

Abstract
The colorimetric properties of resorcinarene solutions had not been investigated since Baeyer’s initial synthesis. We recently reported that solutions containing resorcinarene macrocycles develop color upon heating or standing. In the presence of saccharides, these solutions exhibit significant color changes which are easily seen. We herein present strong evidence that the solution color is due to macrocycle ring opening and oxidation. The optical responses to saccharides are due to complexation of the sugar with the acyclic chromophores. We apply these mechanistic insights toward the challenging problem of the visual detection of neutral oligosaccharides by simple chromogens. In addition, we also report the first single-crystal X-ray crystal structure determination of a rarely observed “diamond” resorcinarene stereoisomer.
Introduction
In 1872, a year after his landmark synthesis of the xanthene dye fluoresceine,1 Baeyer studied the condensation reaction of benzaldehyde and resorcinol in the presence of acid.2 The red solution containing his product mixture changed to violet upon the addition of base. It is now well known that Baeyer’s reaction afforded the first resorcinarenes, cyclic tetramers of resorcinol.3
The broad impact of the resorcinarenes in the fields of molecular recognition, materials science, and supramolecular chemistry has been the subject of extensive study as noted in several recent reviews.3 For instance, they were the first compounds shown to bind sugars in apolar media.4 Because boronic acids are the basis of carbohydrate affinity chromatography, we reasoned that resorcinarenes functionalized with boronic acids would constitute powerful sugar receptors. We thus synthesized 1 and 2a and investigated their properties in the presence of sugars.5
We observed 11 different solution colors of 11 different heated sugar solutions containing 1.6 The analytes included structurally related neutral carbohydrates, glucose phosphates, carboxylic acid, and amino sugars. The solution color changes were rapid, quantifiable, and reproducible. We also showed that solutions containing acetaldehyde-derived 2b exhibited relatively paler color changes in the presence of sugars as compared to those containing boronic acids 1 and 2a or acyclic congener 3a (Figure 1).7 The colorimetric properties of resorcinarene solutions had not received prior attention since Baeyer’s initial report.2
Figure 1.
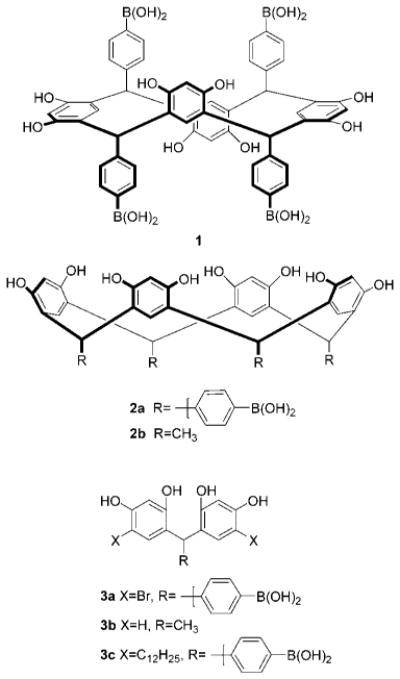
We previously reported that solutions containing resorcinarenes 1, 2a, 2b, and related condensation product 3a exhibit significant color changes in the presence of sugars.
Sugars are a relatively challenging class of compounds to analyze. They exhibit great structural similarity as well as transparency in the visible region. The sensing of specific saccharides could aid the monitoring of disease or industrial fermentation products. The visual determination of saccharides has been of interest for well over a century.
Resorcinol and its derivatives have long been used in simple color tests for sugars. In 1887, Seliwanoff reported a resorcinol color test which was followed by other resorcinol-derived methods.8 These latter and numerous other related reducing sugar assays, while based on simple reagents, typically require toxic materials, harsh and often tedious procedures.9 In the 1990s, significant progress was made toward the improved selective and mild detection of monosaccharides via relatively strong solution color changes observable by visual inspection. The recent advances were due mainly to the pioneering efforts of Shinkai and co-workers. Their studies were based primarily on aniline-functionalized azo dyes containing appended arylboronic acids.10
The field of glycobiology has recently undergone great resurgence due to the exciting therapeutic potential of oligosaccharides.11 A vast number of oligosaccharides are found in glycoproteins and on cell surfaces. Most of the recent progress toward the color detection of sugars has, however, involved monosaccharide analysis.10,12 The great variety of linear and branched oligosaccharides magnifies the problems associated with monosaccharide analysis.
New methods allowing for simple and rapid oligosaccharide detection are in demand. The colorimetric detection of oligosaccharides eluting from chromatographic columns is one of the biggest challenges in sugar analysis.13 The older color tests used for automated HPLC postcolumn detection of monosaccharides fail to effectively directly detect oligosaccharides containing more than three residues. For example, in a representative assay, maltohexaose’s response is only 18% of that observed for the same weight of glucose.13 Colorimetric methods thus typically require prior complete hydrolysis to monosaccharides or covalent attachment of the oligosaccharides to a chromophore, which can lead to diminished separation.13,14
Herein we present the first evidence that xanthenes form and serve as the active chromophores in resorcinarene solutions. We also describe our investigations of the saccharide-induced optical signal transduction mechanism. We extend the scope and utility of our sensing methodology to include the color detection of a homologous series of prototypical neutral oligosaccharides, the linear maltodextrins, containing up to seven glucose residues. We thus address the fundamental problem of diminished optical detection of larger oligosaccharides using simple colorimetric agents.
Results
Formation and Structure of the Chromophore in Resorcinarene Solutions
We reported the synthesis of compounds 1 and 2a in one step in a combined 90% yield via the HCl-promoted condensation of commercially available 4-formylphenyl boronic acid and resorcinol.5 Separation of the two stereoisomers by fractional crystallization afforded white crystalline solids. We obtained X-ray quality crystals of the half-methyl tetraboronate ester of 1 upon slow crystallization from a 9:1 MeOH:EtOH solution. The solid-state architecture was characterized as an infinite, antiparallel two-dimensional network of macrocycles, each of which exhibited 12 intermolecular hydrogen bonds.15
We found that colorless DMSO solutions of freshly crystallized 1 or 2a (5.2 mM), upon standing in solution for several hours or upon heating at 90 °C for 1 min, developed a pinkish-purple color. The color formation was monitored via UV–vis spectroscopy. The appearance of a new λmax at 535 nm was accompanied by a less intense absorbance at 500 nm.6
Initial attempts at understanding the origin of the solution color involved heating solutions of 1 in the dark or in O2 degassed conditions. In both cases, we found that the color intensities diminished, as evidence by both visual inspection and UV–vis spectroscopy.6 For instance, heating a solution of 1 (5.2 mM in DMSO) under O2 degassed conditions led to a 61% decrease in absorbance at 536 nm. Light and O2 apparently promote color formation. In addition, when we acylated the phenolic hydroxyls of 1 and heated a DMSO solution of the resultant octaacetate to reflux, the solution remained colorless.7 The phenolic hydroxyls thus also play a key role in chromophore formation. We reasoned that the chromophore arises via oxidation of a resorcinol moiety to a quinone.6,7
We also heated solutions of resorcinol or benzeneboronic acid separately or as an equimolar mixture using the aforementioned conditions and concentrations, with and without added monosaccharides. We observed only very faint solution colors by visual inspection.6 This result showed that a methine-bridged resorcinol/aldehyde condensation framework was needed for effective chromophore formation and optical sugar detection. Interestingly, methine-bridged condensation product resorcinarene substructures, of which 3a–c are examples, were noted as reaction intermediates in standard xanthene dye syntheses (e.g., the transformation of 4 to 5, n = m = 0, Scheme 1).16
Scheme 1.
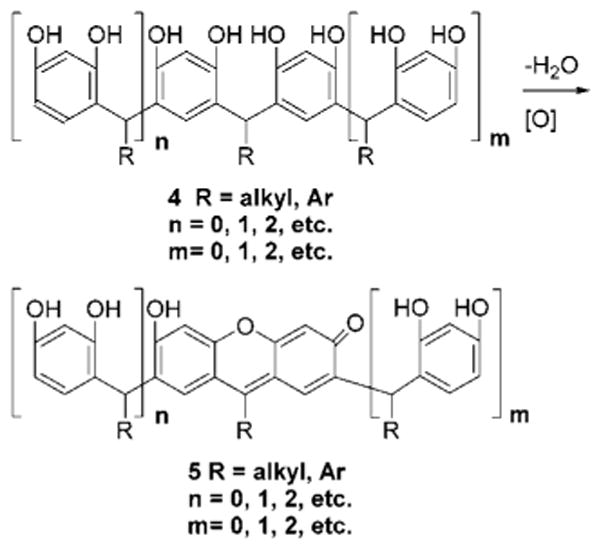
Dehydration and Oxidation of Methine-Bridged Resorcinol Oligomers Leading to a Xanthene
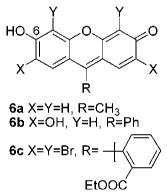
Xanthenes are some of the oldest known synthetic dyes such as fluorescein, rhodamine, 6a and 6b and ethyl eosin (6c), as well as many others. Importantly, the colorimetric properties of xanthenes are a function of the ionization state of the C-6 moiety.17 They typically exhibit two absorbance maxima in the visible region.17 The absorption spectrum of 6b (5.0 × 10−6 M) in 9:1 DMSO:H2O is shown in Figure 2. It exhibits a λmax at 530 nm and a less intense λmax at 500 nm. Interestingly, the λmax absorbance values and spectral features are strikingly similar to those observed for colored DMSO solutions of 1 as well as 2a, 2b, and 3a which we previously reported.7
Figure 2.
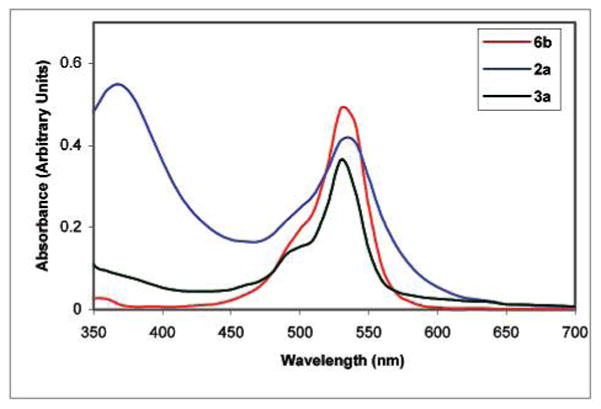
2a (1.0 mg) and 3a (1.0 mg) each in 0.9 mL of DMSO were heated to gentle reflux over 2 min and cooled to room temperature before 0.1 mL of H2O was added to each solution. The final concentrations of 2a and 3a in 9:1 DMSO:H2O are 1.03 × 10−3 and 1.96 × 10−3 M, respectively. A solution of 6b (5.0 × 10−6 M) was prepared at room temperature in 9:1 DMSO:H2O.
Incorporation of a planar xanthene within a resorcinarene macrocycle framework via the transformation shown in Scheme 1 would lead to a considerable increase in strain energy. Simulations (Sybil 6.6) show that an increase in strain energy of 34.2 kcal/mol would occur upon formation of a xanthene substructure within 2b. Prior studies of the related calixarenes (macrocycles formally derived from phenol/formaldehyde condensations) showed that xanthenes did not form in cyclic tetrameric structures.18
Ring opening to acyclic oligomers could thus be a prerequisite for xanthene formation from resorcinarenes. It is known that the condensation reactions producing resorcinarenes are reversible under acidic conditions.3 The detailed mechanism of resorcinarene macrocycle genesis has been studied thoroughly by Weinelt and Schneider.19 They found that 2b and its macrocyclic stereoisomers interconverted via the intermediacy of acyclic oligomers. Their studies included the rapid quenching of condensation reactions between resorcinol and either acetaldehyde or paraldehyde in MeOH in the presence of anhydrous HCl. Because the opening of a resorcinarene ring has only been previously shown to occur upon the addition of strong acid, our hypothesis of acyclic oligomer formation in aqueous or neat DMSO solutions without added acid warrants further analysis.
We had observed that 1H and 13C NMR spectra of DMSO-d6 solutions of 1 (5.2 mM), heated at 90 °C for 3 min (our initial sugar colorimetric detection conditions), exhibited no readily observable change in chemical shifts or peak area integrals as compared to fresh, colorless samples.6 Xanthenes are strongly absorbing materials which need be only produced in trace (ca. 0.5% conversion; see, for example, the concentrations and absorbances shown in Figure 2) amounts to afford solution colors under our conditions.
Evidence for Acid Formation in DMSO Solutions
We find that more vigorous thermolytic conditions are necessary to afford conversion to significant amounts of products. Heating a DMSO (10 mL) solution of freshly recrystallized 2b (100 mg, 18.4 mM) for 8 h at 120 °C followed by analysis via reversed phase HPLC reveals the formation of numerous new products representing a 74% conversion of 2b to products based on relative peak areas.
Strong literature precedent allows us to propose that acyclic oligomers arise from 2b via the in situ formation of strong acids. It is known that acid production from DMSO is promoted by the presence of O2 and peroxides.20 In addition, in situ acid formation has been attributed as the cause of certain oxidations in DMSO.20b We have noted the effect of O2 on resorcinarene solution color intensity (vide supra).
Acid formation observed during DMSO decomposition has been inhibited by free radical scavengers.20c Under the same thermolysis conditions as noted above, but in the presence of free radical scavengers (either BHT or phenothiazene, 10 mol %), we observe less than 28% conversion to products by HPLC analysis.
Important further evidence that strong acids form under our conditions was presented in our recent report describing the first X-ray crystal structure of (CH3)3S+CH3SO3−.21 The latter compound was obtained from a thermolysis reaction of 2b in DMSO. It is known that (CH3)3S+CH3SO3− forms, along with CH3SO3H, CH3SO2H, and CH3SOH (and several other products), via the radical- and acid-promoted decomposition of DMSO.22
Macrocycle Bond Breaking and Oxidation of the Acyclic Products
We next turned our attention to identifying the products of macrocycle ring opening. We isolate 7 (Figure 3), a rarely observed “diamond” resorcinarene stereoisomer,23 in 2.3% yield from the thermolysis of 2b in DMSO, via flash column chromatography. The structure of 7 was previously assigned (as the octabutyrate derivative) via NMR evidence during the acid-catalyzed condensation/isomerization studies of Schneider and Weinelt.19
Figure 3.
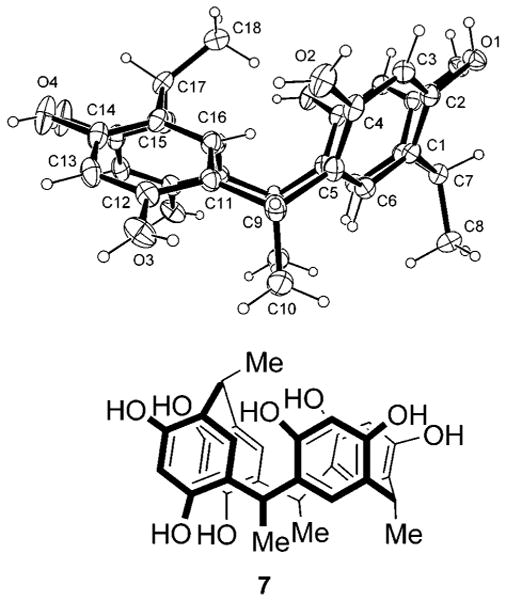
Compound 7 and ORTEP.
Single crystals suitable for X-ray analysis, grown via slow evaporation of a 9:1 CH2Cl2:MeOH solution of 7, allow us to report the first known crystal structure of a resorcinarene diamond isomer. The macrocyclic molecule lies on a mirror plane in the crystal, with three methyl groups (C10, C8, and its mirror-related equivalent) relatively syn. The fourth methyl group, C18, lies on the mirror, and is anti to the other three. The resulting conformation is such that OH groups O1 and O4 both form intramolecular hydrogen-bonding contacts with their mirror-related equivalents on adjacent aromatic rings. The H atoms in these contacts are disordered, with the two O atoms alternately donor and acceptor. The O···O distances are 2.712-(2) Å for O1 and 2.671(2) Å for O4. There are two independent methanol molecules, both lying on mirrors and forming hydrogen bonds with external macrocyclic OH groups O2 and O3. Importantly, stereoisomer 7 can only form from 2b via bond rupture and reformation.19 If 7 were a conformer of 2b, the methyl group (C18) would reside outside, rather than above, the plane of the macrocycle cavity.
We also observe acyclic products during the thermolysis of 2b. A key product, 3b, is found in a broad HPLC fraction eluting from 18 to 20 min. The 1H NMR spectrum of the isolate exhibits several peaks including each of the resonances associated with 3b19 (CH3OD δ 1.46, d, 3J = 7.3 Hz, 4.53, q, 3J = 7.3 Hz, 6.18–6.22, m, 6.89, d, 3J = 8.0 Hz). Overlay of the 1H NMR spectrum of the HPLC isolate with a sample of independently synthesized and isolated 3b confirms the assignment (Figure 4). In addition, the MALDI MS of the HPLC fraction contains a peak at 245.59 amu (246.26 amu calcd). The production of compounds 3b and 7 under our conditions constitutes an important initial link between our investigations and the prior acid-catalyzed macrocycle genesis mechanism studies.19
Figure 4.
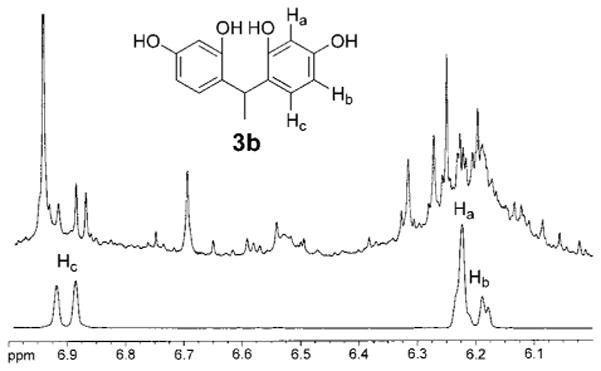
Top: Expansion of a 1H NMR spectrum of semipurified thermolysis reaction products of 2b showing the formation of 3b. Bottom: Expansion of a 1H NMR spectrum of pure 3b.
We also find evidence for higher order oligomer production under our conditions (vide supra) involving thermolysis of 2b in DMSO. At least five sets of doublets appear between 0.72 and 1.53 ppm in the 1H NMR of each of two flash column fractions (TLC Rf = 0.54 and 0.63, 9:1 CH2Cl2:CH3OH, δ 1.53, 1.08, 1.01, 0.97, 0.83, 0.72 ppm, and δ 1.29, 1.15, 1.00, 0.89, 0.84 ppm, CH3OD, respectively). In addition, the MALDI mass spectra (anthracene matrix) of other fractions (Rf = 0.29 and 0.44) exhibits peaks for higher homologues of 3b (entries 1 and 2, Table 1). MALDI MS evidence also suggests the formation of xanthene materials not previously reported in previous fragmentation and equilibration studies of 2b (entries 3–6, Table 1).24,25
Table 1.
MALDI MS Evidence for the Formation of Acyclic Oxidized and Unoxidized Products from the Thermolysis of 2b
| entry | structure | TLC Rf | (m/z) calcd | (m/z) obsd |
|---|---|---|---|---|
| 1 | 4, R = Me, m = 1, n = 0 | 0.29 | 382.41 | 381.89 |
| 2 | 4, R = Me, m = 3, n = 2 | 0.44 | 926.36 | 926.28 |
| 3 | 6a | 0.44 | 226.23 | 225.61 |
| 4 | 5, R = Me, m + n = 4 | 0.26 | 906.01 | 906.33 |
| 5 | 5, R = Me, m + n = 3 | 0.84 | 770.79 | 770.82 |
| 6 | 5, R = Me, n = 1, m = 0 | 0.79 | 362.51 | 361.38 |
To study the formation of the oxidation products, we heat 3b, the parent acyclic unoxidized homologue attained via ring opening of 2b. Heating an air-saturated solution of 3b (0.880 g, 3.576 mmol) dissolved in DMSO (78 mL) at 100 °C for 28 h leads to the formation of several products. The complex 1H NMR of the crude mixture reveals the presence of resorcinol as the predominant (90%) product as well as minor conversion to 2,4-dihydroxyacetophenone 8 (ratio of integrals of resorcinol triplet 6.94 ppm to 8 doublet at 7.76 ppm is 153:1, CH3OD) and very small traces of xanthene 6a (d, 7.65 ppm).
The production of resorcinol and 8 is consistent with the reversible opening and fragmentation of the resorcinarenes in acidic media.19 This result also complements our recent report describing the production of 4-formylphenylboronic acid from 3c.26 Furthermore, in acidic media, the addition of water at the methine carbon of 4 (R = Ar, n = 0, m = 0) followed by elimination has been described as an intermediate step in the synthesis of xanthenes.27
We attain better conversion to xanthene 6a from 3b by limiting thermolysis time to 2 h. The 1H NMR spectrum (DMSO-d6) of the crude reaction mixture clearly shows a doublet at 7.65 ppm characteristic of 6a with improved S/N as compared to the 28 h experiment (vide supra). Resonances centered at 5.26, 6.49, and 6.60 ppm are also discernible, overlaying with the 1H NMR of an analytical sample27 of 6a. Because oxidation to xanthenes can be promoted by peroxides and acid,27,28 we should attain better conversion to xanthenes upon addition of these latter reagents. Indeed, we find that heating a solution of 3b (50 mg, 0.203 mmol), H2SO4 (0.15 mL), and K2S2O8 (1.0 mg) in 1.5 mL of MeOH at reflux for 2 h produces the most significant conversion (4% yield) of 3b to 6a we have observed to date.
We have shown that xanthenes form in solutions containing resorcinarene macrocycles (Scheme 1). The O2-induced radical decomposition of DMSO leads to in situ strong acid formation. The acid catalyzes a reverse condensation reaction to afford acyclic oligomers. The acyclic oligomers undergo oxidation also via the action of acid and peroxide.
Solution Color Changes Promoted by Mono- and Oligosaccharides in the Presence of Resorcinol Condensation Products
The linear maltodextrins 9a–f are a prototypical class of readily available neutral oligosaccharides composed of glucose residues with α-1,4-glycoside linkages. We heat solutions (9:1 DMSO:H2O, 1 mL) containing specific maltodextrins 9a–f (2 mg) in the presence of 2a (5 mg, 5.2 mM) until a temperature of 120 °C is reached in 3.0 min. In these studies, the resorcinarene is initially preheated alone for 15 min in 9:1 DMSO:H2O at reflux prior to sugar addition to maximize chromophore formation.
Upon cooling the respective solutions containing mixtures of 2a and each of 9a–f, we observe solution colors of deep orange-yellow, yellow-gold, pale orange, orange, pale pink, and peach for maltose 9a through maltoheptaose 9f, respectively (Figure 5). The UV–vis spectra of each of the sugar-containing solutions exhibit increased absorbances at 460 nm (and decreased absorbances at 530 nm) as compared to a solution of 2a heated without sugar (Figure 6). Heated solutions containing 1 and 9a–f also exhibit absorbance maxima at 460 nm that are greater than that of 1 heated alone. The boronic acid-containing resorcinol condensation products thus allow for potentially useful colorimetric detection of larger oligosaccharides at 460 nm.
Figure 5.
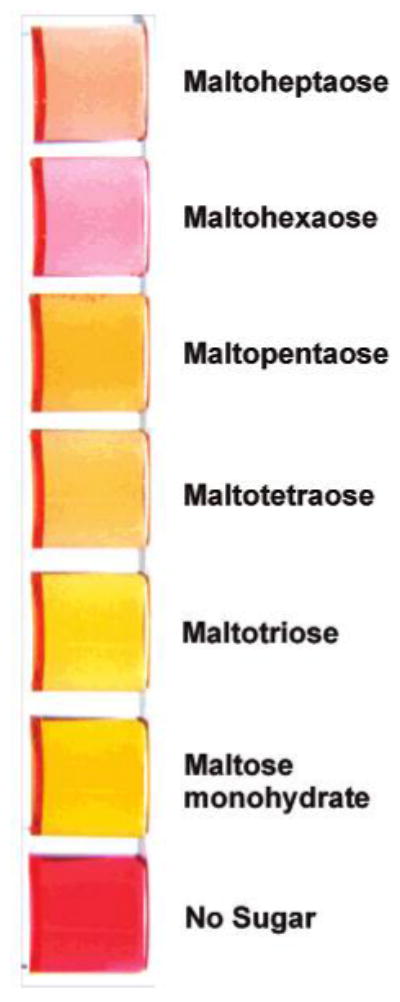
Colored solutions of 2a after heating alone or with maltodextrins 9a–f.
Figure 6.
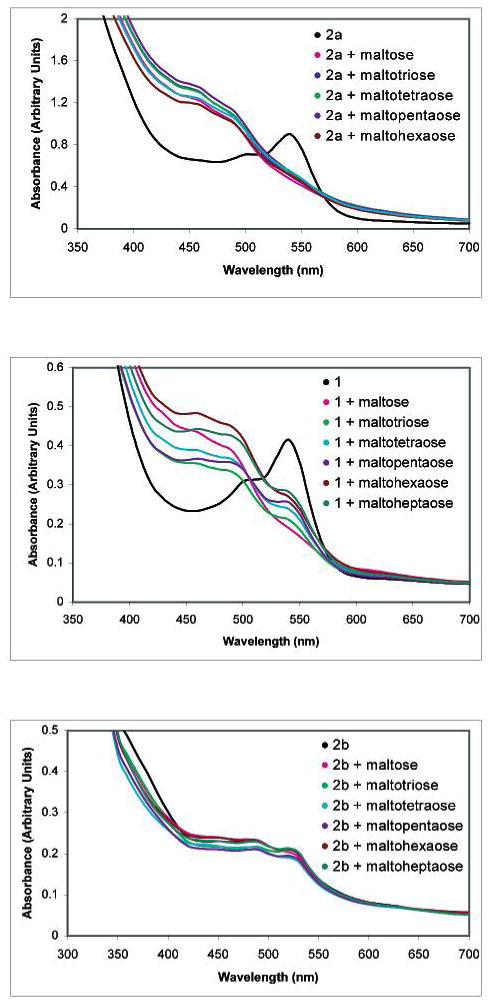
UV–vis spectra of solutions of 2a, 1, and 2b heated alone or in the presence of 9a–f. The boronic acid-containing receptors 1 and 2a exhibit larger absorbance responses to oligosaccharides.
In analogous experiments to those above, but employing 2b instead of 1 or 2a, we find the absorbance changes in the oligosaccharide-containing solutions are greatly diminished as compared to the solution containing 2b alone. The presence of boronic acid moieties is apparently a key to attaining enhanced absorbance values with larger oligosaccharides. This result is in accord with our prior studies involving the comparison of monosaccharide-induced solution color changes in solutions containing 1, 2b, and 3a.7
Sugar Solution Color Changes in the Presence of Resorcinol Condensation Products not Possessing Boronic Acids
To understand the different optical responses to sugars observed with boronic acid- and non-boronic acid-containing compounds, we first examine solutions containing 2b and reducing and nonreducing sugars. We find that nonreducing sugars do not promote significant optical changes when heated in the presence of 2b. For example, when we heat a solution of 2b (5.2 mM) in 9:1 DMSO:H2O (1 mL) in the presence of nonreducing sugars, either methyl-α-glucopyranoside (10, 15.6 mM) or α-methyl-mannopyranoside (11, 15.6 mM), for 2.5 min to gentle reflux, each solution color and absorption spectrum is unchanged from the heated solution of 2b without sugar.
Reducing sugars undergo fragmentation to chain-shortened carbohydrates including aldonic acids, in the presence of peroxide.29 The 1H NMR spectra of respective DMSO-d6 solutions containing three reducing sugars (28 mM), maltose (9a), D-(−)-fructose, and D-(+)-glucose, heated until gentle reflux (2.5 min), exhibit at least five new aldehyde resonances between 9 and 10 ppm as well as a broad proton resonance in the carboxylic acid region at 11.7 ppm.30 None of these observed resonances is present in the NMR spectrum of heated neat DMSO-d6. The region between 9 and 15 ppm in the spectra of the two nonreducing sugars 10 and 11 exhibits only very weak resonances which are also observed in the spectrum of heated neat DMSO-d6. We thus hypothesize that the color changes observed in 2b solutions are due to the new products formed upon reducing sugar thermolysis. When unheated solutions of either maltose (9a), D-fructose, or D-glucose are added to a colored solution of 2b at room temperature, no color change is observed.
If reducing sugar reaction products are a cause of the solution color changes in the presence of the resorcinol condensation products, then the addition of preheated reducing sugars to preheated colored solutions of 2b should produce a color change. We heat DMSO (0.9 mL) solutions of maltose (9a), D-fructose, and D-glucose each to a gentle reflux, cool to room temperature, and add 0.1 mL of H2O. We add each of the solutions to three preheated 9:1 DMSO:H2O solutions of 2b (5.2 mM). The three sugar (15.6 mM) solutions exhibit color changes observable by visual inspection and exhibit UV–vis spectra that are different from that of a heated solution containing only 2b.31
We propose therefore that acyclic, xanthene-containing oligomers, which we have demonstrated to be the active chromophores in resorcinarene solutions, function as colorimetric receptors for sugar-derived acids. To prove this hypothesis, we chose to study the interaction of 6b with N-acetylneuraminic acid (the most naturally abundant sialic acid, 12), an important cell surface residue, in buffered media. Compound 6b is a xanthene dye with hydroxyls adjacent to the C-6-OH and carbonyl moieties. The structure of 6b is analogous to oxidized acyclic oligomers derived from resorcinarenes, in that it contains proximal potential hydrogen-bonding sites for charged carboxylates (Figure 7).
Figure 7.
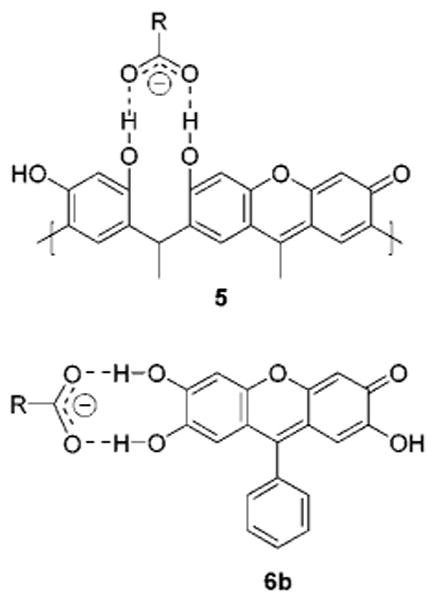
Charged hydrogen bonding between carbohydrates and proximal hydroxyls of 5 and 6b.
Addition of varying amounts of sialic acid 12 (1.9 × 10−4–1.5 × 10−3 M) to a peach-colored solution of compound 6b (1.0 × 10−5 M) in a 9:1 DMSO:H2O solution (HEPES buffer, 5.0 × 10−3 M final concentration, adjusted to pH = 7.5 in H2O before mixing with DMSO) at room temperature results in a solution color change to yellow. The color changes monitored by UV–vis spectroscopy are concentration dependent (Figure 8). We calculate an apparent equilibrium constant, Kexp(app) = 271.3 M−1, with a reproducibility of ±6% (triplicate runs) for the binding of 6b to 12. We observe a color change to pink under the same conditions when NaOAc (2.2 × 10−4 M) instead of 12 is added to a solution of 6b. This latter result suggests that the carboxylate moiety of the sugar is responsible for the change in solution color.32
Figure 8.
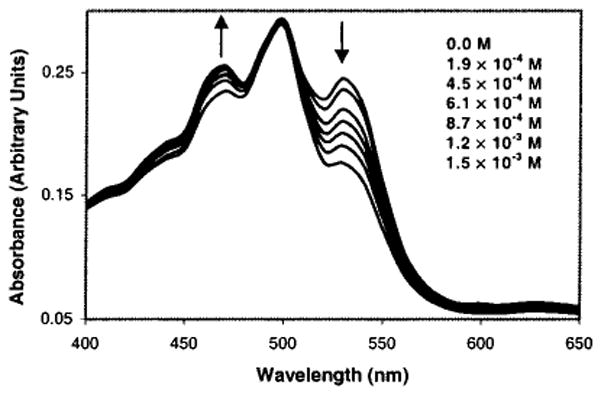
UV–vis spectra of 6b (1.0 × 10−5 M) in the absence and presence of 12 (1.9 × 10−4–1.5 × 10−3 M) in 9:1 DMSO:H2O (pH 7.5 HEPES buffer). Inset shows the concentration of 12.
IR absorption shifts serve as evidence supporting a hydrogen-bonding interaction between the carboxylate moiety of 12 and the hydroxyls of 6b. The FT-IR spectrum of a 1.0 × 10−2 M solution of 12 in DMSO exhibits a carbonyl absorption at 1726 cm−1. Upon addition of an equimolar amount of 6b, a new absorption appears at 1661 cm−1 accompanied by significant weakening of the absorption at 1726 cm−1. This is in keeping with hydrogen bonding of 6b to the sialic acid carbonyl oxygen.33 Further support for hydrogen bonding is the fact that solution color changes upon sialic acid addition to 6b are no longer observed when the proportion of H2O to DMSO is ≥50%.34
Aldonic acids, formed from reducing sugars in situ upon heating in DMSO, thus promote solution color changes via a charged hydrogen-bonding interaction with the xanthene chromophore. This latter interaction perturbs the ionization state of the C-6 hydroxyl of the dye. In contrast, heating reducing sugars in the presence of boronic acids should disfavor aldonic acid formation since anomeric hydroxyls are typically those involved in boronate ester formation. We therefore propose that the heightened optical responses exhibited in colored solutions derived from boronic acid-containing resorcinol condensation products (Figure 6) are due, in large part, to sugar boronate formation.
Sugar Solution Color Changes in the Presence of Resorcinol Condensation Products Possessing Boronic Acids
It is known that boronic acid-appended dyes can produce color changes in the presence of saccharides.10 Importantly, when saccharides form cyclic boronates, the Lewis acidity of boron is enhanced.35 Thus, upon saccharide binding, sp2-hybridized neutral boron is more readily converted to an sp3-hybridized anion via the addition of H2O or HO− as the fourth ligand. It was shown by several researchers that the spectral changes of boronic acid-appended chromophores and fluorophores induced by saccharide binding arise due to the change from a neutral, sp2 boronic acid to an sp3-hybridized anionic boronate-saccharide complex.10,35b We thus set out to determine whether the formation of sp3-hybridized sugar boronates occurs under our experimental conditions.
In the presence of 2a (40 mM), D-fructose-2-13C (1 equiv) in 9:1 DMSO-d6:D2O exhibits several new 13C-2 resonances which correspond to cyclic sugar boronic esters (Figure 9). The 13C chemical shifts (Table 2) are in agreement with the values obtained by Norrild and Eggert for the analogous p-tolylboronic acid sugar complexes.36 Importantly, we observe resonances for three anionic complexes all corresponding to known β-D-fructofuranose esters (Figure 9).
Figure 9.
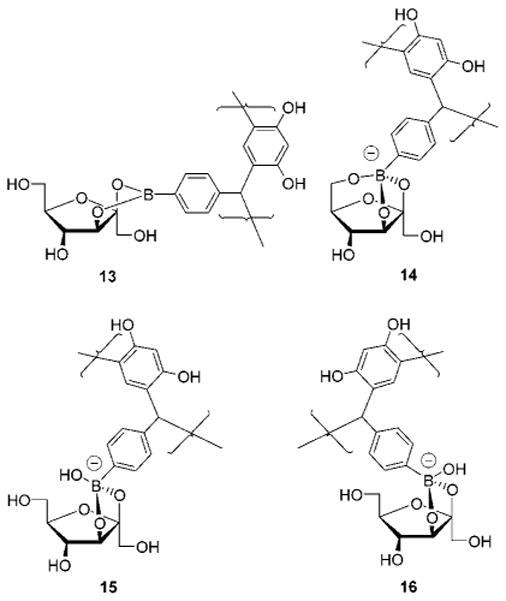
Structures of the four 2a:β-D-fructofuranose complexes (13–16) observed in 9:1 DMSO-d6:D2O. Boronates 15 and 16 are “exo” and “endo” isomers exhibiting 13C-2 chemical shifts of equal intensity at 114.4 and 114.3 ppm, respectively.
Table 2.
| fructose mutarotational isomer bound to boron |
13C-2 chemical shift (ppm)
|
|
|---|---|---|
| literature valuec (p-tolylboronic ester) | experimental valued (2a boronic ester) | |
| β-D-fructofuranose | 113.4a | 113.8 (14) |
| 114.7a | 114.4 (15) | |
| 114.6a | 114.3 (16) | |
| 115.2a | 115.5 (13) | |
| β-D-fructopyranose | 105.0b | 105.0 |
| 107.6b | 107.7 | |
| α-D-fructofuranose | 99.8b | 99.9 |
| α-D-fructopyranose | 92.6b | 93.2 |
In D2O at pD = 11–12.
In DMSO-d6.
Reference 36.
Experimental values are obtained in 9:1 DMSO-d6:D2O.
Further proof that we are forming anionic sugar boronates derives from 11B NMR spectroscopy. The 11B NMR chemical shifts of boronates change as a result of complexation with sugars due to differential electronic shielding of the 11B atom. An upfield shift of the 11B NMR signal accompanies the conversion of sp2-hybridized neutral species into sp3-hybridized boronate anions.37 At pH = 6.5, compound 2a (10 mM) in 1:1 DMSO:H2O (pH value refers to the buffered aqueous portion before mixing) exhibits a single broad resonance at −19.1 ppm which we assign to the neutral sp2-hybridized boronic acid (17, Figure 10). At pH = 11.0, but in the presence of 0.5 equiv of D-fructose, a new resonance appears at −32.9 ppm which intensifies when the amount of D-fructose is increased to 5 equiv. We thus assign the resonance at −32.9 ppm to D-fructose cyclic boronate anion 20. A solution of 2a (20 mM) in DMSO also exhibits a resonance at −32.9 ppm upon D-fructose (3 equiv) addition. The observation of the resonance at −32.9 ppm corresponding to boronate 20 in DMSO is consistent with our 13C NMR results.
Figure 10.
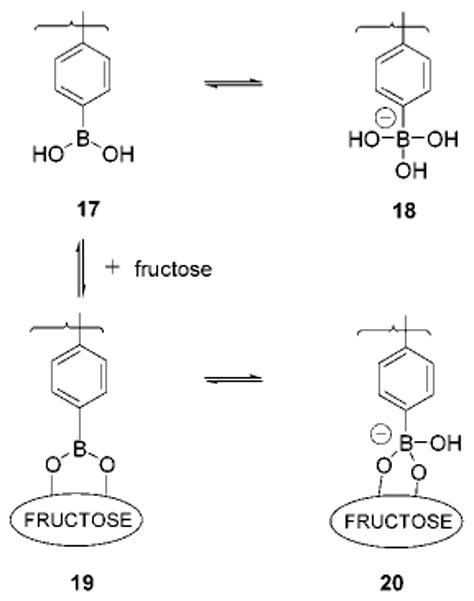
Equilibria for solutions of 2a and D-fructose.
The formation of the sugar boronate anion allows us to establish a mechanism for the sugar-induced color changes with our receptors. Anionic boronate formation, favored in the presence of sugars, leads to the diminished acidity of the C-6 hydroxyl. One way to envision this is via examination of xanthene resonance forms 21 and 22. Structure 22 possesses a more stable cation than does 21, rendering the C-6 hydroxyl of 22 relatively less ionizable.
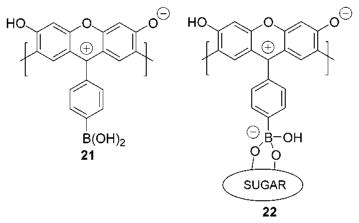
We have reported that colored aqueous DMSO solutions of resorcinol condensation products exhibited enhanced absorbances at 532 nm upon raising solution pH.7 If sugar complexation results in a change of receptor pKa, we should observe a change in receptor absorbance in pH-controlled media in the presence of sugars. Figure 11 shows the results of a pH versus absorbance titration monitored at 534 nm. As expected, the absorbance of colored 9:1 DMSO:H2O solutions containing 2a exhibits a sharp intensity onset beginning at ca. pH = 9. When the same pH titration experiment is run in the presence of D-fructose, the absorbance increase at and beyond pH = 9 is significantly lessened. This is evidence of the diminished ionizability of the C-6 hydroxyl induced by sugar complexation.
Figure 11.
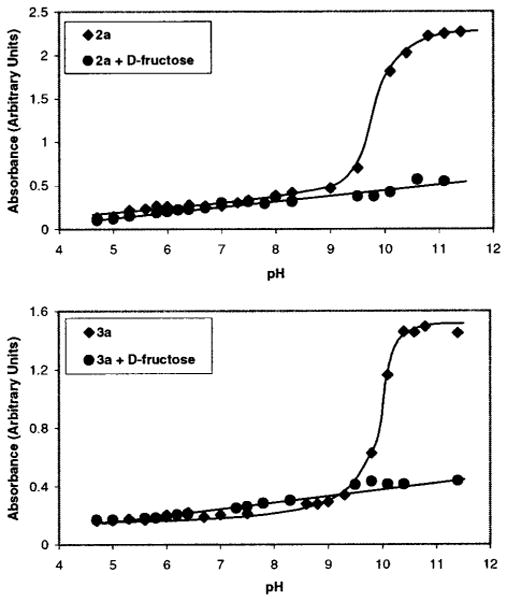
Spectrophotometric study (λ = 534 nm) of colored solutions containing 2a (top, 3.5 × 10−4 M) and 3a (bottom, 3.5 × 10−4 M) alone and with D-fructose (3.5 × 10−2 M) in buffered 9:1 DMSO:H2O (pH values refer to the H2O portion before mixing with DMSO).
A similar pH titration profile is observed for colored solutions of 3a alone and with added D-fructose (Figure 11), demonstrating that this finding appears general to these materials. Solutions containing 2a or 3a each exhibit color changes from dark brown to pale orange when D-fructose is added beyond pH = 9 (of the aqueous portion of the solution mixture). We are thus able to observe monosaccharide-induced solution color changes at room temperature in buffered media using these methods.38
Oligosaccharides should also promote solution color changes via boronate ester formation with chromophore-appended boronic acids. Prior studies (using fluorescence detection) showed that binding constants observed for the interaction of oligosaccharides and fluorophore-appended arylboronic acids increased with larger oligosaccharide size.37 This has been attributed to sugar binding via boronate formation involving the hydroxyls of the oligosaccharide reducing termini. The binding is enhanced by secondary sugar-dye contacts including hydrophobic and CH–π interactions. Larger oligosaccharides have greater flexibility and can thus more readily fold back and interact with the dye molecule when a boronate ester is formed.
The linear maltodextrins 9a–f (0–7.5 × 10−2 M) are dissolved in 0.5 mL of carbonate buffer (2.5 × 10−2 M, pH = 11) and mixed with 0.5 mL of DMSO colored preheated solutions of 2a (5.0 × 10−4 M), respectively, to furnish a 1:1 DMSO:H2O solvent system. The solution colors change from orange to yellow upon sugar addition. We thus detect neutral oligosaccharides in buffered media at room temperature by visual inspection. The apparent equilibrium constant (Kexp(app)) is determined by UV–vis spectroscopy. The expected trend37 of enhanced binding affinity correlating with longer oligosaccharide length is observed (Figure 12). We conclude that the significant solution color changes observed for the oligosaccharides (9a–f) are due, in large part, to the formation of cyclic sugar boronate esters.
Figure 12.
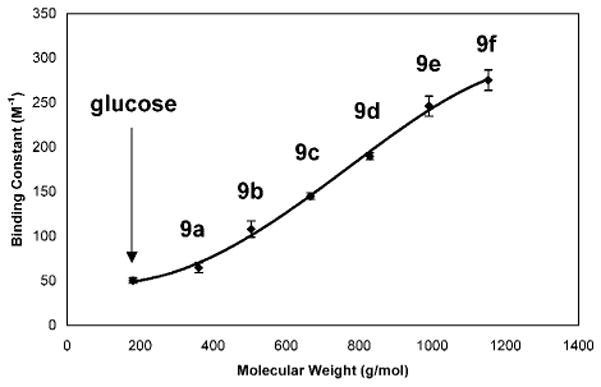
Plot of binding constant (three trials, monitored at 460 nm) vs molecular weight. Increasing molecular weight beginning with α-D-glucose through the series of linear maltodextrins (9a–f) results in an increase of binding constants (65.0, 108.3, 145.0, 189.7, 246.0, and 275.0 M−1) in colored solutions containing 2a. The binding constants are reproducible within ±10%.
The acyclic oligomeric xanthenes existing in colored resorcinarene solutions function as receptors. They produce solution color changes in the presence of saccharides due to factors including cyclic boronate formation and hydrogen bonding. Each of these latter binding motifs can perturb the ionization state of the C-6 hydroxyl, the key moiety responsible for modulating the color of xanthene dyes.17 Acid-containing analytes, whether added directly or derived via in situ formation from the oxidation of reducing sugars, produce color changes via hydrogen bonding. Boronate ester formation, however, is the predominant factor leading to solution color changes with neutral sugars. The important problem of diminished optical response correlating with increasing oligosaccharide chain length13,14 may be overcome via the use of boronic acid-containing chromophores. This is apparently due to the enhanced binding affinities of larger oligosaccharides for boronic acids.37
Conclusion
We have presented strong evidence that the color observed in Baeyer’s initial resorcinarene condensation reaction2 was due to the presence of xanthenes. In other words, he had created fluorescein1 analogues at apparently low levels. Interestingly, Fischer, who won the Nobel Prize for sugar research, presented his doctoral thesis to Baeyer in 1874 on fluorescein and orcinol dyes.39
We have shown that the colored products existing in solutions of resorcinarene macrocycles can serve as colorimetric indicators. The main findings of the present study include (i) the determination of the origin and structure of the active chromophores, (ii) elucidation of mechanisms associated with the solution color changes induced by sugars, and (iii) optical signaling for larger oligosaccharides.
The investigation of the colorimetric and fluorimetric properties of resorcinarenes, xanthenes, and related chromophoric materials is ongoing in our laboratory. Our efforts are now guided by the mechanistic studies described herein. Xanthene dyes containing well-positioned boronic acid or related binding moieties should find application as powerful receptors for saccharides and other polar analytes such as carboxylates and phosphates.
The resorcinarenes, however, do offer potential advantages as compared to functionalized dye materials. A great attraction is their ease of synthesis in one step on ca. 200 g scale.3d Having addressed many of the main mechanistic issues associated with the colorimetric sugar detection process, we are now also focusing on the study and optimization of important applied sensing parameters such as detection selectivity, sensitivity, and reversibility in aqueous and biological media. We are additionally developing new automated detection methods and expanding the detection capabilities of our receptors to include various analytes of biomedical significance.
Experimental Section
General
Matrix assisted laser desorption ionization mass spectra were acquired using a Bruker Proflex III MALDI mass spectrometer with either anthracene or dithranol matrixes. FT-IR spectra were recorded at room temperature on a Perkin-Elmer 1760X FT-IR spectrophotometer. UV–visible spectra were recorded at room temperature on a Spectramax Plus (Molecular Devices). Analytical thin-layer chromatography (TLC) was performed using general purpose silica gel on glass (Scientific Adsorbants). Flash chromatography columns were prepared with silica gel (Scientific Adsorbants, 32–63 μm particle size, 60 Å). Analytic and preparative-scale HPLC were performed on a CM4000 multiple solvent delivery system (Milton Roy) and a Spectromonitor 5000 photodiode array detector (LDC Analytical) using a Dynamax 60 Å C18 (21.4 mm ID × 25 cm L) with a flow rate of 5 mL/min and a gradient of 50% water/MeOH to 100% MeOH in 20 min unless otherwise stated. The following compounds were prepared according to literature methods: 1,5 2a,5 2b,40 3a,7 3b,19 and 6a.16 Isotopically labeled d-fructose-2-13C was purchased from Isotec. All other chemicals were purchased from Sigma or Aldrich and used without further purification. Proton NMR spectra were acquired in either CD3OD, CH3OD, or DMSO-d6 on a Bruker DPX-250, DPX-400, or AMX-500 spectrometer. All δ values are reported with (CH3)4Si at 0.00 ppm or DMSO at 2.45 ppm as references.
Boron NMR Spectra
11B (I = 3/2) NMR data were collected at room temperature using a Chemagnetics Infinity solid-state spectrometer at a frequency of 128.35 MHz. All experiments were performed static (nonspinning), in the liquid-state, using a Chemagnetics 5 mm MAS probe and quartz NMR tubes. Data were recorded (64 k data points) following a 1.5 μs pulse (45° flip angle; aqueous boric acid) over a spectral width of 1.0 MHz. Each spectrum is the sum of 50 000 transients. Spin–lattice relaxation time (T1) was not measured, but no saturation was observed using a 1.0 s pulse delay. All spectra were externally referenced using aqueous boric acid by setting the single 11B resonance to −18.8 ppm.41 Experiments were first attempted on several commercial high-resolution, solution-state spectrometers from both major vendors with no success. We attribute this lack of success to a combination of factors including (a) high 11B background signals from the probe, even with quartz tubes, (b) long receiver dead times as compared to the perceived short transverse relaxation times (T2) of the sample, and (c) insufficient transmitter power required to uniformly irradiate large spectral windows to detect spectral components with a line width as large as 10 kHz.
X-ray Crystallographic Data
Intensity data were collected on a Nonius Kappa CCD diffractometer equipped with Mo Kα radiation and a graphite monochromator. The sample was cooled to 120 K by an Oxford Cryosystems Cryostream chiller. Data collection parameters and crystallographic data are provided in Supporting Information. Absorption and decay effects were negligible. The structure was solved by direct methods, using SIR97,42 and refined using SHELXL97.43 H atoms were observed in difference maps, but were constrained to be in idealized positions in the refinement. OH hydrogen atoms are all disordered into two sites, all of which were treated as half populated. O–H distances were constrained to be 0.84 Å, but otherwise, these H positions were refined.
pH Titration Experiments
Compounds 2a or 3a (3.9 × 10−4 M) in DMSO (stock solutions) are heated to gentle reflux and cooled to room temperature. Additionally, 6.3 mg of D-fructose is dissolved in 0.1 mL of H2O containing buffer (2.5 × 10−1 M) and mixed with 0.9 mL of a receptor stock solution aliquot. The final concentrations: 2a or 3a = 3.5 × 10−4 M, D-fructose = 3.5 × 10−2 M, buffer = 2.5 × 10−2 M. For the pH titration experiments, buffers of pH from 4.5 to 5.0 were prepared in H2O containing AcONa and titrated with HCl. Buffers of pH from 5.5 to 8.5 were prepared in H2O containing KH2-PO4 and titrated with NaOH. Buffers with higher pH were prepared in H2O containing NaHCO3 titrated with NaOH. For the study of sialic acid binding to 6b in H2O at pH 7.5, HEPES dissolved in H2O was titrated with NaOH.
Binding Constant Determinations
The colored solutions of 6b or 2a in 9:1 DMSO:H2O each exhibit a linear dependence of absorbance on concentration in the ranges measured: 3.9 × 10−6–3.1 × 10−5 M (6b) and 5.0 × 10−4–1.0 × 10−2 M (2a). Apparent equilibrium constant (Kexp(app))37 data for the interaction of sugars and receptors derive from UV–vis spectroscopy. The equilibrium constants are evaluated via Benesi–Hildebrand treatment44 (eqs 1 and 2) using UV–vis absorbance changes observed at 460 nm for the linear maltodextrin series in colored solutions of 2a (complexation coverage = 50–60%). A 1:1 stoichiometry is observed for the interaction of 12 and 6b by the continuous variation method (Job plot)44 at [6b] + [12] = 1.0 × 10−5 M (constant). At 530 nm, 12 and 6b solutions exhibit a complexation coverage = 25%. In each case, the double reciprocal plots according to eq 1 afforded very good linearity with a correlation coefficient r ≥ 0.99.
| (1) |
| (2) |
A0 and A are UV–vis absorbances at 460 nm of chemosensor in the absence and presence of a sugar, respectively, while A∞ is that of the complex of sugar and chemosensor.
Supplementary Material
Acknowledgments
We gratefully acknowledge the National Institutes of Health (GM-61915), the Arnold and Mabel Beckman Foundation through the Beckman Young Investigator Award Program, and the donors of the Petroleum Research Fund administered by the American Chemical Society (36497-AC4) for support of this research.
Footnotes
Supporting Information Available: Data collection parameters and crystallographic data for compound 7 (PDF/CIF). This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Baeyer A. Chem Ber. 1871;5:255. [Google Scholar]
- 2.(a) Baeyer A. Ber Dtsch Chem Ges. 1872;5:25. [Google Scholar]; (b) Baeyer A. Ber Dtsch Chem Ges. 1872;5:280. [Google Scholar]
- 3.(a) Schneider HJ, Schneider U. J Inclusion Phenom. 1994;19:67. [Google Scholar]; (b) Cram DJ, Cram JM. Container Molecules and Their Guests. The Royal Society of Chemistry; Cambridge, U.K: 1994. [Google Scholar]; (c) Sherman JC. Tetrahedron. 1995;51:3395. [Google Scholar]; (d) Timmerman P, Verboom W, Reinhoudt DN. Tetrahedron. 1996;52:2663. [Google Scholar]; (e) Jasat A, Sherman JC. Chem Rev. 1999;99:931. doi: 10.1021/cr960048o. [DOI] [PubMed] [Google Scholar]; (f) Rudkevich DM, Rebek J. Eur J Org Chem. 1999;9:1991. [Google Scholar]
- 4.Aoyama Y, Tanaka Y, Sugihara S. J Am Chem Soc. 1989;111:5397. [Google Scholar]
- 5.Lewis PT, Davis CJ, Saraiva M, Treleaven WD, McCarley TD, Strongin RM. J Org Chem. 1997;62:6110. [Google Scholar]
- 6.Davis CJ, Lewis PT, McCarroll ME, Read MW, Cueto R, Strongin RM. Org Lett. 1999;1:331. doi: 10.1021/ol990105a. [DOI] [PubMed] [Google Scholar]
- 7.Lewis PT, Davis CJ, Cabell LA, He M, Read MW, McCarroll ME, Strongin RM. Org Lett. 2000;2:589. doi: 10.1021/ol9903990. [DOI] [PubMed] [Google Scholar]
- 8.For example: Seliwanoff T. Chem Ber. 1887;20:181.Bial M. Dtsch Med Wochenschr. 1902:253.Kulka RG. Biochem J. 1956;63:542. doi: 10.1042/bj0630542.
- 9.Chaplin MF. Monosaccharides. In: Chaplin MF, Kennedy JF, editors. Carbohydrate Analysis A Practical Approach. Oxford University Press; Oxford: 1994. pp. 1–40. [Google Scholar]
- 10.Review: James TD, Samankumara Sandanayake KRAS, Shinkai S. Angew Chem, Int Ed Engl. 1996;35:1910.More recent examples of boronic acid-based dyes and optical sensing of neutral sugars: Koumoto K, Takeuchi M, Shinkai S. Supramol Chem. 1998;9:203.Koumoto K, Shinkai S. Chem Lett. 2000:856.Ward CJ, Patel P, Ashton PR, James TD. Chem Commun. 2000:229.DiCesare N, Lakowicz JR. Org Lett. 2001;3:3891. doi: 10.1021/ol016813p.
- 11.Recent examples: Bertozzi CR, Kiessling LL. Science. 2001;291:2357. doi: 10.1126/science.1059820.Axford J. Trends Immunol. 2001;22:237. doi: 10.1016/s1471-4906(01)01890-7.and references therein.
- 12.Solution color changes in response to monosaccharides, maltose, maltotriose, and maltoteraose using a resorcinarene-methyl red dye competition process: Rusin O, Kral V. Tetrahedron Lett. 2001;42:4235.
- 13.Kennedy JF, Pagliuca G. Oligosaccharides. In: Chaplin MF, Kennedy JF, editors. Carbohydrate Analysis A Practical Approach. Oxford University Press; Oxford: 1994. pp. 46–62. [Google Scholar]
- 14.Review: LoGuidice JM, Lhermitte M. Biomed Chromatogr. 1996;10:290. doi: 10.1002/(SICI)1099-0801(199611)10:6<290::AID-BMC623>3.0.CO;2-H.
- 15.Davis CJ, Lewis PT, Billodeaux DR, Fronczek FR, Escobedo JO, Strongin RM. Org Lett. 2001;3:2443. doi: 10.1021/ol0160194. [DOI] [PubMed] [Google Scholar]
- 16.Sen RN, Sinha NN. J Am Chem Soc. 1923;45:2984. [Google Scholar]
- 17.Gupta SN, Linden SM, Wrzyszczynski A, Neckers DC. Macromolecules. 1988;21:51. [Google Scholar]
- 18.Agbaria K, Biali SE. J Org Chem. 2001;66:5842. doi: 10.1021/jo0103468. [DOI] [PubMed] [Google Scholar]
- 19.Weinelt F, Schneider HJ. J Org Chem. 1991;56:5527. [Google Scholar]
- 20.(a) Gillis BT, Beck PE. J Org Chem. 1963;28:1388. [Google Scholar]; (b) Santususso TM, Swern D. Tetrahedron Lett. 1974:4255. [Google Scholar]; (c) Emert J, Goldenberg M, Chiu GL. J Org Chem. 1977;42:2012. [Google Scholar]
- 21.Fronczek FR, Johnson RJ, Strongin RM. Acta Crystallogr. 2001;E5:o447–o448. [Google Scholar]
- 22.(a) Chen C-T, Yan S-J. Tetrahedron Lett. 1969:3855. [Google Scholar]; (b) Head DL, McCarty CG. Tetrahedron Lett. 1973:1405. [Google Scholar]
- 23.For the structures of the different possible resorcinarene stereoisomers, see ref 3d.
- 24.In the previous work (ref 19), acyclic oligomeric products (3b and two stereoisomeric trimeric compounds, three resorcinol rings, 4, R = Me, m = 1, n = 0, Scheme 1) were isolated and characterized. Higher order acyclic oligomers (e.g., pentamers and hexamers) were also observed as major reaction products. Methyl 1H NMR resonances, appearing as several doublets between 0.7 and 2.0 ppm (CH3OD) that corresponded to neither 3b, 4 (R = Me, m = 1, n = 0), nor resorcinarene macrocycles, thus were assigned to acyclics with five or more resorcinol moieties.
- 25.Flash column chromatography and TLC analysis of the thermolysis products of 2b are complicated by the multiple product formation and fraction streaking.
- 26.Fronczek FR, St Luce NN, Strongin RM. Acta Crystallogr. 2001;C57:1423. doi: 10.1107/s0108270101015621. [DOI] [PubMed] [Google Scholar]
- 27.Sen RN, Sarkar NN. J Am Chem Soc. 1925;47:1079. [Google Scholar]
- 28.Deno NC, Booker EL, Kramer KE, Saines G. J Am Chem Soc. 1969;91:5237. [Google Scholar]
- 29.Arts SJHF, Mombarg EJM, van Bekkum H, Sheldon RA. Synthesis. 1997:597. [Google Scholar]
- 30.Analysis of NMR integral ratios reveals that 59% of starting D-fructose is converted to new products under these conditions. The new product mixture contains 27% aldehydes and 0.9% carboxylic acids.
- 31.We heat solutions of D-(−)-fructose, 9a, or D-(+)-glucose in DMSO to a gentle reflux for 2 min and cool to room temperature. A solution of 2b in DMSO is also heated to gentle reflux and cooled to room temperature. The three sugar solutions are added to the solution of 2b, respectively, followed by the addition of water to afford mixtures in 9:1 DMSO:H2O. The final concentration is 5.2 × 10−3 M for 2b and 1.6 × 10−2 M for all sugars. Each solution is stirred for 6 min prior to UV–vis analysis. The absorbance decrease (as compared to the solution of 2b without sugar) at 530 nm for the solution containing D-(+)-glucose is (17.9 ± 1.0)%, (23.7 ±1.3)% for D-(−)-fructose, and (9.1 ± 2.0)% for 9a (three trials).
- 32.Having shown that acyclic aldonic acids, rather than uncharged sugars, produce a solution color change at room temperature in the presence of our active, non-boronic acid-containing chromophores, we next chose to generalize this finding to include sialic acid (12) because it is a prototypical, readily available, and bioactive cyclic sugar molecule possessing a carboxylate moiety. In addition, in our first communication in this study (ref 6), we used boron-containing 1 to produce significant solution color changes with 12 as well as other carboxylate-, phosphate-, and amine-functionalized saccharides. The results reported herein thus show that the boronic acid moieties may not be required (see also ref 7) to produce solution color changes in the presence of charged sugars. The fact that acetate also induces a color change at room temperature in buffered media demonstrates that simpler carboxylic acids could also be determined under these conditions. Research is in progress in our laboratory to develop selective sensing conditions for bioactive charged phosphates and carboxylates, including sialic acids, on the basis of these findings.
- 33.The IR absorption changes we observe are in keeping with those reported by Zhang and co-workers who have recently studied the formation of hydrogen bonds between anthracene carboxylic acid and xanthene dyes using several techniques including IR and 1H NMR: Zhang H, Zhang M, Shen T. Dyes Pigm. 1999;43:15.
- 34.In addition, 6b (10 mM) in DMSO-d6 was titrated with 0.13–2.6 equiv of 12. The 1H NMR spectrum shows that a 6b hydroxyl resonance exhibits a successive downfield chemical shift from 10.53 to 10.70 ppm upon increasing the concentration of 12.
- 35.(a) Lorand JP, Edwards JD. J Org Chem. 1959;24:769. [Google Scholar]; (b) Yoon J, Czarnik AW. J Am Chem Soc. 1992;114:5874. [Google Scholar]
- 36.Norrild JC, Eggert H. J Chem Soc, Perkin Trans. 1996;2:2583. [Google Scholar]
- 37.Nagai Y, Kobayashi K, Toi H, Aoyama Y. Bull Chem Soc Jpn. 1993;66:2965. [Google Scholar]
- 38.Derivative analysis reveals slope maxima for the titration curves of colored solutions of 2a and 3a occur at pH values corresponding to 9.45 and 9.80, respectively. However, these values have no physical significance towards assigning a respective pKa to the materials since the pH values on the x-axis of Figure 11 refer only to the aqueous sugar-containing solutions prior to mixing with solutions derived from heated 2a and 3a in DMSO. A known method for the determination of resorcinarene pKa values in a mixed aqueous solvent system (Schneider H-J, Güttes D, Schneider U. J Am Chem Soc. 1988;110:6449.) more appropriate for potentiometric titrations was applied to colored solutions of 2a and 3 with and without added fructose. Unfortunately, solubility problems precluded us from obtaining pKa data via this method. Although to date we have not been able to determine pKa values, the strikingly similar effect of fructose on the UV–vis spectra of distinct but related materials (colored solutions of 2a and 3a) shows that the sugar-induced perturbation of the chromophore ionization state is apparently significant and reproducible within this class of materials.
- 39.Fischer E. PhD Dissertation. Strasbourg University; Strasbourg, France: 1874. [Google Scholar]
- 40.(a) Niederl JB, Vogel H. J Am Chem Soc. 1940;62:2512. [Google Scholar]; (b) Erdtmann H, Hoegberg S, Abramsson S, Nilsson B. Tetrahedron Lett. 1968:1679. [Google Scholar]; (c) Nilsson B. Acta Chem Scand. 1968:22732. doi: 10.3891/acta.chem.scand.22-0518. [DOI] [PubMed] [Google Scholar]; (d) Cram DJ, Karbach S, Kim HE, Knobler CB, Maverick EF, Eriscon JL, Helgeson RC. J Am Chem Soc. 1988;110:2229. [Google Scholar]
- 41.Onak TP, Landesman H, Williams RE, Shapiro I. J Phys Chem. 1959;63:1533. [Google Scholar]
- 42.Altomare A, Burla MC, Camalli M, Cascarano GL, Giacovazzo C, Guagliardi A, Moliterni AGG, Polidori G, Spagna R. J Appl Crystallogr. 1999;32:115. [Google Scholar]
- 43.Sheldrick G. SHELXL97. University of Göttingen; Germany: 1997. [PubMed] [Google Scholar]
- 44.Benesi H, Hildebrand JH. J Am Chem Soc. 1949;71:2703. doi: 10.1021/ja01192a003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.