

Abstract
Double-stranded RNA is a common pathogen-associated molecular pattern that is recognized by cellular Toll-like receptor 3 (TLR3) and used by virus-infected cells to activate specific transcription factors and trigger induction of antiviral genes. Here, we report a new branch of TLR3-signaling that does not lead to gene induction, but affects many cellular properties, such as cell migration, adhesion and proliferation. We demonstrated that the migration of multiple cell lineages was affected by dsRNA-treatment or influenza virus infection, in a TLR3-dependent fashion. Surprisingly, for this effect of TLR3 signaling, the adaptor proteins, TRIF and MyD88, were not required. The effects of the new pathway were mediated by the proto-oncoprotein c-Src, which bound to TLR3 after dsRNA-stimulation of cells. The response was biphasic: upon dsRNA-treatment we observed an immediate increase in cell motility followed by its strong inhibition. Our results indicate that the first phase was mediated by dsRNA-induced phosphorylation and activation of Src whereas the second phase resulted from the sequestration of activated Src in lipid rafts, thus decreasing its active cytoplasmic pool. As expected, two other functions of Src, its effect on cell adhesion and cell proliferation, were also inhibited by dsRNA-treatment. These results demonstrate that activated TLR3 can engage Src to trigger multiple cellular effects and reveal a possible link between innate immune response and cell growth regulation. This study also provides a rare example of TLR-mediated cellular effects that do not require gene induction and the first example of an adaptor-independent effect of any TLR.
Introduction
The innate immune system is the first line of defense against microbial infections (1, 2); it is also used to detect danger signals from endogenous ligands generated by cells under stress or damaged tissues (3). Toll-like receptors (TLR) are the major class of proteins that initiate innate immune signaling. The members of the mammalian TLR family recognize diverse pathogen-associated patterns and trigger the intracellular inflammatory responses. While TLRs play an essential role in host defense by initiating innate immunity, inappropriate activation of TLR-mediated signaling is detrimental to the host, leading to chronic inflammatory disease and inflammation-associated cancers (4). TLR3 is a sensor of double-stranded (ds) RNA and has been studied in the context of immediate innate responses to virus infection (5). DsRNA is often produced in virus-infected cells and it triggers the synthesis of interferon. Thus, traditionally TLR3 has been viewed as the critical sensor of virus infection and the initiator of resultant innate immune response (5). Although in many cases TLR3 contributes to the host defense, it may also contribute to pathogenesis. West Nile virus causes encephalitis and surprisingly TLR3−/− mice are more resistant to lethal infection by this virus than Wt mice (6). Although in the absence of TLR3, virally induced cytokine production is impaired and peripheral viral load is higher, in the brains of TLR3−/− mice viral load, inflammation and neuropathology are reduced. The observed difference between Wt and TLR3−/− mice disappeared when the virus was administered not peripherally but intra-cerebrally. These results demonstrate that TLR3-dependent inflammatory response to West Nile virus infection is needed for efficient viral entry to the brain and consequent neuronal injury. TLR3 mediates IL-8 release in HIV myopathy and TLR3 deficiency was observed in patients with HSV encephalitis (7–9). Recent unexpected results demonstrate a much broader physiological role of TLR3 in uninfected cells, where cellular RNA is the trigger (3); it may be endocytosed after release by tissue injury or necrotic cells. A strong association was found between protective action against macular degeneration and a specific natural mutation in TLR3 (10, 11). In another study, TLR3 signaling was shown to suppress angiogenesis (12). In an experimental septic peritonitis model it was shown that TLR3 amplifies the immune response and serves as the sensor of necrosis (13). Finally, in the DSS-induced acute colitis model, subcutaneous administration of dsRNA provides protection in TLR3-dependent fashion (14). These studies demonstrate that TLR3 has an important role in many inflammatory diseases.
TLR3 is primarily located on the endosomal membrane, not plasma membrane, and endocytosis of extracellular dsRNA is required for its recognition by TLR3 (15, 16). DsRNA-binding to TLR3 triggers its dimerization, an essential step for signaling (17, 18). TLR3 uses the adaptor protein TRIF to connect to the protein kinases TAK1, IKK and TBK1/IKKε; the transcription factors IRF3, NF-κB and AP-1 are activated and consequently, transcription of hundreds of cellular genes, including the IFN genes, is induced (19). We have observed that IRF3 mediated gene induction is inhibited by a tyrosine-kinase inhibitor. This observation has led to the identification of two essential tyrosine residues in the TIR domain of TLR3 whose mutations blocked the signaling pathways (20). These residues are phosphorylated in response to dsRNA and presumably serve as the recruiting stations for other signaling proteins; both are required for full activation of the transcription factors IRF3 and NF-κB (21).
Johnsen et al. have observed that c-Src binds to activated TLR3 causing its own activation (22). Src was discovered as the oncogene encoded by avian sarcoma virus (23). Later on it became apparent that the viral Src gene (v-Src) is derived from the cellular proto-oncogene c-Src (24). c-Src is a non-receptor tyrosine kinase that regulates many important cellular functions such as migration, adhesion and differentiation (25). Its activity is modulated by phosphorylation of specific Tyr residues; phosphorylation of Tyr416 upregulates its enzyme activity whereas that of Tyr527 by the tyrosine kinase, Csk, renders the enzyme inactive (25, 26). In resting cells, almost all Src is phosphorylated at Tyr527 and hence inactive (27); but signal-induced phosphorylation of Tyr416 overrides this inhibition. Src can also be activated by its conformational change caused by disruption of the intramolecular interaction between its SH2 and SH3 domains often mediated by their binding to phosphotyrosines of other proteins, such as growth factor receptors. There are three predominant members of the Src family, Src, Yes and Fyn; they share some properties but not others.
Here, we report the existence of a new branch of TLR3 signaling that affects many properties of dsRNA-treated cells, such as cell migration, adhesion and proliferation. Surprisingly, this branch of TLR3 signaling was TRIF-independent but it required the presence of Src. Almost all cellular effects of all TLRs are entirely mediated by the products of genes whose transcription is induced by ligand-elicited signals, but the Src-mediated effects of TLR3 did not require gene induction. These observations underscore a novel facet of TLR signaling.
Materials and Methods
Reagents
Anti-Flag M2 antibody, anti-Flag M2 affinity gel, and anti-actin antibody were purchased from Sigma (St. Louis, MO, USA). Antibody against Cbp/PAG was purchased from Novus Biologicals (Littleton, CO, USA); Src and phospho-Y416-Src were from Santa Cruz Biotechnology (Santa Cruz, CA, USA) and Cell Signaling (Cambridge, MA, USA), respectively; Flotillin-1 and Focal Adhesion Kinase (FAK) were from BD Biosciences (San Jose, CA, USA); Phospho-Y576/577 FAK was from Cell Signaling (Cambridge, MA, USA); Anti-mouse P54 antibody was raised at the Hybridoma Core, the Lerner Research Institute (Cleveland, OH, USA). Polyinosinic-polycytidylic acid, (poly(I:C)) was purchased from GE Healthcare (Piscataway, NJ, USA), polyinosinic acid (pI) from Sigma, polyadenylic acid (pA) from GE Healthcare, and salmon sperm DNA were from Invitrogen (Carlsbad, CA, USA). Influenza virus A (Udorn strain) was kindly provided by Robert M. Krug (University of Texas at Austin, Texas, USA). Murine Src cDNA was a gift from George R. Stark (Cleveland Clinic, OH, USA).
Cell culture
Human embryonic kidney, HEK293 cells (ATCC, Vanassas, VA, USA), HT1080 cells (George Stark, Cleveland Clinic, OH, USA), IRF3−/− MEFs (mouse embryo fibroblasts) (T. Taniguchi, University of Tokyo, Tokyo, Japan), SYF−/− MEFs, missing genes for Src, Yes and Fyn (ATCC, Vanassas, VA, USA), Src-restored SYF−/− MEFs (SYF-Src MEFs), TLR3−/− MEFs, TRIF−/− MEFs (K. Fitzgerald, University of Massachusetts Medical School, Worcester, MA, USA), TRIF−/− MyD88−/− MEFs (S. Akira, Osaka University, Osaka, Japan), TRAF3−/− MEFs (Xiaoxia Li, Cleveland Clinic, OH, USA) and their corresponding wild type (Wt) MEFs were all maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% FBS (Atlanta Biologicals, Lawrenceville, GA, USA), 100 units/ml penicillin, 100 μg/ml streptomycin. TLR3-expressing HEK293 cells (Wt11 cells) were described elsewhere (21). Human umbilical vein endothelial cells (HUVEC, #C2519A) (Lonza, Walkersville, MD, USA) were cultured in EGM-2 BulletKit (CC-3162, Lonza, Walkersville, MD, USA). Primary TLR3−/− and TLR3+/+ MEFs were generated from TLR3−/− embryos and TLR3+/+ embryos, respectively. Human airway epithelial cell line, BEAS-2B, was grown in F-12K Nutrient Mixture supplemented with 10% FBS, 100 units/ml penicillin, 100 μg/ml streptomycin, and 10 mM HEPES. Cultures of primary mouse mesangial cells, bone marrow-derived macrophages, and primary thioglycollate-elicited peritoneal macrophage were described previously (28, 29).
Immunoblotting and immunoprecipitation
Immunoblotting was performed as described elsewhere (21). Briefly, cells were lysed in buffer A (150 mM NaCl, 20 mM HEPES pH 7.4, 1.5 mM MgCl2, 2 mM DTT, 2 mM EGTA, 10 mM NaF, 12.5 mM β-glycerophosphate, 1 mM Na3VO4 and complete EDTA-free protease inhibitor (Roche Applied Science, Indianapolis, IN, USA) with 0.5% (v/v) Triton X-100. For preparation of whole cell extract including lipid rafts, buffer A plus 1% SDS and sonication were used. For immunoprecipitation, cells were lysed in buffer A with 0.5% Triton X-100. The lysates were precleared with 10 μl mouse IgG agarose (Sigma, St. Louse, MO, USA) for 1 h. Precleared lysates were incubated overnight with 20 μl anti-Flag M2 beads (Sigma). After incubation, beads were washed and the indicated co-precipitated proteins were detected by immunoblotting.
Wound healing assay
Wound healing assay has been described elsewhere (30). Briefly, confluent cell cultures were grown on 6-well plates. Wounds were scratched with the tip of a micropipette, with or without pretreatment with poly(I:C) (100 μg/ml). After washing with warm media, cells were maintained in regular media with 10% FBS, with or without poly(I:C) for 4 h, 8 h, or 24 h. To analyze cell migration, a picture for each scratch was taken at the same area of cells at 0 h and the end of the incubation time. At least 8 fields for each condition were taken, and the numbers of migrating cells into scratched fields were calculated. The number of migrating cells without any stimulation was defined as 100 unless mentioned otherwise.
Time-Lapse Imaging of Migrating Cells
Cells were grown in a 6-well plate for overnight. After scratching cells pretreated with poly(I:C) (100 μg/ml) for 4h or untreated cells, unattached cells were rinsed off, and the remaining cells with or without poly(I:C) were placed into a temperature- andCO2-controlled incubator (at 37°C, 5% CO2) on the stage of a Leica DMIRE2 inverted microscope (Leica, Bannockburn, Ill, USA), equipped with electronically controlled shutters and filter wheel. Phase contrast images were captured under a 10x objective with a camera controller C4742_95 (Hamamatsu, Bridgewater, NJ, USA), run by Improvision Openlab software, version 3.1.5, every 5 min for 24h. The area occupied by migration cells was calculated from each image in Image-Pro Plus (Media Cybernetics, Bethesda, MD, USA) in Excel (Microsoft, Redmond, WA, USA). At least three independent experiments were carried out for each experimental condition.
Transwell migration assay
Transwell migration assays were performed as reported (30). 2.0 × 104 cells/well were seeded into the top chambers of a 24-well, 8-μm pore-size micropore polycarbonate membrane filter (Corning Incorporated, Corning, NY, USA). After 2 h incubation, poly(I:C) was added into both upper chambers and lower chamber. The lower chambers were filled with DMEM containing 10% FBS as a chemoattractant and incubated for 24h at 37°C, while the upper chambers did not contain FBS. Cells remaining on the upper surface were carefully removed with a cotton swab, and the membranes were fixed with cold methanol for 5 min, followed by staining with 1% crystal violet. Migration was quantified by counting the migrated cells in five random 20x magnification fields per filter. At least three independent experiments were carried out. The number of migrating cells without any stimulation was defined as 100 unless mentioned otherwise.
Cell proliferation assay and cell adhesion assay
For cell proliferation assay, 5 × 105 cells were plated in 6-well plate, treated with or without poly(I:C) (100 μg/ml) for 24 h, and then the cell number was counted. The doubling time was calculated in Doubling Time Online Calculator (http://www.doubling-time.com/compute.php). The average of doubling time of untreated cells was defined as 100 in each group of cells. At least 6 wells for each condition were carried out.
For cell adhesion assay, cells were plated in 96-well plates precoated with fibronectin (1 mg/ml) in the presence or the absence of poly(I:C) (100 μg/ml), after 1.5 h adherent cells were measured by colorimetric assay using crystal violet. The optical density of adherent untreated cells was set as 100. At least 12 wells for each condition were tested.
In vitro endothelial cell network formation (tube formation) assay
The assay was performed following the manufacturer’s protocol (In vitro angiogenesis assay kit, #ECM625, Millipore, Billerica, MA). Briefly, HUVECs, pretreated with poly(I:C) for 4 h or untreated, were plated in ECMatrix™-coated chamber slides (1 × 105/chamber), and incubated for 12 h. At least 8 randomly chosen fields were used for imaging. The number of branch points was counted.
Lipid raft purification
Raft fractions from TLR3-expressing HEK293 cells were prepared essentially as described elsewhere (31). Cells were lysed for 30 min on ice in buffer A, containing 0.25% Triton X-100. Lysates were mixed at a ratio of 1:1 with 80% (weight/volume) sucrose in buffer A and were transferred to ultracentrifuge tubes. The resulting lysates in 40% sucrose were overlayered with 30% and 5% sucrose in buffer A to achieve a discontinuous gradient. After ultracentrifugation for 17 h at 4°C at 200,000 g in a swinging-bucket rotor (SW41) in an Optima L-100 XP ultracentrifuge (Beckman Coulter, Brea, CA, USA), 3 ml of the lipid raft material was recovered from the interface of the 5% and 30% sucrose layers, was diluted with 9 ml of chilled buffer A and was pelleted by centrifugation for 4 h at 4°C at 150,000 g. The raft pellet was reconstituted in 100 ml of 50 mM ammonium bicarbonate pH 8.0. The Triton X-100-soluble fraction was collected in 100 ml from the last 1 ml of the 40% sucrose layer at the bottom of the ultracentrifuge tube. For determining the kinetics of Src translocation, fractions of 1.2 ml were collected from the top of the gradient after the 17 h ultracentrifugation, and subjected to immunobloting.
Statistics
Statistical analyses were performed using Prism 4 (GraphPad Software, La Jolla, CA, USA). Error bars represent SEM, and differences were analyzed using ANOVAs with Bonferroni test.
Results
TLR3 signaling regulates cell migration in a biphasic manner
In a series of experiments, we investigated the requirements for Src-binding to TLR3, its phosphorylation and the consequence on cell migration. Binding of Src to TLR3 was monitored by immunoprecipitation of one of the partner proteins and Western blotting for the other. In TLR3-expressing HEK293 (Wt11) cells, association of the two proteins could be observed only after TLR3-stimulation by dsRNA-treatment; the TLR3-associated Src was phosphorylated at Tyr416 (Fig 1A). As expected, in the parental HEK293 cells, which do not express TLR3, dsRNA-treatment did not cause Src phosphorylation (Fig 1B). Focal adhesion kinase (FAK), one of the major substrates of Src which is a tyrosine kinase, was phosphorylated at Tyr 576/577 only after TLR3 stimulation in Wt11 cells (Fig 1C). Similar results were obtained with primary mouse cells; in bone marrow derived macrophages, stimulation of endogenous TLR3 caused Src and FAK phosphorylation (Fig 1D). Cell migration is one of major cellular functions regulated by Src and FAK, and critical for mammalian development as well as inflammatory responses to tissue injury and infections. We inquired whether the rate of migration of cells was affected by dsRNA-treatment. In the wound healing assay used for measuring cell migration, confluent monolayers of cells were scratched and cells migrating into the scratched area from both edges were counted. When mouse bone-marrow derived primary macrophages were treated with the synthetic dsRNA, polyinosinic-polycytidylic acid (poly(I:C)), the rate of cell migration increased in the initial period of 4 h, but the number of cells that migrated over 24 h were fewer in the treated cells (Fig 1E), suggesting that dsRNA elicited a biphasic response in cell migration, increasing it in the beginning and decreasing it later. The biphasic response was more obvious with a modified protocol (Fig 1F). The cells were pretreated with dsRNA for 4 h followed by scratching to test only the late effect of dsRNA on cell migration (black bar); cells were directly scratched without the pretreatment to test only the early phase (gray bar). Migration of untreated cells over 4 h were set at 100 % (white bar). This protocol (no treatment or pre-treatment from −4 h) was adopted for subsequent experiments.
FIGURE 1.

TLR3 signaling regulates cell migration in a biphasic manner. A, Flag-TLR3-expressing HEK293 (Wt11) cells were treated with poly(I:C) (100 μg/ml) for the indicated times, cell lysates were immunoprecipitated with Flag antibody, and analyzed for pY416Src, Src, or TLR3 by Western Blot. B, HEK293 (293) or Wt11 cells were treated with poly(I:C) for 1 h when cell lysates were analyzed for pY416Src and actin by Western Blot. C, Wt11 cells were treated with poly(I:C) for 1 h, when cell lysates were analyzed for pY576/577FAK or FAK by Western Blot. D, Mouse bone marrow-derived macrophages (BMDMs) were treated with poly(I:C) for 1h, when the cell lysates were analyzed for pY416Src and pY576/577FAK by Western Blot. E, Mouse BMDMs were treated with poly(I:C) and their migration over 4 h or 24 h was measured by wound healing assay. For each time point, the number of cells that migrated in the untreated plates was considered as 100% and all other values were normalized to this. F, BMDMs were pre-treated with poly(I:C) for 4 h or left untreated, followed by wound healing assay with poly(I:C) treatment for another 4 h.
TLR3 signaling suppresses cell migration in multiple lineages
To test the generality of the migratory response to dsRNA, we used several cell types of different lineages. We also added an alternative assay, the transwell migration assay, to measure cell migration. In this assay, cells were plated on the upper chamber of two-chambered wells and they were treated with dsRNA, if desired. Then a serum gradient was set up between the upper (low) and the lower (high) chambers. As a result, cells migrated through holes in the upper chamber and stuck to the lower side of the upper chamber bottom. Both wound-healing assay and transwell migration assay showed that dsRNA-pretreatment inhibited both types of migrations (chemokinesis and chemotaxis) over 8 h, of mouse embryonic fibroblasts (MEFs) (Fig 2A) and peritoneal macrophages (Fig 2B). The kinetics of migration of mouse kidney mesangial cells (MMCs), important for glomerular repair following mesangial cell injury (32, 33) was measured by the wound-healing assay using time-lapse photography (Video 1 and Video 2), the areas occupied by MMCs at a given time (Fig 2C), and actual average migration speeds (Fig 2D) were calculated. As shown in Fig 2C and 2D, dsRNA treatment profoundly inhibited cell migration and the cumulative effect was more marked at later time points, significantly reduced cell migration speed. To assess the physiological significance of the newly discovered effect of dsRNA on cell migration, we performed a different assay. Migration of vascular endothelial cells is required for microtube network formation, a prerequisite for angiogenesis. Migration of human umbilical vein endothelial cells, which express TLR3, was inhibited by dsRNA-treatment; consequently, microtube formation by these cells was also inhibited (Fig 2E). When the effect on network formation was quantified by counting the number of branch points, we observed about 80% inhibition in dsRNA-treated cells.
FIGURE 2.

TLR3 signaling suppresses cell migration in multiple cell lineages. A, Mouse embryonic fibroblasts (MEFs) were pre-treated with poly(I:C) for 4 h before measuring migration over 8 h using wound healing (white bars) and transwell migration (gray bars) assays. B, Migration of mouse peritoneal macrophages were measured by wound healing (white bars) or transwell (gray bars) assays as described in (A). C, Mouse kidney mesangial cells (MMCs) were pre-treated with poly(I:C) for 4 h, when wound healing assay was performed with poly(I:C) treatment for the indicated times. The areas occupied by migrating cells from the video images were calculated. D, Migration speed of multiple cells from the video images were calculated in the absence or the presence of poly(I:C). E, HUVECs, pretreated with poly(I:C) for 4h or left untreated, were plated on ECMatrix™-coated chamber slides in the absence or the presence of poly(I:C) for 12 h. Representative fields from each condition are shown (left panel). The number of branch points in more than 8 fields in each condition was counted (right panel).
Only TLR3 ligands suppress cell migration
For triggering TLR3 signaling, exogenous dsRNA, added to the culture medium, needs to be endocytosed by cells, so that the ligand can reach TLR3 which resides primarily on the endosomal membrane. When endocytosis was blocked by treating cells with Chloroquine (10 μM), the effect on cell migration was abolished (Fig 3A), presumably because dsRNA failed to reach TLR3. Next, we asked whether signaling by other nucleic acid recognizing TLRs can also cause the effect of cell migration. Polyinosinic acid (pI), which can also activate TLR3 (34), was as effective as poly(I:C) (pIC) (Fig 3B, dark gray bar and light gray bar). But polyadenylic acid (pA), a single-stranded RNA which activates TLR7 (Fig 3B, left black bar), or salmon sperm DNA (dsDNA), a double-stranded DNA which activates TLR9 (Fig 3B, right black bar), did not have any effects on cell migration. DsRNA is often considered as a chemical signature of virus infection and many, if not all, known dsRNA-signaling pathways are activated by infection with appropriate viruses. To test whether the effect of cell migration is caused by virus-infection as well, we used influenza A virus and the human bronchial epithelial cell line, BEAS-2B. Because virus infection causes cytopathic effects at later times after infection, we chose to examine, in this case, the early effects of enhanced migration. Indeed, infection with influenza A virus enhanced cell migration (Fig 3C). Next, we asked whether RIG-I-like helicase (RLH), a cytosolic sensor for dsRNA, suppresses cell migration. Poly(I:C) transfection by FuGENE6 did not change migration of MMCs (Fig 3D). To confirm this result, we used Sendai virus (SeV), which activates RLH. As we expected, SeV did not affect cell migration, but enhanced IL-6 production into medium (Fig 3E). These results demonstrated that the effect was not only dsRNA-specific, but also TLR3-specific.
FIGURE 3.

Only TLR3 ligands suppress cell migration. A, MMCs were pretreated with poly(I:C) (50 μg/ml) in the absence or the presence of chloroquine (10 μM) for 4h, wound healing assay was performed over 16 h. B, MMCs were pretreated with poly(I:C) (pIC), polyinosinic acid (pI), polyadenylic acid (pA), or salmon sperm DNA (dsDNA), each at 100 μg/ml, wound healing assay was performed over 24 h. C, BEAS-2B (human bronchial epithelial cells) were used for wound healing assay in the absence or the presence of Influenza virus A (FluA) at MOI: 3 for 4 h. D, MMCs were treated (TLR3, 100 μg/ml) or transfected (RLH, 3 μg/ml) with poly(I:C), wound healing assay was performed over 24 h. E, MMCs were infected with SeV (MOI:10) for 4 h, and wound healing assay was performed over 24 h (white bars). IL-6 induction was measured in the culture media by ELISA after the wound healing assay (gray bars).
Cell migration effect is TLR3-dependent, but independent of IRF3, TRIF, MyD88 or TRAF3
Using MEFs from TLR3−/− mice, we confirmed that the effect of dsRNA on cell migration was mediated by TLR3 (Fig 4A). As expected, Src phosphorylation is also dependent on TLR3 (Fig 4B). All known effects of TLR3-signaling are carried out by the products of genes whose transcription is induced by dsRNA. Because a large number of these genes use IRF3 as the key transcription factor, we inquired whether IRF3 was required for the effects on cell migration; migration of IRF3−/− MEF was strongly inhibited by dsRNA (Fig 4C) indicating that IRF3 was not required for this effect. TLR3-mediated activation of all transcription factors and MAP kinases require the obligatory adaptor protein, TRIF, which is the conduit between TLR3 and the signaling complex. Surprisingly, dsRNA treatment inhibited migration of TRIF−/− cells efficiently (Fig 4D), although, as expected, gene induction did not occur in these cells, as tested by Western blot analysis of P54, a TLR3-inducible protein (Fig 4E), and microarray analysis of the induced RNAs (data not shown). TRIF deficiency also did not affect the recruitment of Src by TLR3, as tested by co-IP of Src with TLR3 in TRIF−/− MEF cells (Fig 4F). As expected, continued gene expression was not needed for dsRNA-stimulated Src phosphorylation; inhibiting new mRNA or protein synthesis did not impair the process (Fig 4G). The universal TLR adaptor protein, MyD88, was also not required; cells missing both TRIF and MyD88 were responsive to the effect of dsRNA on cell migration (Fig 4H). Similarly another signaling protein, TRAF3, which is known to be required for gene induction by TLR3 via TRIF, was not required for the effect of TLR3 on cell migration (Fig 4I). These results demonstrated that the observed effect of dsRNA on cell migration, was not mediated by new gene induction and more strikingly, although TLR3 was required for the effect, its obligatory adaptor protein, TRIF, was not required, indicating the existence of a new branch of TLR3 signaling.
FIGURE 4.

Cell migration effect is TLR3 dependent, but independent of IRF3, TRIF, MyD88 or TRAF3. A, Wt or TLR3−/− MEFs were treated with poly(I:C) for 12 h and migration during the last 8 h was measured by wound healing assay. B, Wt or TLR3−/− MEFs were treated with poly(I:C) (100 μg/ml) for 1 h, then pY416Src, Src, or actin were analyzed by Western Blot. C, Wt or IRF3−/− MEFs were treated with poly(I:C) and cell migration was analyzed as described in (A). D, TRIF−/− MEFs were incubated with poly(I:C) for 12 h and migration during the last 8 h was measured by wound healing (white bars) or transwell migration (gray bars) assays. E. Wt or TRIF−/− MEFs were treated with poly(I:C) for 8 h, induction of P54 was analyzed by Western Blot. F. Flag-TLR3 (human) expressing Wt or TRIF−/− MEFs were incubated with poly(I:C) for 1 h, cell lysates were immunoprecipitated with Flag antibody and the immunoprecipitates were analyzed for Src or TLR3 by Western Blot. G, Wt11 cells were pretreated with cycloheximide (CHX) (10 μg/ml) or actinomycin D (Act D) (1 μg/ml) for 30 min, and treated with poly(I:C) (100 μg/ml) for the next 1 h. Cell lysates were analyzed for pY416Src or actin by Western Blot. H, Wt or TRIF−/− MyD88−/− MEFs were treated with poly(I:C) and cell migration was analyzed by wound healing assay as described in (A). I, Wt or TRAF3−/− MEFs were treated with poly(I:C) and cell migration was analyzed by wound healing assay as described in (A).
Other Src-related phenotypes are also regulated by TLR3 signaling
Activation of Src causes changes in the rates of cell adhesion and proliferation; activated Src promotes them and its absence inhibits these processes. To examine the functional consequences of TLR3-mediated Src activation we measured the efficiency of adhesion of dsRNA-treated and untreated MEFs. At early time (1.5 h), the treatment enhanced adhesion by about 75% (Fig 5A), whereas 4 h of pretreatment inhibited adhesion by about 50% (Fig 5B). As expected, there was no early or late effect of dsRNA-treatment on TLR3−/− MEFs which adhered as efficiently as untreated Wt MEFs. For measuring cell proliferation, which takes a long time, we measured only the late effect. Plated cells were treated with dsRNA for 24 h, at which point the number of cells was counted. DsRNA increased the doubling time of Wt MEFs by about 40%, whereas there was no effect on the rate of replication of TLR3−/− MEFs (Fig 5C).
FIGURE 5.
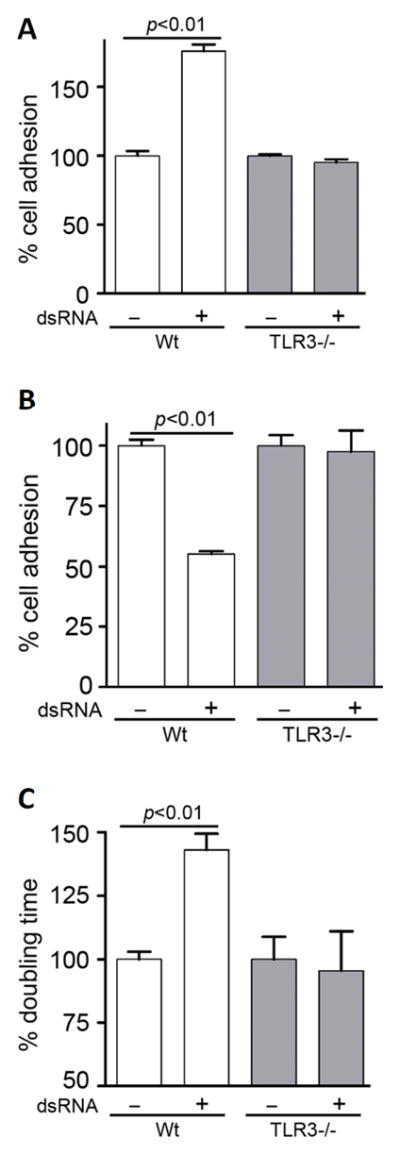
Other Src-related phenotypes are also regulated by TLR3 signaling. A, Wt or TLR3−/− MEFs were seeded in 96-well plates pre-coated with fibronectin (1 mg/ml) in the absence or the presence of poly(I:C), after 1.5 h adherent cells were measured by colorimetric assay. The optical density of adherent untreated cells was set as 100 and all other values were normalized to this. B, Wt or TLR3−/− MEFs were pre-treated with poly(I:C) for 4 h before plating them, cell adhesion was analyzed as described in (A). C, Wt or TLR3−/− MEFs (0.5 × 106/well) were seeded in 6-well plates, treated with poly(I:C) for 24 h when the number of cells were counted and the doubling time was calculated. The doubling times of untreated Wt MEFs (21.04 h) and untreated TLR3−/− MEFs (23.95 h) were defined as 100 and all other values were normalized to these.
Src mediates the effects on cell migration, cell adhesion, and cell proliferation
Src is the founding member of a family of protein tyrosine kinases; two other prominent members of this family are Yes and Fyn. When we tested the effect of dsRNA on cell migration, MEFs missing Src, Yes and Fyn (SYF−/− MEFs) did not respond at all (Fig 6A, gray bars). Moreover, even the untreated mutant cells migrated poorly as compared to untreated Wt cells. These results indicated that one or more of the three proteins, Src, Yes and Fyn, regulate cell migration as well as mediate the effect of dsRNA. To test if Src was the critical protein, using a lentiviral vector, we restored Src expression in SYF−/− MEFs (SYF-Src MEFs) (Fig 6A, black bars, and Fig 6B). Src expression imparted high motility to the cells and more importantly, restored the effects of dsRNA. Similarly, in cell adhesion assay (Fig 6C) and cell proliferation assay (Fig 6D), SYF−/− MEFs did not show any suppression effect by dsRNA, while SYF-Src MEFs displayed the suppression effects on cell adhesion and cell proliferation. These results demonstrated that Src, but not other members of the family, is involved in TLR3 signaling.
FIGURE 6.

Src mediates the effects on cell migration, cell adhesion, and cell proliferation. A, Wt, Src/Yes/Fyn triple knock-out (SYF−/−), or Src complemented SYF−/− (SYF-Src) MEFs were treated with poly(I:C) for 12 h, and cell migration during the last 8 h was measured by wound healing assay. The number of migrating untreated Wt MEFs was defined as 100 and all other values were normalized to this. B, Cell lysates from Wt, SYF−/−, SYF-Src MEFs were analyzed for Src by Western Blot. C, Wt, SYF−/−, or SYF-Src MEFs were analyzed for cell adhesion assay upon poly(I:C) treatment, as described in Fig. 5B. D, Wt, SYF−/−, or SYF-Src MEFs were used for cell proliferation assay upon poly(I:C) treatment, as described in Fig. 5C.
Src phosphorylation leads to its subcellular redistribution
Once we established the Src connection, we set out to explain the observed biphasic response of cell functions to TLR3 stimulation. Since pY416-Src is known to stimulate cell migration, the early stimulatory effect of dsRNA on cell migration could be readily explained but the later inhibitory effect was more puzzling. Closer examination of Fig 4B revealed that the level of Src was decreased after 1 h of dsRNA-treatment of Wt MEFs (middle panel). This decrease was more pronounced in MMCs (Fig 7A); although the residual Src was highly phosphorylated (upper panel), the level of total Src was greatly reduced in dsRNA-treated cells. This observation suggested that phosphorylation of Src was leading to its degradation, a conclusion that was proved to be wrong. Instead of testing 0.5% Triton X100-soluble cell extracts, as used in the experiments shown in Fig 4B and Fig 7A, when we tested total cell extracts, the level of Src did not change upon dsRNA-stimulation (Fig 7B). This observation suggested that activated Src might be translocating from the soluble fraction to unsoluble fraction such as lipid rafts. Because it has been reported in the literature that phosphorylated Src sometimes accumulates in lipid rafts and thus removed from the active pool, we tested this possibility. Indeed, when lipid rafts and cytoplasmic fractions were separated, almost all Src appeared in the raft fraction of stimulated cells (Fig 7C). We examined more closely the kinetics of activated Src translocation to lipid rafts, by sucrose gradient centrifugation analysis of cell extracts that separates lipids rafts from the soluble fractions (Fig 7D). Cells were treated with dsRNA for various lengths of time, cell extracts were fractionated and different fractions were tested for the presence of Src; FAK is served as a marker for the cytoplasm and Flotillin-1 for lipid rafts. In untreated cells, all Src was in the soluble fractions, but soon after dsRNA-treatment Src started to appear in the raft fractions (1 h, Fig 7D); by 4 h, almost all of the Src pool was concentrated in the lipid rafts. To further explore the mechanism, we tested whether Src interacted with Csk-binding protein/Phophoprotein Associated with Glycosphingolipid enriched microdomains (Cbp/PAG), which is exclusively localized to lipid rafts and important for the negative regulation of Src (35). The physical interaction of Src and Cbp/PAG was increased after dsRNA treatment for 4 h (Fig 7E). The above analyses provide the mechanistic basis for our observation that early after dsRNA-treatment cell migration increases whereas at later time, cell migration decreases. The biphasic response is regulated by Src phosphorylation and Src translocation from the cytoplasm to lipid rafts.
FIGURE 7.

Src phosphorylation leads to its subcellular redistribution. A, MMCs were treated with poly(I:C) for 1 h and the levels of pY416Src, Src, and actin in the 0.5% Triton X100-soluble fraction were analyzed by Western Blot. B, MMC lysates were prepared by solubilizing in 1% SDS containing buffer followed by sonication, the levels of Src and actin in cell lysates were analyzed by Western Blot. C, Wt11 cells were treated with poly(I:C) for 4 h, the raft fractions and the soluble fractions were separated by sucrose-gradient ultracentrifugation and analyzed for Src levels by Western Blot. Flotillin-1 was used as a marker for lipid raft fractions. D, Wt11 cells were incubated with poly(I:C) for the indicated time (right), the cell extracts were analyzed by sucrose gradient ultracentrifugation that separates lipid rafts from soluble fractions; the gradient fractions (1.2 ml each) were collected from the top to the bottom of ultracentrifuge tube, and subjected to Western Blot for Src, Flotillin-1 and FAK (a soluble fraction marker). E, MMCs were treated with poly(I:C) for 4 h, cell lysates were immunoprecipitated with Cbp/PAG and the immunoprecipitates were analyzed for Src by Western Blot.
Discussion
Our current understanding of the early events in TLR3 signaling is depicted in Fig 8. The results presented here clearly identified a new branch of TLR3-signaling. It is known that dsRNA-binding to TLR3 ectodomain occurs in the endosomal lumen and it leads to a conformational change and dimerization of TLR3 (15–18). Like conventional signaling leading to gene induction, the new branch also requires activation of TLR3 by dsRNA and the presence of Src, but from that point onwards the two branches bifurcate. Autophosphorylation of c-Src activates it and causes enhanced cell migration, cell adhesion and cell proliferation. But the stimulating effects are transient; sequestration of phospho-Src to lipid rafts quickly reduces the functional pool of Src and inhibits its cellular activities. The observed sequestration of Src may also be responsible for the known transiency of transcriptional signaling by TLR3. For all innate immune signaling the activating signals are usually transient and for TLR3, Src may be the conduit of this effect. Consequently, in the continuous presence of dsRNA, the lasting effect is inhibition of the cell functions. Our genetic analyses showed that all these effects are both TLR3 and Src-dependent. Interestingly, a recent report indicates that Src also participates in dsRNA – mediated RIG-I signaling by binding to the RING domain of TRAF3 (36). Our results show that in the TLR3 pathway the involvement of Src is not TRAF3 dependent (Fig 4I). Moreover, TRIF and the universal adaptor, MyD88, which is required for signaling by all other TLRs, were also dispensable for Src signaling (Fig 4H). Until now, all known activities of TLRs are dependent on cognate TIR-containing adaptor proteins; here, we provide the first example of a cellular effect of any TLR signaling that is adaptor-independent.
FIGURE 8.

A new branch of TLR3 signaling. The Src-dependent branch of TLR3 signaling pathway is distinct from the gene induction branch. DsRNA-stimulated dimerization of TLR3 leads to Src recruitment, its auto-phosphorylation leading to transient functional activation, followed by sequestration in the lipid rafts as an inactive pool, causing inhibition of cell migration, proliferation and adhesion.
c-Src is myristoylated and bound to cell membrane, a property that may facilitate its association with the cytoplasmic tail of another membrane-anchored protein, TLR3. Similar association of Src with the cytoplasmic domains of several growth factor receptors is well-documented (37, 38). The level of c-Src is upregulated in many human cancers (25, 39). In intestinal epithelial cells, Src regulates cell growth and differentiation and its activity is elevated in the majority of malignant and pre-malignant tumors of colon. Migration of these cells is regulated by c-Src through the phosphorylation of villin, an actin modifying protein (40). The lipid-raft anchored Csk-adaptor, Cbp/PAG, controls the oncogenicity of c-Src. Upon phosphorylation, Cbp binds to activated Src and sequesters it in lipid rafts causing functional inactivation of c-Src (35). Similar inactivation of activated Src can also be mediated by ubiquitination and degradation (41). Our results indicate that the lipid-raft sequestration strategy is used for eliminating Src that is activated by TLR3 signaling.
We have performed several experiments to evaluate the physiological significance of the Src-mediated effect of TLR3 signaling on cell migration. We used Influenza virus infection of bronchial epithelial cells, the physiological targets, to provide a link between virus infection, TLR3 and cellular migration (Fig 3C). Similarly, migration of mouse primary macrophages was affected by influenza virus infection in a TLR3-dependent fashion (data not shown). These results established that virus infection had similar effects on cell migration, as dsRNA. In viral pathogenesis, infiltrating immune cells play dual roles: they help clear the infected cells but they also produce local inflammation. The biphasic effects of TLR3 activation on cell migration may promote the former but block the latter. For example, early enhanced migration of macrophages at the site of infection may promote elimination of infected cells whereas later inhibition of migration may limit inflammation caused by accumulation of a large number of cytokine-producing cells there. Although, the primary function of TLR3 is thought to be in mounting innate immune defense against virus infection, genetic data indicate its critical importance in many physiological processes that are unrelated to virus infection. It is tempting to speculate that some of these effects may be mediated by the Src branch, and not the TRIF branch, of TLR3 signaling. Appropriate cell migration is required for network formation by vascular endothelial cells leading to micro-vessel formation and angiogenesis. Our demonstration that this process is inhibited by TLR3 signaling (Fig 2E) provides a molecular basis for the reported inhibition of angiogenesis by dsRNA in vivo (12, 42, 43). Our observation on the effect on macrophage migration by TLR3/Src signaling may be directly relevant to the reported beneficial effects of dsRNA administration on acute colitis development (14).
The demonstration of gene expression-independent cellular effects of TLR3 signaling raises the possibility that other TLRs may have similar direct cellular effects. One such effect on phagosome maturation has been reported for TLR2/TLR4 signaling (44); this effect is mediated by the activation of p38 MAP kinase in a MyD88-dependent fashion. In contrast, the TLR3 effects on cell migration were independent of TRIF, which mediates all other known effects of TLR3. Our study demonstrates that, through TLR3, virus infection or other sources of dsRNA can affect Src functions, thus revealing a new connection between innate immunity and a proto-oncogene that regulates normal and cancer cell growth.
Supplementary Material
Acknowledgments
This investigation was supported by the National Institutes of Health grants, CA062220 and CA068782.
We thank Saumendra Sarkar for participation in the early phase of this project, Neetu Gupta, Judy Drazba, Steven N. Emancipator, Hung-Yin Kao and Xiwen Cheng for scientific discussion and experimental advice, and Difernando Vanegas for technical assistance.
Footnotes
Disclosures
The authors have no financial conflicts of interest.
References
- 1.Janeway CA, Jr, Medzhitov R. Innate immune recognition. Annu Rev Immunol. 2002;20:197–216. doi: 10.1146/annurev.immunol.20.083001.084359. [DOI] [PubMed] [Google Scholar]
- 2.Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124:783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 3.Kariko K, Ni H, Capodici J, Lamphier M, Weissman D. mRNA is an endogenous ligand for Toll-like receptor 3. J Biol Chem. 2004;279:12542–12550. doi: 10.1074/jbc.M310175200. [DOI] [PubMed] [Google Scholar]
- 4.Rakoff-Nahoum S, Medzhitov R. Toll-like receptors and cancer. Nat Rev Cancer. 2009;9:57–63. doi: 10.1038/nrc2541. [DOI] [PubMed] [Google Scholar]
- 5.Alexopoulou L, Holt AC, Medzhitov R, Flavell RA. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature. 2001;413:732–738. doi: 10.1038/35099560. [DOI] [PubMed] [Google Scholar]
- 6.Wang T, Town T, Alexopoulou L, Anderson JF, Fikrig E, Flavell RA. Toll-like receptor 3 mediates West Nile virus entry into the brain causing lethal encephalitis. Nature medicine. 2004;10:1366–1373. doi: 10.1038/nm1140. [DOI] [PubMed] [Google Scholar]
- 7.Schreiner B, Voss J, Wischhusen J, Dombrowski Y, Steinle A, Lochmuller H, Dalakas M, Melms A, Wiendl H. Expression of toll-like receptors by human muscle cells in vitro and in vivo: TLR3 is highly expressed in inflammatory and HIV myopathies, mediates IL-8 release and up-regulation of NKG2D-ligands. Faseb J. 2006;20:118–120. doi: 10.1096/fj.05-4342fje. [DOI] [PubMed] [Google Scholar]
- 8.Casrouge A, Zhang SY, Eidenschenk C, Jouanguy E, Puel A, Yang K, Alcais A, Picard C, Mahfoufi N, Nicolas N, Lorenzo L, Plancoulaine S, Senechal B, Geissmann F, Tabeta K, Hoebe K, Du X, Miller RL, Heron B, Mignot C, de Villemeur TB, Lebon P, Dulac O, Rozenberg F, Beutler B, Tardieu M, Abel L, Casanova JL. Herpes simplex virus encephalitis in human UNC-93B deficiency. Science. 2006;314:308–312. doi: 10.1126/science.1128346. [DOI] [PubMed] [Google Scholar]
- 9.Zhang SY, Jouanguy E, Ugolini S, Smahi A, Elain G, Romero P, Segal D, Sancho-Shimizu V, Lorenzo L, Puel A, Picard C, Chapgier A, Plancoulaine S, Titeux M, Cognet C, von Bernuth H, Ku CL, Casrouge A, Zhang XX, Barreiro L, Leonard J, Hamilton C, Lebon P, Heron B, Vallee L, Quintana-Murci L, Hovnanian A, Rozenberg F, Vivier E, Geissmann F, Tardieu M, Abel L, Casanova JL. TLR3 deficiency in patients with herpes simplex encephalitis. Science. 2007;317:1522–1527. doi: 10.1126/science.1139522. [DOI] [PubMed] [Google Scholar]
- 10.Yang Z, Stratton C, Francis PJ, Kleinman ME, Tan PL, Gibbs D, Tong Z, Chen H, Constantine R, Yang X, Chen Y, Zeng J, Davey L, Ma X, Hau VS, Wang C, Harmon J, Buehler J, Pearson E, Patel S, Kaminoh Y, Watkins S, Luo L, Zabriskie NA, Bernstein PS, Cho W, Schwager A, Hinton DR, Klein ML, Hamon SC, Simmons E, Yu B, Campochiaro B, Sunness JS, Campochiaro P, Jorde L, Parmigiani G, Zack DJ, Katsanis N, Ambati J, Zhang K. Toll-like receptor 3 and geographic atrophy in age-related macular degeneration. N Engl J Med. 2008;359:1456–1463. doi: 10.1056/NEJMoa0802437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Edwards AO, Chen D, Fridley BL, James KM, Wu Y, Abecasis G, Swaroop A, Othman M, Branham K, Iyengar SK, Sivakumaran TA, Klein R, Klein BE, Tosakulwong N. Toll-like receptor polymorphisms and age-related macular degeneration. Invest Ophthalmol Vis Sci. 2008;49:1652–1659. doi: 10.1167/iovs.07-1378. [DOI] [PubMed] [Google Scholar]
- 12.Kleinman ME, Yamada K, Takeda A, Chandrasekaran V, Nozaki M, Baffi JZ, Albuquerque RJ, Yamasaki S, Itaya M, Pan Y, Appukuttan B, Gibbs D, Yang Z, Kariko K, Ambati BK, Wilgus TA, DiPietro LA, Sakurai E, Zhang K, Smith JR, Taylor EW, Ambati J. Sequence- and target-independent angiogenesis suppression by siRNA via TLR3. Nature. 2008;452:591–597. doi: 10.1038/nature06765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cavassani KA, Ishii M, Wen H, Schaller MA, Lincoln PM, Lukacs NW, Hogaboam CM, Kunkel SL. TLR3 is an endogenous sensor of tissue necrosis during acute inflammatory events. J Exp Med. 2008;205:2609–2621. doi: 10.1084/jem.20081370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vijay-Kumar M, Wu H, Aitken J, Kolachala VL, Neish AS, Sitaraman SV, Gewirtz AT. Activation of toll-like receptor 3 protects against DSS-induced acute colitis. Inflamm Bowel Dis. 2007;13:856–864. doi: 10.1002/ibd.20142. [DOI] [PubMed] [Google Scholar]
- 15.Uematsu S, Akira S. Toll-like receptors and Type I interferons. J Biol Chem. 2007;282:15319–15323. doi: 10.1074/jbc.R700009200. [DOI] [PubMed] [Google Scholar]
- 16.de Bouteiller O, Merck E, Hasan UA, Hubac S, Benguigui B, Trinchieri G, Bates EE, Caux C. Recognition of double-stranded RNA by human toll-like receptor 3 and downstream receptor signaling requires multimerization and an acidic pH. J Biol Chem. 2005;280:38133–38145. doi: 10.1074/jbc.M507163200. [DOI] [PubMed] [Google Scholar]
- 17.Leonard JN, Ghirlando R, Askins J, Bell JK, Margulies DH, Davies DR, Segal DM. The TLR3 signaling complex forms by cooperative receptor dimerization. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:258–263. doi: 10.1073/pnas.0710779105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu L, Botos I, Wang Y, Leonard JN, Shiloach J, Segal DM, Davies DR. Structural basis of toll-like receptor 3 signaling with double-stranded RNA. Science. 2008;320:379–381. doi: 10.1126/science.1155406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sen GC, Sarkar SN. The interferon-stimulated genes: targets of direct signaling by interferons, double-stranded RNA, and viruses. Curr Top Microbiol Immunol. 2007;316:233–250. doi: 10.1007/978-3-540-71329-6_12. [DOI] [PubMed] [Google Scholar]
- 20.Sarkar SN, Smith HL, Rowe TM, Sen GC. Double-stranded RNA signaling by Toll-like receptor 3 requires specific tyrosine residues in its cytoplasmic domain. J Biol Chem. 2003;278:4393–4396. doi: 10.1074/jbc.C200655200. [DOI] [PubMed] [Google Scholar]
- 21.Sarkar SN, Peters KL, Elco CP, Sakamoto S, Pal S, Sen GC. Novel roles of TLR3 tyrosine phosphorylation and PI3 kinase in double-stranded RNA signaling. Nat Struct Mol Biol. 2004;11:1060–1067. doi: 10.1038/nsmb847. [DOI] [PubMed] [Google Scholar]
- 22.Johnsen IB, Nguyen TT, Ringdal M, Tryggestad AM, Bakke O, Lien E, Espevik T, Anthonsen MW. Toll-like receptor 3 associates with c-Src tyrosine kinase on endosomes to initiate antiviral signaling. Embo J. 2006;25:3335–3346. doi: 10.1038/sj.emboj.7601222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bishop J, Varmus HE. Functions and origins of retroviral transforming genes. In: Weiss NTRA, Varmus HE, Coffin JM, editors. The molecular biology of tumor viruses. Cold Spring Harbor Laboratory; Cold Spring Harbor, NY: 1982. pp. 999–1108. [Google Scholar]
- 24.Takeya T, Hanafusa H. Structure and sequence of the cellular gene homologous to the RSV src gene and the mechanism for generating the transforming virus. Cell. 1983;32:881–890. doi: 10.1016/0092-8674(83)90073-9. [DOI] [PubMed] [Google Scholar]
- 25.Frame MC. Src in cancer: deregulation and consequences for cell behaviour. Biochim Biophys Acta. 2002;1602:114–130. doi: 10.1016/s0304-419x(02)00040-9. [DOI] [PubMed] [Google Scholar]
- 26.Frame MC, V, Fincham J, Carragher NO, Wyke JA. v-Src’s hold over actin and cell adhesions. Nat Rev Mol Cell Biol. 2002;3:233–245. doi: 10.1038/nrm779. [DOI] [PubMed] [Google Scholar]
- 27.Zheng XM, Resnick RJ, Shalloway D. A phosphotyrosine displacement mechanism for activation of Src by PTPalpha. Embo J. 2000;19:964–978. doi: 10.1093/emboj/19.5.964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chattopadhyay S, Marques JT, Yamashita M, Peters KL, Smith K, Desai A, Williams BR, Sen GC. Viral apoptosis is induced by IRF-3-mediated activation of Bax. Embo J. 2010;29:1762–1773. doi: 10.1038/emboj.2010.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fensterl V, White CL, Yamashita M, Sen GC. Novel characteristics of the function and induction of murine p56 family proteins. J Virol. 2008;82:11045–11053. doi: 10.1128/JVI.01593-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Asanuma K, Yanagida-Asanuma E, Faul C, Tomino Y, Kim K, Mundel P. Synaptopodin orchestrates actin organization and cell motility via regulation of RhoA signalling. Nat Cell Biol. 2006;8:485–491. doi: 10.1038/ncb1400. [DOI] [PubMed] [Google Scholar]
- 31.Gupta N, Wollscheid B, Watts JD, Scheer B, Aebersold R, DeFranco AL. Quantitative proteomic analysis of B cell lipid rafts reveals that ezrin regulates antigen receptor-mediated lipid raft dynamics. Nat Immunol. 2006;7:625–633. doi: 10.1038/ni1337. [DOI] [PubMed] [Google Scholar]
- 32.Hugo C, Shankland SJ, Bowen-Pope DF, Couser WG, Johnson RJ. Extraglomerular origin of the mesangial cell after injury. A new role of the juxtaglomerular apparatus. J Clin Invest. 1997;100:786–794. doi: 10.1172/JCI119592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Haseley LA, Hugo C, Reidy MA, Johnson RJ. Dissociation of mesangial cell migration and proliferation in experimental glomerulonephritis. Kidney Int. 1999;56:964–972. doi: 10.1046/j.1523-1755.1999.00641.x. [DOI] [PubMed] [Google Scholar]
- 34.Marshall-Clarke S, Downes JE, Haga IR, Bowie AG, Borrow P, Pennock JL, Grencis RK, Rothwell P. Polyinosinic acid is a ligand for toll-like receptor 3. J Biol Chem. 2007;282:24759–24766. doi: 10.1074/jbc.M700188200. [DOI] [PubMed] [Google Scholar]
- 35.Oneyama C, Hikita T, Enya K, Dobenecker MW, Saito K, Nada S, Tarakhovsky A, Okada M. The lipid raft-anchored adaptor protein Cbp controls the oncogenic potential of c-Src. Mol Cell. 2008;30:426–436. doi: 10.1016/j.molcel.2008.03.026. [DOI] [PubMed] [Google Scholar]
- 36.Johnsen IB, Nguyen TT, Bergstroem B, Fitzgerald KA, Anthonsen MW. The tyrosine kinase c-Src enhances RIG-I (retinoic acid-inducible gene I)-elicited antiviral signaling. J Biol Chem. 2009;284:19122–19131. doi: 10.1074/jbc.M808233200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Veracini L, Franco M, Boureux A, Simon V, Roche S, Benistant C. Two distinct pools of Src family tyrosine kinases regulate PDGF-induced DNA synthesis and actin dorsal ruffles. J Cell Sci. 2006;119:2921–2934. doi: 10.1242/jcs.03015. [DOI] [PubMed] [Google Scholar]
- 38.Egloff AM, Grandis JR. Targeting epidermal growth factor receptor and SRC pathways in head and neck cancer. Semin Oncol. 2008;35:286–297. doi: 10.1053/j.seminoncol.2008.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Summy JM, Gallick GE. Src family kinases in tumor progression and metastasis. Cancer Metastasis Rev. 2003;22:337–358. doi: 10.1023/a:1023772912750. [DOI] [PubMed] [Google Scholar]
- 40.Mathew S, George SP, Wang Y, Siddiqui MR, Srinivasan K, Tan L, Khurana S. Potential molecular mechanism for c-Src kinase-mediated regulation of intestinal cell migration. J Biol Chem. 2008;283:22709–22722. doi: 10.1074/jbc.M801319200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Harris KF, Shoji I, Cooper EM, Kumar S, Oda H, Howley PM. Ubiquitin-mediated degradation of active Src tyrosine kinase. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:13738–13743. doi: 10.1073/pnas.96.24.13738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Berge M, Bonnin P, Sulpice E, Vilar J, Allanic D, Silvestre JS, Levy BI, Tucker GC, Tobelem G, Merkulova-Rainon T. Small interfering RNAs induce target-independent inhibition of tumor growth and vasculature remodeling in a mouse model of hepatocellular carcinoma. The American journal of pathology. 2010;177:3192–3201. doi: 10.2353/ajpath.2010.100157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cho WG, Albuquerque RJ, Kleinman ME, Tarallo V, Greco A, Nozaki M, Green MG, Baffi JZ, Ambati BK, De Falco M, Alexander JS, Brunetti A, De Falco S, Ambati J. Small interfering RNA-induced TLR3 activation inhibits blood and lymphatic vessel growth. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:7137–7142. doi: 10.1073/pnas.0812317106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Blander JM, Medzhitov R. Regulation of phagosome maturation by signals from toll-like receptors. Science. 2004;304:1014–1018. doi: 10.1126/science.1096158. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.