other factors is located between −6.0 and −1.4 kb from the transcription start site (13, 14). In addition to cooperative interactions between GR and Stat5a at the promoter, C/EBPβ potentiates Stat5a-mediated transactivation of β-casein gene in a manner dependent on a functional GR. It has been proposed that GR relieves an inhibitory conformation of C/EBPβ within in its N-terminal transactivation domain (15). Thus, Stat5a, GR, and C/EBPβ cooperate to mediate maximal expression of β-casein and are thought to act as a unit for recruitment of coactivators such as p300 with histone acetyl transferase activity required for gene activation through acetylation of histones and chromatin remodeling.
In addition to positive interacting factors the repressors, YY1, silencing mediator of retinoid and thyroid receptor, and histone deacetylase 3 (HDAC3) have been implicated to play a role in hormone regulation of β-casein gene expression. YY1 interacts constitutively with the promoter in the absence of hormone, and its dissociation induced by PRL and glucocorticoids is required for activation of β-casein. The YY1 binding site is low affinity, and readily displaced by Stat5 and C/EBPβ at adjacent sites (16, 17). Recent chromatin immunoprecipitation (ChIP) assays demonstrated a correlation between the dynamics of YY1 and HDAC3 dissociation from the β-casein promoter upon treatment of cells with PRL plus glucocorticoid, suggesting that YY1 and HDAC3 form a functional complex (18). Silencing mediator of retinoid and thyroid receptor has been reported to interact constitutively with Stat5a, and its dissociation has been suggested to also be required for cytokine-induced activation of Stat5 (19). Thus, hormone activation of β-casein gene transcription involves release of inhibitory factors as well as recruitment of transcriptional activators.
Progesterone has multiple roles in the mammary gland during pregnancy. In early pregnancy, it stimulates proliferation and expansion of epithelial cells and alveolar morphogenesis. In mid-to-late pregnancy, it inhibits milk protein production and tight junction closure until parturition; the latter inhibitory action is important for preventing reflux of any accumulated milk into the interstitial space (20–24). In the mouse, a dramatic decline in circulating progesterone at parturition leads to increased milk protein production and tight junction closure (25–27). Progesterone receptor (PR) expression in the mammary gland also declines progressively during pregnancy, suggesting that reduced PR at parturition also contributes to the onset of lactation or “secretory activation” (28). In humans, progesterone levels remain high during the onset of labor. However, lactation is delayed for 2–3 d when a postpartum drop in circulating progesterone also occurs (22). Thus, release from inhibition by progesterone appears to be a general mechanism required for secretory activation of the mammary gland at the transition from pregnancy to lactation.
The suppressive effect of progesterone on milk protein gene expression was first shown in vivo, where ovariectomy of pregnant animals resulted in transient lactogenesis and accumulation of β-casein mRNA and protein. This effect was prevented by the administration of progesterone at the time of ovariectomy but not by other hormones, including estrogen, PRL, or glucocorticoids (13, 20, 21, 24, 25, 29). Experiments with organ cultures from mammary gland explants of pregnant mice similarly demonstrated that progesterone inhibited β-casein mRNA accumulation stimulated by PRL (30–32). We subsequently showed in cell culture systems that PR in a progestin-agonist-dependent manner inhibited PRL- and glucocorticoid-induced transcriptional activation of β-casein by interfering with the PRLR/Jak-2/Stat5a signaling pathway (33). Inhibition by progesterone was observed in multiple cell lines either by transient cotransfection with β-casein reporter genes, PR and reconstituted PRL, and GR signaling pathways or with ectopically expressed PR in HC-11 cells and primary mouse mammary epithelial cell cultures using endogenous β-casein and PRL/GR pathways (33).
In this study, we used HC-11 cells and ChIP assays to examine the influence of progesterone and PR on cooperative interactions and dynamics of the various transcription factors and coregulatory proteins that interact with the promoter and enhancer of the endogenous β-casein gene. Results show that PR recruited to the promoter and enhancer in a progesterone-dependent manner inhibited the assembly of an activating transcription complex and dissociation of corepressors induced by PRL and glucocorticoids. Additionally, progestin-dependent PR recruitment to the β-casein gene produced repressive histone modifications at the promoter and enhancer. These studies define a novel mechanism of progesterone repression of gene transcription of a developmentally important target gene in the mammary gland.
Results
HC-11 cells as an experimental model to study PR regulation of differentiated mammary epithelium
Study of PR signaling in differentiated mammary epithelial cells in culture has been hampered by the well-known rapid loss of expression of endogenous PR in primary cultures and lack of expression of PR in established nontumorigenic breast epithelial cell lines. HC-11 is a mouse mammary epithelial cell line capable of undergoing differentiation in vitro. These cells contain an intact PRL receptor/Jak2/Stat5 signaling pathway and functional GR, and expression of the endogenous β-casein gene can be induced by synergistic actions of PRL and glucocorticoids (1). The dynamics of association and dissociation of Stat5a, GR, and C/EBPβ with the β-casein promoter and enhancer in response to PRL and glucocorticoids has been characterized by ChIP assay (34). A rapid (5–15 min) recruitment of Stat5a, GR, and C/EBPβ at the promoter was observed in response to treatment with PRL plus hydrocortisone (HC) accompanied by recruitment of p300, RNA polymerase II (RNA pol II), acetylation of histone H3, and dissociation of YY1. Similar results were observed for binding to the enhancer except that GR recruitment was most efficient by treatment with HC alone, whereas C/EBPβ recruitment was maximal when HC-11 cells were treated with PRL alone (34).
To examine the influence of progesterone on interactions of transcriptional regulatory proteins with the β-casein gene, PR was expressed ectopically in HC-11 by transduction with a recombinant adenovirus encoding full-length PR-B. The optimal condition for viral transduction was determined by varying the multiplicity of infection (MOI) of the adenovirus. At MOI of 25–50, PR was expressed in the majority of cells with a predominantly nuclear immunostaining pattern (Fig. 1A) and as an intact protein at levels equal to or less than endogenous PR in T47D breast cancer cells as determined by immunoblotting (Fig. 1B). In previous studies with ectopically expressed PR in HC-11 cells, we showed that the progestin R5020 inhibited PRL/glucocorticoid induction of a β-casein-LUC reporter gene as well as endogenous β-casein mRNA expression as measured by RT-PCR (33). This effect was mediated equally by mouse or human PR (hPR) and by the A or B isoform of PR (33). Because of the efficiency of the hPR-specific antibody [1294 monoclonal antibody (mAb)] for ChIP assays, hPR-B was used in the present experiments (18, 33, 35, 36). To confirm and extend previous results using quantitative real-time PCR, a robust induction of endogenous β-casein mRNA by treatment with PRL and glucocorticoids was observed with nontransduced HC-11 cells lacking PR and in transduced cells expressing PR (Fig. 1C). By contrast, no induction of β-casein was observed by treatment with R5020 either in the absence or presence of PR. A small reduction of β-casein expression was observed in cells expressing PR in the absence of R5020 (26–46%) as compared with cells lacking PR. A much more dramatic statistically significant reduction (73–82%) was obtained by treatment with R5020, and this did not occur with nontransduced cells lacking PR (Fig. 1C). These results demonstrate that progesterone inhibits PRL/glucocorticoid induction of endogenous β-casein gene expression in a manner dependent on PR and that progesterone alone has no inductive activity. Thus, ectopic expression of PR by adenovirus in HC-11 cells provides an appropriate cell culture system to examine interactions of PR with the endogenous β-casein gene in the context of natural chromatin.
Fig. 1.

Ectopic PR expressed in HC-11 cells mediates progestin-dependent inhibition of β-casein mRNA expression induced by PRL and glucocorticoids. A, a–d, Immunocytochemistry detection of PR in HC-11 cells infected with varying MOI (a, 0; b, 10; c, 25; and d, 50) of recombinant adenovirus encoding PR-B. B, PR expression detected by Western blotting of HC-11 cells infected with adenovirus encoding PR-B vs. T47D breast cancer cells; 100 μg of protein in cell lysates were analyzed, and β-actin was used as a loading control. C, Hormone effects on β-casein RNA expression in HC-11 cells. Cells were mock transduced (no PR) or were transduced at an MOI of 25 or 50 with recombinant adenovirus expressing PR and treated for 24 h with ethyl alcohol (EtOH), R5020 (100 nm), PRL (3 μg/ml)/HC (1 μg/ml), or PRL (3 μg/ml)/HC (1 μg/ml) + R5020 (100 nm). RNA was isolated and assayed by quantitative real-time PCR as described in Materials and Methods. Graphical data represent relative β-casein mRNA normalized to GAPDH as the mean ± sem from three independent experiments, n = 3. *, P < 0.05 for differences between no PR and PR.
PR recruitment to the β-casein promoter and enhancer impairs PRL/glucocorticoid-induced binding of Stat5a
We previously showed by ChIP assay of HC-11 cells that PR was recruited to the proximal promoter of β-casein in a progestin-dependent manner at 1 h of hormone treatment, whereas Stat5a was recruited by 1 h treatment with PRL + HC (33). No recruitment of either PR or Stat5a was detected by cotreatment of cells for 1 h with PRL/HC + R5020, suggesting a mutual interference between activated PR and Stat5a (33). However, interactions of steroid receptors and other transcription factors with regulatory gene elements are known to be dynamic (37), and we did not analyze interactions with the distal enhancer (33). Therefore, we have now examined a time course of PR and Stat5a recruitment to both the promoter and enhancer in response to various hormone treatments. Primers for PCR amplification of the proximal promoter span a 170-bp region (−190 to −20 from the transcription start site) containing binding sites for Stat5a, C/EBPβ, YY1, and GRE/progestin response element (PRE) half-sites, whereas a 400-bp region (−6400 to −6000) of the distal enhancer was amplified that contains binding sites for Stat5, C/EBPβ, and other factors (Fig. 2A). Little to no recruitment of PR or Stat5a to the promoter or enhancer was detected in the absence of hormone. PR and Stat5a were rapidly recruited to both sites within 5 min of treatment with their cognate activating hormone(s) (Figs. 2 and 3). Recruitment at the promoter was maximal for Stat5a at 5 min of PRL/HC treatment, remained at this level for 4 h, and returned to the basal no hormone-treated level by 24 h (Fig. 2B). The kinetics and efficiency of Stat5a interaction at the enhancer in response to PRL/HC was similar (Fig. 2B). A similar dynamic of PR interaction in response to R5020 was observed, except the efficiency of PR binding to the enhancer was less than the promoter (Fig. 3A). Although the enhancer lacks obvious GRE/PRE binding sites for PR, the progestin-dependent recruitment of PR to the enhancer is specific.
Fig. 2.
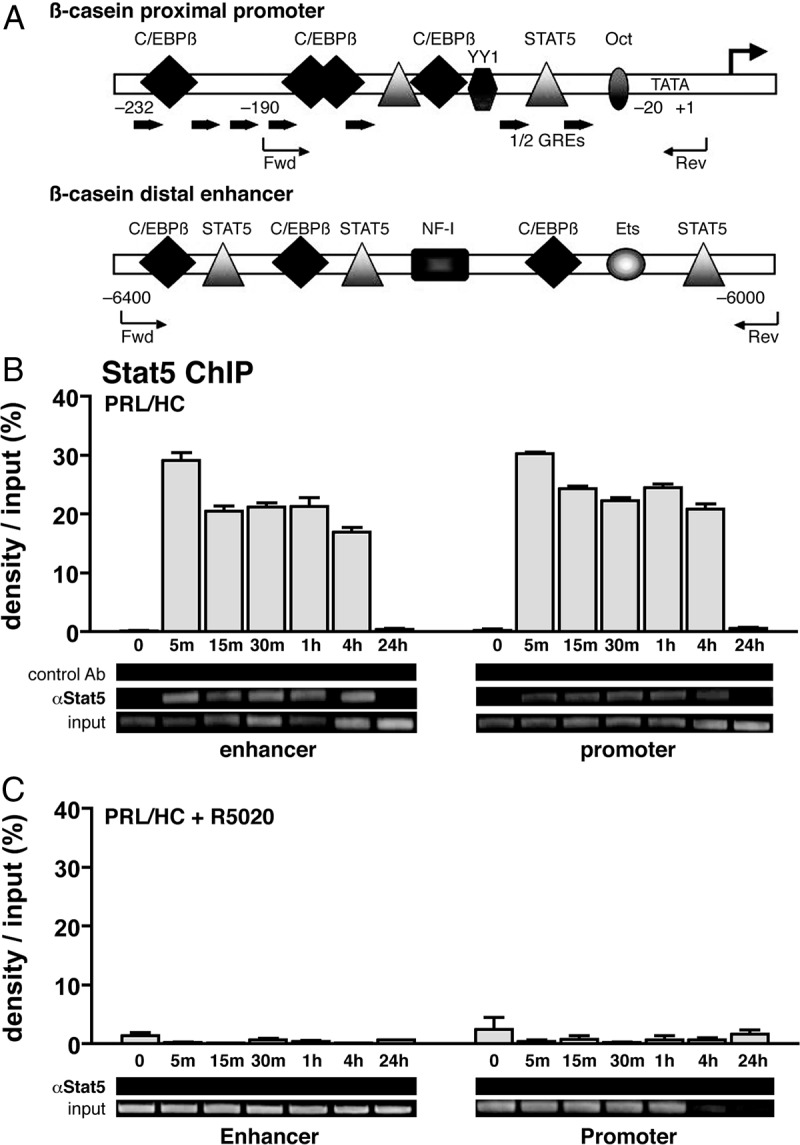
Recruitment of Stat5a to the β-casein promoter and distal enhancer in HC-11 cells is impaired by progesterone. HC-11 cells were transduced with 50 pfu/cell of adenovirus encoding hPR-B and then treated for 1 h with either: ethyl alcohol (EtOH) (−control), ovine PRL (3 μg/ml)/HC (1 μg/ml), R5020 (100 nm), or ovine PRL/HC + R5020 (same concentrations as given alone). Cells were then harvested, lysed, and processed for ChIP with antibodies to Stat5 (L-20) assay using PCR primers flanking the Stat5 response element (S5RE) and GRE half-sites within the proximal promoter of β-casein (−190 to −20) or to the distal enhancer region (−6400 to 6000). A, Schematic representation of β-casein proximal promoter and distal enhancer with forward (Fwd) and reverse (Rev) primer sets used for PCR denoted. B, Stat5a binding to the proximal promoter and distal enhancer in response to treatment with of PRL/HC for different time points between 5 min and 24 h. C, Stat5a binding to the proximal promoter and distal enhancer in the presence of PRL/HC and R5020 for time points between 0 and 24 h. Quantification of ChIP assays from multiple experiments was performed by agarose gel electrophoresis of PCR-amplified input and immunoprecipitated DNA and image scanning analysis of band intensitives with Syngene Genetools software. Data represent the average density of immunoprecipitated DNA as a percentage of input DNA on the agarose gels adjusted after subtracting signals obtained with control IgG. The values are averages ± sem from three independent experiments. Insets are representative agarose gels of PCR products from one of the three experiments. Oct, Octamer transcription factor; Ets, Ets transcription factor; NF-1, nuclear factor 1.
Fig. 3.

PRL/glucocorticoid treatment has no effect on assembly of PR with the β-casein promoter and enhancer but accelerates disassembly. HC-11 cells were transduced with 50 pfu/cell of adenovirus encoding hPR-B and then treated for 1 h as in Fig. 2 with either: ethyl alcohol (EtOH) (−control), ovine PRL/HC, R5020, or ovine PRL/HC + R5020. Cells were harvested and processed for ChIP assay as in Fig. 2 except using antibody to PR (1294). A, PR binding to the promoter and enhancer in response to R5020 at different time points between 5 min and 24 h. B, PR binding to the promoter and enhancer in the presence of PRL/HC and R5020 at time points between 0 and 24 h. Quantification of ChIP assays from multiple experiments was performed as in Fig. 2. Values are averages ± sem from three independent experiments.
The dynamics of PR and Stat5a interaction with β-casein was dramatically altered in the presence of both PRL/HC and R5020 as compared with treatment by single activating cognate hormones for PR or Stat5a. No Stat5a recruitment at the enhancer or promoter was detected at any time point (5 min to 24 h) after cotreatment of cells with PRL/HC + R5020 (Fig. 2C). In contrast, PR recruitment at the enhancer and promoter at early times of treatment with PRL/HC + R5020 (5–30 min) was similar to that with R5020 alone (Fig. 3B). However, PR interaction returned to basal nonhormone-treated control levels by 60 min of treatment with PRL/HC + R5020, whereas maximal association of PR in the presence of R5020 alone was sustained for more than 4 h (compare Fig. 3A and Fig. 3B). These data indicate that PRL/HC had no effect on progestin-induced binding of PR to the promoter and enhancer but did accelerate the dissociation of PR from these sites. In contrast PR/progestin completely blocked binding of Stat5a induced by PRL/HC (Fig. 2C).
To determine whether PR recruitment to the β-casein gene is required for progestin-dependent inhibition of Stat5 binding, two additional experiments were performed. First, we analyzed the ability of a PR DNA-binding domain (DBD) mutant (C587A) expressed from an adenoviral vector in HC-11 cells to repress recruitment of Stat5 to the β-casein gene. The PR DBD mutant lacks DNA binding activity for PRE and was shown previously to be unable to mediate progestin-dependent repression of PRL/HC induction of β-casein gene transcription (33). By ChIP assay, the PR DBD mutant (C587A) was not recruited to either the promoter or the enhancer of β-casein in the absence or presence of R5020 (Fig. 4A). It also failed to mediate a progestin-dependent repression of Stat5 recruitment induced by PRL/glucocorticoid (Fig. 4B), as compared with wild-type PR (Fig. 2). Secondly, we analyzed the dose dependency of the repressive action of 5020 to determine whether partial recruitment of PR would correlate with partial inhibition of Stat5 binding to the β-casein gene in ChIP assay. HC-11 cells expressing PR-B were treated for 30 min with PRL (3 μg/ml) + HC (1 μg/ml) in the presence of varying concentrations of R5020 (0–100 nm). This time point was chosen because PR remains maximally associated with the β-casein promoter and enhancer at 30 min in the presence of PRL/HC and a saturating dose of R5020 (Fig. 3). As anticipated, PRL/HC in the absence of R5020 failed to stimulate recruitment of PR. In the presence of PRL/HC, a dose-dependent recruitment of PR was detected at the promoter and enhancer over a range of 10−10 to 10−7 m R5020. A proportional reduction of Stat5a binding was observed in response to increasing doses of R5020 that paralleled the proportional increase of PR binding at both the enhancer and promoter (Fig. 5, A and B). These results, taken together, support the conclusion that the repressive effect of progestin on PRL/HC induction of β-casein requires physical recruitment of PR and that PR binding either competes with, or displaces, Stat5a binding at the promoter and enhancer.
Fig. 4.

Mutating the DBD of PR eliminates progestin inhibition of Stat5a binding to the β-casein promoter and enhancer. HC-11 cells were transduced with 50 pfu/cell of adenovirus encoding hPR-B containing a point mutation in the zinc finger (C587A) region of the DBD and then treated for 1 h with either: ethyl alcohol (EtOH) (−control), ovine PRL (3 μg/ml), R5020 (100 nm), or ovine PRL (3 μg/ml) + R5020 (100 nm). Cells were processed for ChIP assay using antibodies to PR (1294) or to Stat5 (L-20). A, hPR-B (C587A) binding at the proximal promoter and distal enhancer. B, Stat5a binding at the proximal promoter and distal enhancer. Graphical representation of ChIP data was performed as in Figs. 2 and 3. The values are averages ± sem from three experiments. Insets are representative agarose gels of PCR products from one of three replicate experiments.
Fig. 5.

PRL/glucocorticoid-induced recruitment of Stat5a is blocked by progestin in a dose-dependent manner. HC-11 cells were transduced with 50 pfu/cell of adenovirus encoding hPR-B and treated for 30 min with ovine PRL (3 μg/ml)/HC (1 μg/ml) alone or with PRL/HC + varying concentrations of R5020 (10−12 to 10−7 m) as indicated in the figure. Cells were processed for ChIP assay as in Figs. 2 and 3. A, left panel, PR-B binding to the proximal promoter; right panel, Stat5a binding to the proximal promoter. B, left panel, hPR-B binding to the distal enhancer; right panel, Stat5a binding to the distal enhancer. White open bars represent treatment with PRL/glucocorticoid alone; gray bars, represent cotreatment with R5020 and PRL/HC. Quantification of ChIP assays from multiple experiments was performed as in Fig. 2. The values are averages ± sem from three independent experiments.
PR recruitment impairs assembly of a Stat5, GR, C/EBPβ, p300, and RNA pol II transcriptional activator complex and inhibits histone acetylation at the β-casein promoter/enhancer
Maximal hormone activation of β-casein gene transcription requires cooperative interactions of at least three sequence-specific activators, including Stat5a, GR, and C/EBPβ, and they act as a unit to recruit coactivators required for chromatin remodeling and assembly of RNA pol II (8, 15, 34). Therefore, we used ChIP assays to examine the influence of progestin on recruitment of these other activating factors. To examine effects on GR, HC-11 cells were treated for 30 min with R5020 (100 nm), HC (1 μg/ml), R5020 (100 nm) + HC (1 μg/ml), HC (1 μg/ml) + PRL (3 μg/ml), or all three hormones together. We chose a 30-min treatment because previous ChIP assays showed maximal GR recruitment by HC at this time (34). No PR interaction with either the promoter or enhancer was detected in cells treated with HC or PRL + HC, under conditions where R5020 stimulated an efficient interaction of PR. Cotreatment of cells with R5020 + HC or R5020 + PRL did not promote further interaction of PR over that obtained with R5020 alone (Supplemental Fig. 1, published on The Endocrine Society's Journals Online web site at http://mend.endojournals.org). Thus, PR recruitment was progestin specific and no cross-reactive effect of HC or PRL on binding of PR to the β-casein gene was observed. GR was recruited to the proximal promoter by HC, and adding PRL had no further effect. GR was also recruited to the distal enhancer by HC, although to a lower degree than the proximal promoter (Fig. 6A). R5020 inhibited binding of GR to the promoter and enhancer induced either by HC, or by PRL/HC, whereas R5020 alone was unable to stimulate interaction of GR with β-casein gene (Fig. 6A). Using an antibody to total C/EBPβ, we observed that PRL/HC treatment for 30 min resulted in recruitment of C/EBPβ to the proximal promoter with little to no binding at the enhancer, even though there are potential C/EBPβ response element sequences within the enhancer region (Fig. 6A). This selective recruitment of C/EBPβ to the promoter is consistent with previous studies (34). R5020 alone failed to recruit C/EBPβ but prevented C/EBPβ association with the proximal promoter induced by PRL/HC (Fig. 6A). Thus PR occupied by progestin prevented the assembly of all three sequence-specific activators, Stat5a, GR, C/EBPβ, required for maximal hormonal induction of β-casein transcription.
Fig. 6.

Progesterone blocks recruitment of the transcriptional activation complex at the promoter and enhancer of β-casein gene induced by PRL and glucocorticoids. HC-11 cells were transduced with 50 pfu/cell of adenovirus encoding hPR-B and then treated with ethyl alcohol (EtOH) (−control), R5020 (100 nm), HC (1 μg/ml), HC (1 μg/ml) + R5020 (100 nm), PRL (3 μg/ml) + HC (1 μg/ml), or PRL (3 μg/ml) + HC (1 μg/ml) + R5020 (R) at 100 nm for the indicated times (15 or 30 min). Cells were harvested and processed for ChIP assay as in Fig. 2 using antibodies to GR, C/EBPβ, p300, acetylated H3, and total RNA pol II. A, GR, C/EBPβ, p300, acetyl-H3, and RNA pol II interaction with the proximal promoter and distal enhancer. Graphical quantitation of the ChIP data from replicate experiments was performed as in Fig. 2. Values are averages ± sem from three independent experiments. B, Stat5a, PR, GR, C/EBPβ, p300, or RNA pol II interaction with the intervening region between the promoter and enhancer of β-casein (−3000 bp from the transcription start site). Gel insets are from a single representative experiment that was repeated three times.
p300 is a histone acetyl transferase-containing coactivator for Stat5, GR, and C/EBPβ and is known to be involved in hormone induction of β-casein gene expression. Treatment of HC-11 cells with PRL/HC resulted in an efficient interaction of p300 with the proximal promoter and distal enhancer (Fig. 6A). Addition of R5020 alone did not stimulate interaction of p300 with the β-casein gene and when given as a cotreatment prevented recruitment of p300 induced by PRL/HC (Fig. 6A). Consistent with p300 results, R5020 alone had no effect on acetylation of histone H3 but inhibited acetylation induced by PRL/HC. Treatment with PRL/HC for 30 min resulted in recruitment of RNA pol II to the proximal promoter and distal enhancer, and this was also inhibited by cotreatment with R5020 (Fig. 6A). As controls for DNA specificity of the ChIP assay, no association of PR, Stat5a, GR, C/EBPβ, or p300 was detected with a region of the β-casein gene (200-bp amplicon), located between the enhancer and promoter (∼3000 bp from the transcription start site), that lacks known response elements for any of these factors (Fig. 6B). However, RNA pol II was recruited to this region in response to PRL/HC treatment but not by the progestin R5020 (Fig. 6B). These data taken together indicate that PR recruitment to the promoter and enhancer of the β-casein gene impairs PRL/HC-induced assembly of the activator complex required for transcription of the β-casein gene.
PR recruitment prevents derepression of the β-casein gene induced by PRL/glucocorticoids
In confirmation of previous studies (34), a constitutive association of the corepressor YY1 was observed with the proximal promoter of the β-casein gene in the absence of hormone, and this was rapidly (30 min) lost upon treatment with PRL/HC (Fig. 7). R5020 treatment alone had no effect on constitutive interaction of YY1, but it blocked the ability of PRL/HC to induce YY1 dissociation (Fig. 7). HDAC3 interaction was detected with the proximal promoter but not with the enhancer in the absence of hormone, and it was also dissociated by a 30-min treatment with PRL/HC. R5020 alone had no effect on constitutive association of HDAC3 with the promoter, and prevented its dissociation induced by PRL/HC (Fig. 7). Methylation of lysine 9 (K9) of histone H3 is a modification commonly observed with transcriptionally repressed chromatin (38, 39). By ChIP assay, dimethylation of K9-H3 in the absence of hormone was detected at the proximal promoter and distal enhancer (Fig. 7). Upon treatment with PRL/HC for 30 min, dimethylation of K9-H3 decreased to undetectable levels, indicating that a rapid demethylation of this repressive histone mark correlates with transcriptional activation of the β-casein gene. Similar to results with YY1 and HDAC3, R5020 alone had no effect on dimethylation of K9-H3, whereas it inhibited the ability of PRL/HC to induce demethylation (Fig. 7). These results demonstrate that in addition to inhibiting assembly of a transcriptional activator complex, physical recruitment of PR by progestin prevents relief of repression of the β-casein gene induced by PRL and glucocorticoids.
Fig. 7.
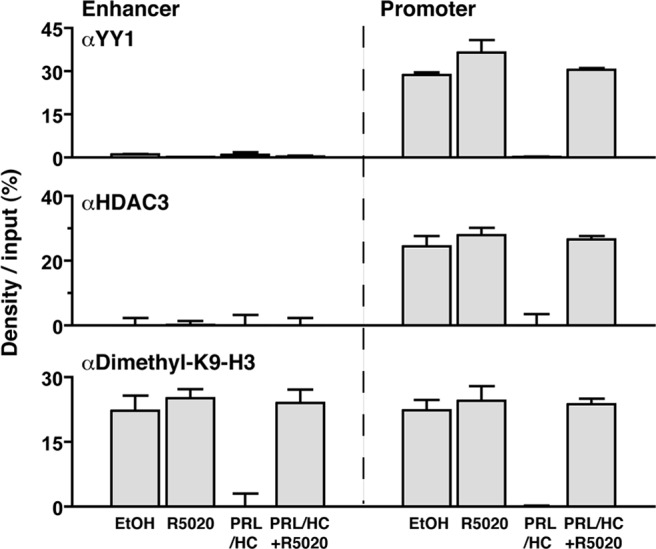
PR recruitment blocks dissociation of corepressors and demethylation of K9–H3 induced by PRL/glucocorticoids. HC-11 cells were transduced with 50 pfu/cell of adenovirus encoding hPR-B and treated with ethyl alcohol (EtOH) (−control), R5020 (100 nm), PRL (3 μg/ml)/HC (1 μg/ml), or PRL (3 μg/ml)/HC (1 μg/ml) + R5020 (100 nm) for 30 min. Cells were then harvested and processed for ChIP assay using antibodies specific to YY1 (sc-281), HDAC3 (07-522), or dimethyl-K9–H3 (07-441). Graphical quantitation of ChIP data from replicate experiments was performed as in Fig. 2. Values are averages from three independent experiments ± sem.
PR interaction with the β-casein gene promotes repressive chromatin modifications
The inhibitory effect of progesterone on PRL/HC-induced dissociation of YY1/HDAC3 and demethylation of K9-H3 could be indirect through PR impairing the assembly of the Stat5a/GR/C/EBPβ complex required to mediate the effects of PRL/HC. However, it should be noted that at the 30-min time point in the presence of all three hormones, only PR was recruited to the promoter and enhancer by ChIP assay, suggesting that it could also be acting by promoting a repressed chromatin state (Figs. 2 and 3). To explore this possibility further, we performed a washout experiment designed to determine whether short-term binding of PR to the enhancer/promoter has prolonged effects on Stat5 binding and histone modifications. HC-11 cells were treated for 1 h with R5020 followed by washing with culture medium lacking R5020 to promote dissociation of ligand and early expulsion of PR from the β-casein gene. At different times after the washout, cells were then treated with a short pulse (15 min) of PRL/HC and processed for ChIP assay. As shown in Fig. 8A, left panel, the washout accelerated dissociation of PR from the promoter and enhancer. As compared with cells treated continuously with progestin where PR remains bound for more than 4 h (Fig. 3), most detectable PR binding was gone by 15 min after the washout (Fig. 8A, left panel). No PRL/HC induction of Stat5 binding was detected until 1 h after the washout, and maximal recruitment of Stat5a occurred between 4 and 24 h after washout (Fig. 8A, right panel). By comparison, maximal recruitment of Stat5a in nonpretreated cells occurred within 5 min of PRL/HC addition (Fig. 2B). After the washout, PRL/HC-induced acetylation of histone H3 and demethylation of K9-H3 were also delayed. In nonpretreated cells, demethylation of K9-H3 and acetylation of H3 induced by PRL/HC took place between 15 and 30 min (Figs. 6A and 7), whereas these events were unaffected by PRL/HC at 1 h and required up to 4 h or more for maximal effects after washout of the cells (Fig. 8B). These results indicate that PR recruitment to the β-casein promoter and enhancer promotes sustained effects that are inhibitory to binding of Stat5a and to histone modifications induced by PRL/HC even after PR has been dissociated.
Fig. 8.

Progestin-dependent recruitment of PR promotes a prolonged repressive chromatin state for Stat5 binding and histone modifications. HC-11 cells were transduced with 50 pfu/cell of adenovirus encoding hPR-B and then treated with R5020 (100 nm) for 1 h. After 1 h, cells were either harvested for ChIP assay (+control) or were washed three times (washout) with serum-free medium and incubated at 37 C for indicated times after washout (from 5 min to 24 h) with a short pulse of PRL (3 μg/ml)/HC (1 μg/ml) for 15 min. A, left panel, PR interaction with the β-casein promoter at times after washout as indicated; right panel, Stat5a interaction with the β-casein promoter at times after washout as indicated. B, left panel, Dimethylation of K9-H3 of the β-casein promoter at times after washout as indicated; right panel, Acetylation of histone H3 of the β-casein promoter at times after washout as indicated. White open bars (+) represent 1-h R5020 treatment alone, and shaded bars represent times after R5020 washout before a 15-min treatment with PRL/HC. ChIP assays at each time point were conducted for PR, Stat5, dimethyl-K9-H3, and acetylated histone H3, and quantification from multiple experiments was performed as in Fig. 2. The values are averages ± sem from three independent experiments.
Discussion
Milk protein gene transcription is a tightly regulated process, involving cooperative interactions of multiple transcription factors and coregulatory proteins with promoter and enhancer regions, remodeling of chromatin, and assembly of the preinitiation complex in a spatiotemporal fashion (3, 30). Milk protein genes need to be silenced or minimally expressed during most of the adult female life and turned on quickly and robustly at the transition between late pregnancy and lactation (22, 23). In this study, we examined the mechanism of the repressive effect of progesterone on transcriptional activation of the β-casein gene induced by lactogenic hormones in differentiated mammary epithelial HC-11 cells. Although this experimental system required ectopic expression of PR by recombinant adenoviral vectors, PR expression was carefully controlled to be less than or equal to endogenous PR in the majority of cells, and effects on β-casein expression and recruitment of factors by ChIP assay were highly dependent on progesterone. Also, under the same HC-11 cell culture conditions, we observed that R5020 induced the expression of an endogenous target gene, inhibitor of differentiation 4, that together with PRL, gave a synergistic induction that involved cobinding of PR and Stat5 to enhancer regions of the inhibitor of differentiation 4 gene (40). Progestin induction of receptor activator of nuclear factor κ-B ligand gene expression and PR binding to receptor activator of nuclear factor κ-B ligand enhancers was also observed in HC-11 cells expressing PR from adenovirus vectors (Obr, A. E., and D. P. Edwards, unpublished observations). Thus, both progestin-dependent gene induction and repression have been observed in HC-11 cells under these conditions, indicating the physiological relevance of our results on gene repression. Our results support the conclusion that inhibition of PRL/HC-induced activation of β-casein gene transcription by progesterone is mediated by a physical recruitment of PR to the promoter and enhancer that blocks assembly of an activation complex consisting of Stat5, GR, C/EBPβ, and p300. In addition, recruited PR exerts a repressive effect on chromatin by maintaining interactions of the corepressors YY1 and HDAC3 with the β-casein gene, deacetylation of histones, and dimethylation of K9 of histone 3 at the promoter and enhancer.
A competitive mechanism for how PR blocks Stat5 binding was indicated by the time course and progesterone dose-response experiments (Figs. 2, 3, and 5). PR and GR are capable of binding to the same or similar DNA response elements. Thus, PR recruitment to the β-casein gene presumably reflects PR binding to the GRE half-sites in the proximal promoter. We previously showed, by in vitro DNA binding assays, that PR is able to bind directly to the two GRE half-sites flanking the proximal most Stat5 response element of β-casein (33). Results from transient cell transfection assays, with β-casein reporter genes containing point mutations in the GRE, further showed the importance of PR-DNA binding and the GRE half-sites for functional inhibitory response to progestin (33). PR DNA binding was also shown in the present study with a PR DBD mutant to be required for progestin-mediated inhibition of Stat5 recruitment (Fig. 4). These data taken together suggest that PR binds to one or both GRE/PRE half-sites and interferes with Stat5a binding to its adjacent response element. Interesting questions raised by these experiments are why GR and PR, which are so closely related, have opposite functional effects on PRL induction of β-casein, and why PR appears to effectively compete with GR for binding to β-casein in ChIP assays. Domain swapping experiments showed that the N terminus of GR was responsible for the positive synergistic activation of β-casein by glucocorticoids and PRL and that this response was abrogated by swapping the N terminus of GR with that of PR (41). However, the region of PR responsible for mediating repression of β-casein, and whether a swap of that domain can confer a functional inhibitory response to GR, has not been analyzed. Because PR binds efficiently in vitro to the GRE half-sites and to the β-casein promoter in vivo as detected by ChIP, it is possible that PR simply binds with higher affinity to the same or overlapping sites as GR. Alternatively, if GR acts predominantly through tethering with Stat5a as suggested by some studies (10), then PR could also interfere with GR binding indirectly through blocking Stat5a-DNA binding. Further studies will be needed to determine the interplay and mechanistic differences between PR and GR interactions with the β-casein gene.
The transcription factor C/EBPβ is necessary for full hormonal activation of β-casein gene transcription, but it requires an active GR to work in this capacity (8, 15, 42). The proximal promoter contains binding sites for C/EBPβ (34), and as expected, PRL + HC treatment induced recruitment of C/EBPβ to the proximal promoter. This interaction was also abolished by addition of progestin (Fig. 6A). It is not known from these results whether inhibition of C/EBPβ recruitment is direct through interaction of C/EBPβ with its proximal promoter binding site or indirect by inhibiting GR binding, which is required to recruit C/EBPβ. We were unable to detect binding of C/EBPβ to the distal enhancer, even though by sequence analysis, there are potential binding sites present. However, the C/EBPβ interaction was analyzed only at 30 min of hormone treatment, and it is possible the kinetics of binding at the distal enhancer are different than the promoter.
How PR interacts with the distal enhancer to inhibit recruitment of other activators is not known. There are no sequences in the enhancer corresponding to a PRE, and PR is recruited less efficiently to the enhancer than the promoter, indicating the presence of a lower affinity binding site in the enhancer. Thus, PR interaction with the enhancer occurs either through binding to a weak noncanonical PRE, by tethering to another protein bound to the enhancer, or by long-range DNA looping between the promoter and enhancer. In support of a DNA looping mechanism, we previously observed by use of the chromosome conformation capture assay (43) that PRL/HC treatment induced a physical interaction between the promoter and enhancer of the β-casein gene in HC-11 cells, and this long-range interaction was inhibited by progestin-induced recruitment of PR (18). Whether PR binding to the enhancer, as a secondary site, also requires long-range looping interactions between the promoter and the enhancer remains to be determined.
We observed a PRL/HC-dependent recruitment of RNA pol II at the enhancer and promoter that was blocked at both sites by progestin. The role of RNA pol II binding at the enhancer is not known. Studies on the interaction of the androgen receptor (AR) coactivator complex with the distal enhancer and promoter of the prostate-specific antigen gene showed by chromosome conformation capture assay that recruitment of AR in response to androgen promoted a physical association of pol II with the enhancer (∼4 kb upstream of the transcription start site) and the promoter (−400 bp). ChIP assays designed to detect factor loading at the enhancer, promoter, and an intervening region between the promoter and enhancer showed that RNA pol II, but not the AR complex, tracked from the enhancer to the promoter along the intervening region in an androgen-dependent manner (37). We also observed RNA pol II interaction with an intervening sequence between the enhancer and promoter of β-casein gene and that this binding was enhanced by treatment with PRL/HC (Fig. 6B). In contrast, no binding was detected in the intervening region with any of the sequence-specific transcription of factors analyzed, including PR, GR, Stat5 and C/EBPβ, and p300 (Fig. 6B). These results suggest that RNA pol II interaction with the β-casein gene undergoes a hormone-dependent sliding from the enhancer to the promoter.
Results in this article support the concept that PRL/HC-induced activation of β-casein gene transcription involves both recruitment of a positive acting transcription factor/coactivator complexes and relief from repression exerted by YY1/HDAC3 and dimethylation of K9-H3. The corepressors YY1 and HDAC3 were constitutively bound to the β-casein promoter in the absence of hormone, and both were rapidly dissociated by treatment with PRL/HC. The similar dynamics of YY1 and HDAC3 in response to hormone, taken together with the fact that the enhancer lacks binding sites for YY1, and YY1/HDAC3 bind only to the promoter, suggests that YY1 and HDAC3 act together as a repressor complex. The β-casein enhancer and promoter exhibited dimethylation of K9 of histone H3, a modification typically associated with repressive chromatin templates (38, 39, 44), and treatment with PRL/HC also resulted in a rapid loss of this repressive histone modification. Interaction of YY1/HDAC3 and dimethylation of K9-H3 were maintained when PR was recruited by R5020. The results with dimethylation of K9-H3 indicate that PRL/HC induction of β-casein involves activation and/or recruitment of a histone lysine demethylase. Lysine-specific demethylase 1 and other demethylases of the Jumonji C domain family have been demonstrated to function as transcriptional coactivators for androgen and estrogen receptors by mediating ligand-dependent derepression of target genes through demethylation of repressive histone marks (45–47). It will be important in future studies to determine more directly the mechanisms and role of histone lysine demethylases in hormonal regulation of β-casein gene transcription and to identify which members of the lysine-specific demethylase 1 and/or Jumonji C family members are involved (48). In addition to blocking assembly of an activator complex and maintaining corepressor interactions, PR recruited to the β-casein gene appears to actively promote repressive chromatin modifications. Transient recruitment of PR and its premature expulsion by washout of excess free steroid resulted in a delayed binding of Stat5, acetylation of histone H3, and demethylation of K9-H3 induced by PRL/HC (Fig. 8). These results suggest that interaction of PR with the β-casein gene promotes a sustained repressive modification of the chromatin template that is incompatible with binding Stat5a and histone acetylation. An ability of PR to actively promote repressive chromatin modifications may explain in part the question of how PR that is not expressed in all mammary epithelial cells can repress β-casein in cells that lack PR (31). The present data raise the possibility that PR may induce repressive epigenetic changes that are sustained during mammary gland development in epithelial cells that lose PR expression.
Mechanisms responsible for direct gene repression mediated by steroid hormone receptors have not been as well defined as those for hormone induction. Another biologically important repressive action of PR is to inhibit nuclear factor kB (NF-kB)-dependent transcription of inflammatory-response pathway genes in breast cancer cells. Progesterone was reported to inhibit IL-1β induction of cyclooxygenase-2 by impairing recruitment of the p65 subunit of NF-kB to the proximal and distal response elements of cyclooxygenase-2 gene (49). Progesterone inhibition of IL-1β induction of a different set of target genes was shown to occur through a physical recruitment of PR that does not impair binding of NF-kB but instead interferes with downstream recruitment of RNA pol II and initiation of transcription (35). Additionally, inhibition of some NF-kB target genes required hormone agonist and the agonist AF2 conformation of PR (chemokine ligand 2/CCL20), whereas others were inhibited either by PR agonist or antagonist [IL-8 and chemokine (C-X-C motif) ligand] (35). In cell cotransfection assays with β-casein-luciferase reporter genes, we observed that the PR antagonist RU486 inhibited PRL/HC induction as efficiently as the progestin agonist R5020 (33). Because RU486 induces binding of PR to target DNA, it suggests that physical recruitment of PR is a key event required for inhibition of PRL/HC induction of β-casein transcription and that the agonist conformation may not be necessary. Why the PR-agonist complex when bound to the β-casein gene does not induce gene transcription is not known. ChIP assays with steroid receptor coactivators (SRC) revealed that SRC-2, but not SRC-1 and SRC-3, was recruited to the β-casein promoter and enhancer in response to progestin (data not shown). This is of interest, because SRC-2 was shown through use of alternate protein interaction surfaces to function as a corepressor for GR-mediated inhibition of NF-kB-induced transaction of the IL-8 gene (50, 51). However, small hairpin RNA knockdown of SRC-2 in HC-11 cells did not alter the ability of PR to mediate repression of the β-casein gene, indicating that SRC-2 does not function as a corepressor in this context (data not shown). SRC-2 recruitment likely reflects an agonist conformation of PR bound to the β-casein gene. Because DNA in a sequence-specific manner can act as a ligand to affect protein conformation of steroid receptors (50–52), it suggests that PR bound to β-casein may adopt a conformation distinct from that of receptor bound to target genes that are activated by progesterone. As further support of this idea, PR was reported to recruit SRC-1 and SRC-3, but not SRC-2, to the progesterone-activated mouse mammary tumor virus promoter (53), distinctive from the pattern of SRC recruitment by PR to β-casein. Clearly, further studies are required to define other specific protein and DNA interactions required for progestin-dependent PR-mediated repression of the β-casein gene.
Materials and Methods
Hormones
R5020 (promegestone; 17α,21dimethyl-19-norpregna-4,9-diene-3,20-one) was obtained from DuPont-New England Nuclear Research Products (Boston, MA), dexamethasone was from Sigma (St. Louis, MO), and ovine PRL was provided by National Institute of Diabetes and Digestive and Kidney Diseases (Bethesda, MD).
Antibodies
1294/H9 is a mouse IgG1 mAb produced against purified hPR that recognizes both the A and B isoforms of PR (36). Polyclonal anti-GR (PA1-512) antibody was purchased from Affinity Bioreagents (Golden, CO). Polyclonal antiacetyl-histone H3 (06-599) antibody, antidimethyl-histone H3 (07-441), and anti-HDAC3 (07-522) were obtained from Upstate Biotechnology (Charlottesville, VA). C-terminal rabbit polyclonal anti-YY1 (C-20) (sc-281), C-terminal rabbit polyclonal anti-p300 (C-20) (sc-585), C-terminal rabbit polyclonal anti-Stat5a (L-20) (sc1081), anti-RNA pol II (N-20) (sc-899), and anti-C/EBPβ (C-19) (sc-150) were obtained from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA).
HC-11 cells
HC-11 mouse mammary epithelial cells were maintained at 37 C and 5% CO2 in RPMI 1640 medium supplemented with 10% bovine calf serum (HyClone, Logan, UT), 2 mm glutamine, 5 μg/ml insulin, 10 ng/ml epidermal growth factor, and 100 U/ml of penicillin and streptomycin. After cells reached confluency (2 × 107), they were grown for an additional 72 h in medium transduced with recombinant adenovirus expressing hPR-B (25–50 plaque-forming unit (pfu)/cell) and then incubated for another 48 h in differentiation medium (RPMI 1640 supplemented with 0.5 m glutamine, 5 μg/ml insulin, 1 μg/ml HC, and 10% dextran-coated charcoal-stripped donor horse serum). At 48 h after differentiation, cells were treated in the same medium with various hormones as indicated in Results and figures, including the synthetic progestin R5020 (5–100 nm) and ovine PRL (3 μg/ml).
Adenovirus transduction to express PR
Recombinant adenoviral transfer vectors containing hPR-B under the control of the cytomegalovirus promoter was constructed by overlap recombination as described previously (33, 54, 55) These are replication-defective viruses lacking the transforming EIA and EIB genes. Purified virus encoding PR-B, termed Ad5dl327cytomegalovirus-PR-B, was grown in large scale and concentrated by CsCl gradient centrifugation. The virus concentration was determined by absorbance at 260 nm, where 1 A260 unit represents 1012 viral particles. The particle-pfu ratio was determined to be close to 100 for all viruses (55).
Immunoblot analysis
Cells were lysed in the following buffer: 50 mm Tris, 2 mm EDTA, 1 mm EGTA, 30 mm KCl, and 1% Igepal, and lysates containing equal amount of total protein were solubilized in 1% sodium dodecyl sulfate (SDS) sample buffer and electrophoresed on 8–12% polyacrylamide SDS gels. Separated proteins were transferred to Immobilon-P polyvinylidene fluoride membranes (Millipore, Bedford, MA) and incubated with appropriate antibodies in 50 mm Tris-HCl (ph7.4), 150 mm NaCl, and 0.1% Tween 20. Antibody interaction was detected by enhanced chemiluminescence (Amersham, Arlington Heights, IL).
Immunocytochemistry
HC-11 cells were grown to confluency as above in four-well chamber slides (no. 354114; BD Falcon, San Jose, CA). At confluency, fresh 10% bovine serum RPMI 1640 media were added to cells containing recombinant adenovirus encoding for hPR-B at various MOI (0–50). Cells were grown for an additional 3 d and then primed for 48 h in differentiation medium. Cells were then fixed for 20 min with 1% paraformaldehyde in PBS and permeabilized in 0.5% Triton X-100. After blocking in 3% BSA, slides were washed in PBS and incubated for 1 h at room temperature with mouse mAb to PR (1294) at 1 μg/ml followed by incubation for 1 h at room temperature with an Alexa Fluor 488 (fluorescein isothiocyanate) secondary antibody to mouse IgG at a 1:800 dilution. Cells were costained with 4′,6-diamidino-2-phenylindole for 5 min at 1:5000 dilution and imaged on a Zeiss Axiovert 200M inverted microscope at ×20 objective (Zeiss, Oberkochen, Germany). Images were captured using AxioVision Release 4.4 software. Metamorph 7.0r4 software was used to quantify percent PR staining. The percentage of cells staining positive for PR was measured as a ratio of fluorescein isothiocyanate positive cells to total cell count using 4′,6-diamidino-2-phenylindole-stained nuclei. Data represent the average of three images taken per slide.
RNA isolation and real-time quantitative PCR analysis
Total RNA was isolated from untreated and hormone-treated HC-11 cells using RNeasy kit (no. 74104; QIAGEN, Valencia, CA), and RNA was reverse transcribed using SuperScript III (no. 18080-093; Invitrogen, Carlsbad, CA). Relative expression levels of β-casein were determined with SYBR Green PCR Master Mix (Applied Biosystems, Foster City, CA) with forward primer 5′-GGTGAATCTCATGGGACAGC-3′ and reverse primer 5′-TGACTGGATGCTGGAGTGAA-3′ using an ABI Prism 7700 Sequence Detector System (PE Applied Biosystems, Foster City, CA) according to manufacturer's instructions. Transcript levels were normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH), and data were analyzed using the Q-Gene software (BioTechniques Software Library) (56).
ChIP assay
ChIP assay was carried out as described previously with slight modification (33). Cell pellets were sonicated using a Branson-450 Sonifer with microtip in 10-sec bursts followed by 1 min of cooling on ice for 10 cycles. Sonicated samples were centrifuged at 14,000 rpm for 15 min at 4 C, and the supernatants were diluted 10-fold in ChIP buffer [140 mm NaCl, 1 mm EDTA, 1% Triton X-100, 0.1% Na-deoxycholate, 0.1% SDS, and 50 mm HEPES (ph 7.8)] containing protease inhibitors. Three percentage of the supernatant was taken for input controls and processed with immunoprecipitated samples at the point of the cross-linking reversal step. Remaining supernatants were incubated at 4 C overnight with appropriate antibodies: 15 μg of 1294 (PR), 25 μg of L-20 (Stat5), 5 μg of PA1-512 (GR), 5 μg of 06-599 (acetyl-histone H3), 5 μg of C-20 (p300), 5 μg of N-20 total RNA (pol II), 5 μg of C-19 (C/EBPβ), 5 μg of C-20 (YY1), 5 μg of 07-522 (HDAC3), 07-441 (dimethyl-Histone H3), or 6 μg of control-unrelated antibody (rabbit antimouse IgG). Immuno-isolated complexes were collected with 25 μl of Protein G Sepharose 4 Fast Flow (17-0618-01; GE Healthcare Biosciences AB, Uppsala, Sweden) for 1 h at 4 C with rotation. Beads were washed consecutively for 5 min each in ChIP buffer and high salt wash buffer (ChIP buffer + 500 mm NaCl) and LiCl wash buffer [20 mm Tris (pH 8.0), 1 mm EDTA, 250 mm LiCl, 0.5% Nonidet P-40, and 0.5% Na-deoxycholate] supplemented with protease inhibitors. Beads were then washed twice for 5 min in TE buffer [10 mm Tris, (pH 8.0) and 1 mm EDTA], and DNA complexes were eluted and formaldehyde cross-links were reversed as described (33). The DNA was PCR amplified and analyzed by electrophoresis. The relative amounts of immuno-isolated and input DNA were determined by comparison with a standard curve generated by serial dilutions (1:100, 1:20, 1:10, 1:5, and 1:1) of input DNA. PCR-amplified DNA was analyzed by electrophoresis on ethidium bromide-stained 2% agarose gels, and relative band intensities were captured using an image scanner (ChemiGenius Bioimaging System; Syngene, Frederick, MD) and system software (GeneSnap 6.08; Syngene), and band densities were quantitated using Syngene Genetools 3.07 software. DNA immunoprecipitated with specific antibody was normalized by first subtracting signals obtained with a control-unrelated IgG and then expressing the normalized value as a ratio to input DNA present on the gels.
Polymerase chain reaction
PCR for ChIP assays were carried out in a Bio-Rad DNA Engine PTC-200 (Bio-Rad Laboratories, Hercules, CA). The following primers were used: β-casein promoter forward, 5′-CCAGCTTCT-GAATTGCTGCC-3′, β-casein promoter reverse, 5′-GTCCTATCAGACTCTGTGAC-3′, β-casein enhancer forward, 5′-AGTCTCAAGGAAATACTGGATCTATTG-3′, β-casein enhancer reverse, 5′-GAGTTTGTGAACCATCTTTTACTAACC-3′, β-casein intermediate region forward, 5′-GCTATGATTCTGGATGTCCTG-3′, β-casein intermediate region reverse, 5′-GAAAGGAAG-GTAGCTAGCAAG-3′, β-casein exon VII forward, 5′-CATATGCTCAGGCTCAAACCATC-TCT-3′, and β-casein exon VII reverse, 5′-GTACTGCA-GAAGGTCTTGGACAGAC-3′. Primers for mouse GAPDH were purchased from Applied Biosystems (no. 4352339E). PCR was performed using 5 μl of DNA (input DNA was diluted 1:4) using 32 cycles of: 30 sec at 95 C, 30 sec at 55 C, and 1 min at 72 C.
Supplementary Material
Acknowledgments
This work was supported by Public Health Service Grants HD038129 (to D.P.E.), CA46938 (to D.P.E.), and CA16303 (to J.M.R.).
Disclosure Summary: D.P.E. received royalties from DAKO Corp. for antibodies. A.C.B., A.E.O., E.B.K., S.L.G., and J.M.R. have nothing to disclose.
NURSA Molecule Pages:
Nuclear Receptors: PR | GR;
Coregulators: HDAC3 | p300;
Ligands: R5020 | Hydrocortisone.
Footnotes
- AR
- Androgen receptor
- C/EBPβ
- CCAAT/enhancer-binding protein-B
- ChIP
- chromatin immunoprecipitation
- DBD
- DNA-binding domain
- GAPDH
- glyceraldehyde-3-phosphate dehydrogenase
- GR
- glucocorticoid receptor
- GRE
- glucocorticoid response element
- HC
- hydrocortisone
- HDAC3
- histone deacetylase 3
- hPR
- human PR
- Jak
- Janus kinase
- K9
- lysine 9
- mAb
- monoclonal antibody
- MOI
- multiplicity of infection
- NF-kB
- nuclear factor kB
- pfu
- plaque-forming unit
- PR
- progesterone receptor
- PRE
- progestin response element
- PRL
- prolactin
- RNA pol II
- RNA polymerase II
- SDS
- sodium dodecyl sulfate
- SRC
- receptor coactivator
- Stat
- signal transducer and activator of transcription
- YY
- Yin and Yang.
References
- 1. Doppler W, Höck W, Hofer P, Groner B, Ball RK. 1990. Prolactin and glucocorticoid hormones control transcription of the β-casein gene by kinetically distinct mechanisms. Mol Endocrinol 4:912–919 [DOI] [PubMed] [Google Scholar]
- 2. Groner B, Altiok S, Meier V. 1994. Hormonal regulation of transcription factor activity in mammary epithelial cells. Mol Cell Endocrinol 100:109–114 [DOI] [PubMed] [Google Scholar]
- 3. Rosen JM, Wyszomierski SL, Hadsell D. 1999. Regulation of milk protein gene expression. Annu Rev Nutr 19:407–436 [DOI] [PubMed] [Google Scholar]
- 4. Beuvink I, Hess D, Flotow H, Hofsteenge J, Groner B, Hynes NE. 2000. Stat5a serine phosphorylation. Serine 779 is constitutively phosphorylated in the mammary gland, and serine 725 phosphorylation influences prolactin-stimulated in vitro DNA binding activity. J Biol Chem 275:10247–10255 [DOI] [PubMed] [Google Scholar]
- 5. Pfitzner E, Jähne R, Wissler M, Stoecklin E, Groner B. 1998. p300/CREB-binding protein enhances the prolactin-mediated transcriptional induction through direct interaction with the transactivation domain of Stat5, but does not participate in the Stat5-mediated suppression of the glucocorticoid response. Mol Endocrinol 12:1582–1593 [DOI] [PubMed] [Google Scholar]
- 6. Liu X, Robinson GW, Wagner KU, Garrett L, Wynshaw-Boris A, Hennighausen L. 1997. Stat5a is mandatory for adult mammary gland development and lactogenesis. Gene Dev 11:179–186 [DOI] [PubMed] [Google Scholar]
- 7. Udy GB, Towers RP, Snell RG, Wilkins RJ, Park SH, Ram PA, Waxman DJ, Davey HW. 1997. Requirement of STAT5b for sexual dimorphism of body growth rates and liver gene expression. Proc Natl Acad Sci USA 94:7239–7244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lechner J, Welte T, Doppler W. 1997. Mechanism of interaction between the glucocorticoid receptor and Stat5: role of DNA-binding. Immunobiology 198:112–123 [DOI] [PubMed] [Google Scholar]
- 9. Lechner J, Welte T, Tomasi JK, Bruno P, Cairns C, Gustafsson J, Doppler W. 1997. Promoter-dependent synergy between glucocorticoid receptor and Stat5 in the activation of β-casein gene transcription. J Biol Chem 272:20954–20960 [DOI] [PubMed] [Google Scholar]
- 10. Stöcklin E, Wissler M, Gouilleux F, Groner B. 1996. Functional interactions between Stat5 and the glucocorticoid receptor. Nature 383:726–728 [DOI] [PubMed] [Google Scholar]
- 11. Wyszomierski SL, Yeh J, Rosen JM. 1999. Glucocorticoid receptor/signal transducer and activator of transcription 5 (STAT5) interactions enhance STAT5 activation by prolonging STAT5 DNA binding and tyrosine phosphorylation. Mol Endocrinol 13:330–343 [DOI] [PubMed] [Google Scholar]
- 12. Vafaizadeh V, Klemmt P, Brendel C, Weber K, Doebele C, Britt K, Grez M, Fehse B, Desriviéres S, Groner B. 2010. Mammary epithelial reconstitution with gene-modified stem cells assigns roles to Stat5 in luminal alveolar cell fate decisions, differentiation, involution, and mammary tumor formation. Stem Cells 28:928–938 [DOI] [PubMed] [Google Scholar]
- 13. Rijnkels M. 2002. Multispecies comparison of the casein gene loci and evolution of casein gene family. J Mammary Gland Biol 7:327–345 [DOI] [PubMed] [Google Scholar]
- 14. Winklehner-Jennewein P, Geymayer S, Lechner J, Welte T, Hansson L, Geley S, Doppler W. 1998. A distal enhancer region in the human β-casein gene mediates the response to prolactin and glucocorticoid hormones. Gene 217:127–139 [DOI] [PubMed] [Google Scholar]
- 15. Wyszomierski SL, Rosen JM. 2001. Cooperative effects of STAT5 (signal transducer and activator of transcription 5) and C/EBPβ (CCAAT/enhancer-binding protein-β) on β-casein gene transcription are mediated by the glucocorticoid receptor. Mol Endocrinol 15:228–240 [DOI] [PubMed] [Google Scholar]
- 16. Bauknecht T, See RH, Shi Y. 1996. A novel C/EBP β-YY1 complex controls the cell-type-specific activity of the human papillomavirus type 18 upstream regulatory region. J Virol 70:7695–7705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Meier VS, Groner B. 1994. The nuclear factor YY1 participates in repression of the β-casein gene promoter in mammary epithelial cells and is counteracted by mammary gland factor during lactogenic hormone induction. Mol Cell Biol 14:128–137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kabotyanski EB, Rijnkels M, Freeman-Zadrowski C, Buser AC, Edwards DP, Rosen JM. 2009. Lactogenic hormonal induction of long distance interactions between β-casein gene regulatory elements. J Biol Chem 284:22815–22824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Nakajima H, Brindle PK, Handa M, Ihle JN. 2001. Functional interaction of STAT5 and nuclear receptor co-repressor SMRT: implications in negative regulation of STAT5-dependent transcription. EMBO J 20:6836–6844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Deis RP, Delouis C. 1983. Lactogenesis induced by ovariectomy in pregnant rats and its regulation by oestrogen and progesterone. J Steroid Biochem 18:687–690 [DOI] [PubMed] [Google Scholar]
- 21. Kuhn NJ. 1969. Specificity of progesterone inhibition of lactogenesis. J Endocrinol 45:615–616 [DOI] [PubMed] [Google Scholar]
- 22. Neville MC, McFadden TB, Forsyth I. 2002. Hormonal regulation of mammary differentiation and milk secretion. J Mammary Gland Biol 7:49–66 [DOI] [PubMed] [Google Scholar]
- 23. Nguyen DA, Parlow AF, Neville MC. 2001. Hormonal regulation of tight junction closure in the mouse mammary epithelium during the transition from pregnancy to lactation. J Endocrinol 170:347–356 [DOI] [PubMed] [Google Scholar]
- 24. Rosen JM, O'Neal DL, McHugh JE, Comstock JP. 1978. Progesterone-mediated inhibition of casein mRNA and polysomal casein synthesis in the rat mammary gland during pregnancy. Biochemistry 17:290–297 [DOI] [PubMed] [Google Scholar]
- 25. Kuhn NJ. 1969. Progesterone withdrawal as the lactogenic trigger in the rat. J Endocrinol 44:39–54 [DOI] [PubMed] [Google Scholar]
- 26. Loizzi RF. 1985. Progesterone withdrawal stimulates mammary gland tubulin polymerization in pregnant rats. Endocrinology 116:2543–2547 [DOI] [PubMed] [Google Scholar]
- 27. Virgo BB, Bellward GD. 1974. Serum progesterone levels in the pregnant and postpartum laboratory mouse. Endocrinology 95:1486–1490 [DOI] [PubMed] [Google Scholar]
- 28. Ismail PM, Li J, DeMayo FJ, O'Malley BW, Lydon JP. 2002. A novel LacZ reporter mouse reveals complex regulation of the progesterone receptor promoter during mammary gland development. Mol Endocrinol 16:2475–2489 [DOI] [PubMed] [Google Scholar]
- 29. Assairi L, Delouis C, Gaye P, Houdebine LM, Bousquet MO, Denamur R. 1974. Inhibition by progesterone of the lactogenic effect of prolactin in the pseudopregnant rabbit. Biochem J 144:245–252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ganguly R, Majumder PK, Ganguly N, Banerjee MR. 1982. The mechanism of progesterone-glucocorticoid interaction in regulation of casein gene expression. J Biol Chem 257:2182–2187 [PubMed] [Google Scholar]
- 31. Terada N, Wakimoto H, Oka T. 1988. Regulation of milk protein synthesis by progesterone in cultured mouse mammary gland. J Steroid Biochem 29:99–104 [DOI] [PubMed] [Google Scholar]
- 32. Teyssot B, Houdebine LM. 1981. Role of progesterone and glucocorticoids in the transcription of the β-casein and 28-S ribosomal genes in the rabbit mammary gland. Euro J Biochem/FEBS 114:597–608 [DOI] [PubMed] [Google Scholar]
- 33. Buser AC, Gass-Handel EK, Wyszomierski SL, Doppler W, Leonhardt SA, Schaack J, Rosen JM, Watkin H, Anderson SM, Edwards DP. 2007. Progesterone receptor repression of prolactin/signal transducer and activator of transcription 5-mediated transcription of the β-casein gene in mammary epithelial cells. Mol Endocrinol 21:106–125 [DOI] [PubMed] [Google Scholar]
- 34. Kabotyanski EB, Huetter M, Xian W, Rijnkels M, Rosen JM. 2006. Integration of prolactin and glucocorticoid signaling at the β-casein promoter and enhancer by ordered recruitment of specific transcription factors and chromatin modifiers. Mol Endocrinol 20:2355–2368 [DOI] [PubMed] [Google Scholar]
- 35. Kobayashi S, Stice JP, Kazmin D, Wittmann BM, Kimbrel EA, Edwards DP, Chang CY, McDonnell DP. 2010. Mechanisms of progesterone receptor inhibition of inflammatory responses in cellular models of breast cancer. Mol Endocrinol 24:2292–2302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Press M, Spaulding B, Groshen S, Kaminsky D, Hagerty M, Sherman L, Christensen K, Edwards DP. 2002. Comparison of different antibodies for detection of progesterone receptor in breast cancer. Steroids 67:799–813 [DOI] [PubMed] [Google Scholar]
- 37. Wang Q, Carroll JS, Brown M. 2005. Spatial and temporal recruitment of androgen receptor and its coactivators involves chromosomal looping and polymerase tracking. Mol Cell 19:631–642 [DOI] [PubMed] [Google Scholar]
- 38. Kondo Y, Shen L, Yan PS, Huang TH, Issa JP. 2004. Chromatin immunoprecipitation microarrays for identification of genes silenced by histone H3 lysine 9 methylation. Proc Natl Acad Sci USA 101:7398–7403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sims RJ, 3rd, Nishioka K, Reinberg D. 2003. Histone lysine methylation: a signature for chromatin function. Trends Genet 19:629–639 [DOI] [PubMed] [Google Scholar]
- 40. Fernandez-Valdivia R, Mukherjee A, Creighton CJ, Buser AC, DeMayo FJ, Edwards DP, Lydon JP. 2008. Transcriptional response of the murine mammary gland to acute progesterone exposure. Endocrinology 149:6236–6250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Doppler W, Windegger M, Soratroi C, Tomasi J, Lechner J, Rusconi S, Cato AC, Almlöf T, Liden J, Okret S, Gustafsson JA, Richard-Foy H, Starr DB, Klocker H, Edwards D, Geymayer S. 2001. Expression level-dependent contribution of glucocorticoid receptor domains for functional interaction with STAT5. Mol Cell Biol 21:3266–3279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Doppler W, Welte T, Philipp S. 1995. CCAAT/enhancer-binding protein isoforms β and δ are expressed in mammary epithelial cells and bind to multiple sites in the β-casein gene promoter. J Biol Chem 270:17962–17969 [DOI] [PubMed] [Google Scholar]
- 43. Dekker J. 2006. The three ‘C’s of chromosome conformation capture: controls, controls, controls. Nat Methods 3:17–21 [DOI] [PubMed] [Google Scholar]
- 44. Vakoc CR, Sachdeva MM, Wang H, Blobel GA. 2006. Profile of histone lysine methylation across transcribed mammalian chromatin. Mol Cell Biol 26:9185–9195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Garcia-Bassets I, Kwon YS, Telese F, Prefontaine GG, Hutt KR, Cheng CS, Ju BG, Ohgi KA, Wang J, Escoubet-Lozach L, Rose DW, Glass CK, Fu XD, Rosenfeld MG. 2007. Histone methylation-dependent mechanisms impose ligand dependency for gene activation by nuclear receptors. Cell 128:505–518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Metzger E, Wissmann M, Yin N, Müller JM, Schneider R, Peters AH, Günther T, Buettner R, Schüle R. 2005. LSD1 demethylates repressive histone marks to promote androgen-receptor-dependent transcription. Nature 437:436–439 [DOI] [PubMed] [Google Scholar]
- 47. Wissmann M, Yin N, Müller JM, Greschik H, Fodor BD, Jenuwein T, Vogler C, Schneider R, Günther T, Buettner R, Metzger E, Schüle R. 2007. Cooperative demethylation by JMJD2C and LSD1 promotes androgen receptor-dependent gene expression. Nat Cell Biol 9:347–353 [DOI] [PubMed] [Google Scholar]
- 48. Lim S, Metzger E, Schüle R, Kirfel J, Buettner R. 2010. Epigenetic regulation of cancer growth by histone demethylases. Int J Cancer 127:1991–1998 [DOI] [PubMed] [Google Scholar]
- 49. Hardy DB, Janowski BA, Chen CC, Mendelson CR. 2008. Progesterone receptor inhibits aromatase and inflammatory response pathways in breast cancer cells via ligand-dependent and ligand-independent mechanisms. Mol Endocrinol 22:1812–1824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Luecke HF, Yamamoto KR. 2005. The glucocorticoid receptor blocks P-TEFb recruitment by NFκB to effect promoter-specific transcriptional repression. Gene Dev 19:1116–1127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Rogatsky I, Luecke HF, Leitman DC, Yamamoto KR. 2002. Alternate surfaces of transcriptional coregulator GRIP1 function in different glucocorticoid receptor activation and repression contexts. Proc Natl Acad Sci USA 99:16701–16706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Meijsing SH, Pufall MA, So AY, Bates DL, Chen L, Yamamoto KR. 2009. DNA binding site sequence directs glucocorticoid receptor structure and activity. Science 324:407–410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Li X, Wong J, Tsai SY, Tsai MJ, O'Malley BW. 2003. Progesterone and glucocorticoid receptors recruit distinct coactivator complexes and promote distinct patterns of local chromatin modification. Mol Cell Biol 23:3763–3773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Clemm DL, Sherman L, Boonyaratanakornkit V, Schrader WT, Weigel NL, Edwards DP. 2000. Differential hormone-dependent phosphorylation of progesterone receptor A and B forms revealed by a phosphoserine site-specific monoclonal antibody. Mol Endocrinol 14:52–65 [DOI] [PubMed] [Google Scholar]
- 55. Schaack J, Langer S, Guo X. 1995. Efficient selection of recombinant adenoviruses by vectors that express β-galactosidase. J Virol 69:3920–3923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Muller PY, Janovjak H, Miserez AR, Dobbie Z. 2002. Processing of gene expression data generated by quantitative real-time RT-PCR. Biotechniques 32:1372–1374, 1376:1378–1379 [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.