

Abstract
51V solid-state NMR (SSNMR) studies of a series of non-innocent vanadium(V) catechol complexes have been conducted to evaluate the possibility that 51V NMR observables, quadrupolar and chemical shift anisotropies, and electronic structures of such compounds can be used to characterize these compounds. The vanadium(V) catechol complexes described in these studies have relatively small quadrupolar coupling constants, which cover a surprisingly small range from 3.4 to 4.2 MHz. On the other hand, isotropic 51V NMR chemical shifts cover a wide range from −200 ppm to 400 ppm in solution and from −219 to 530 ppm in the solid state. A linear correlation of 51V NMR isotropic solution and solid-state chemical shifts of complexes containing non-innocent ligands is observed. These experimental results provide the information needed for the application of 51V SSNMR spectroscopy in characterizing the electronic properties of a wide variety of vanadium-containing systems, and in particular those containing non-innocent ligands and that have chemical shifts outside the populated range of −300 ppm to −700 ppm. The studies presented in this report demonstrate that the small quadrupolar couplings covering a narrow range of values reflect the symmetric electronic charge distribution, which is also similar across these complexes. These quadrupolar interaction parameters alone are not sufficient to capture the rich electronic structure of these complexes. In contrast, the chemical shift anisotropy tensor elements accessible from 51V SSNMR experiments are a highly sensitive probe of subtle differences in electronic distribution and orbital occupancy in these compounds. Quantum chemical (DFT) calculations of NMR parameters for [VO(hshed)(Cat)] yield 51V CSA tensor in reasonable agreement with the experimental results, but surprisingly, the calculated quadrupolar coupling constant is significantly greater than the experimental value. The studies demonstrate that substitution of the catechol ligand with electron donating groups results in an increase in the HOMO-LUMO gap and can be directly followed by an upfield shift for the vanadium catechol complex. In contrast, substitution of the catechol ligand with electron withdrawing groups results in a decrease in the HOMO-LUMO gap and can directly be followed by a downfield shift for the complex. The vanadium catechol complexes were used in this work because the 51V is a half-integer quadrupolar nucleus whose NMR observables are highly sensitive to the local environment. However, the results are general and could be extended to other redox active complexes that exhibit similar coordination chemistry as the vanadium catechol complexes.
Introduction
The characterization of metal complexes containing redox active ligands also referred to as “non-innocent” can be particularly challenging because of the potential of ligand-to-metal charge transfer (LMCT) processes that can prevent unambiguous determination of the oxidation state of the coordinating metal ion.1 Such redox active ligands are ubiquitous in biological chemistry where the ligand supporting a radical is abundant and plays a crucial role in the chemistry of metal complexes such as Fe-porphyrins,2 Fe(III)-transferrin,3 purple acid phosphatase,4 Cu(II) in galactose oxidase5 and Mn(II) in photosystem II.6 A range of redox active ligands are o-quinone/semiquinone/catechol, dithiolene/ene-dithiolate, O2(dioxygen)/O2•−(superoxide)/O22− (peroxide), NO+(nitrosyl cation)/NO• (nitric oxide radical)/NO− (nitroxide anion), and tyrosyl/tyrosilate.1,7–11 The major method for characterization of metal complexes with redox active ligands in the solid state is X-ray crystallography.9,10,12–15 Solution characterization is often done using electrochemistry, UV- vis spectroscopy and EPR spectroscopy.8–10,15 Another characterization method used less frequently for such complexes in solution is multinuclear NMR spectroscopy.16 Solid state methods commonly used include FTIR spectroscopy,12,15,17 but a need exists for alternative characterization methods of these metal complexes with redox active ligands particularly in cases when the material is not crystalline. Solid-state NMR spectroscopy has been used for characterization of complexes containing any NMR active nucleus (e.g. 51V) and is very informative with regard to the electronic properties of the complexes.18–24
Solid-state 51V NMR spectroscopy (SSNMR) is a potent tool that provides important insights into the electronic structure of an active site(s) of vanadium-containing systems (i.e. proteins, bioinorganic solids, and inorganic catalysts).25–46 51V is a half-integer quadrupolar nucleus (I=7/2). Its electric field gradient tensor (EFG) and the chemical shift anisotropy (CSA) tensor are much more sensitive reporters of the electronic structure of vanadium-containing materials than solution-based 51V NMR isotropic chemical shifts.26–38,47,48 The relationship between vanadium coordination environments and its 51V SSNMR observables- quadrupolar and chemical shift anisotropy tensors– can be exploited to characterize the electronic properties of ternary vanadium complexes containing non-innocent ligands.26,37,42,49–51
One common class of non-innocent ligands is the o-dioxolene or also referred to as catechol ligand. Catechols coordinate as a neutral or anionic ligand with an (OO) motif and in a bidentate fashion, as catecholate dianion (electronic spin state S=0), as semiquinonato monoanions (S=1/2) or as a neutral o-benzoquinones (S=0).9,13,15,52–59 Pierpont reported an equilibrium between Mn+1(Cat2−) and Mn+(SQ−) where M is any 3d metal both in solution and in the solid state and these redox isomers are known as valence tautomers.9 The electron transfer properties and valence tautomerism encouraged applications of these systems as data storage media and to sensor technology.9,10 Low energy electronic transition have been reported from the excited state of o-dioxolene based chromophore to the semiconductor surfaces in numerous vital applications, including optoelectronics and solar cells.60,61 Vanadium (oxidation state II to V) catechol complexes that have been reported thus far document the interesting and varied properties of these systems.9,10,16,36,58
A number of vanadium-catechol systems have been reported with interesting structural, electronic, reactivity, magnetic and spectroscopic properties.13,15,62 51V NMR spectroscopy showed an unusual spread in the solution chemical shifts for a series of vanadium(V)-catechol complexes documenting that this method is sensitive to subtle changes in the electronic structure of these complexes.16 One vanadium-catechol compound, [(3,5-di-tertiary-butylcatecholato)-{N-(2-methylpyridine)-3-methoxysalicylideneaminato} oxovanadium(V)] (SJZ00108)36 shown in Figure 1 has an unusual electronic environment which has been investigated by 51V SSNMR spectroscopy. This complex has a 51V isotropic chemical shift of 426 ppm and is outside of the chemical shift range generally observed for vanadium complexes (Figure 2). To build a general understanding of the NMR properties of catechol-based vanadium compounds and while basing these studies on the results of our earlier report, we have conducted 51V SSNMR investigations of a series of known o-dioxolane-oxovanadium(V) complexes with (ONN & OO) ancillary ligand donor sets.
Figure 1.
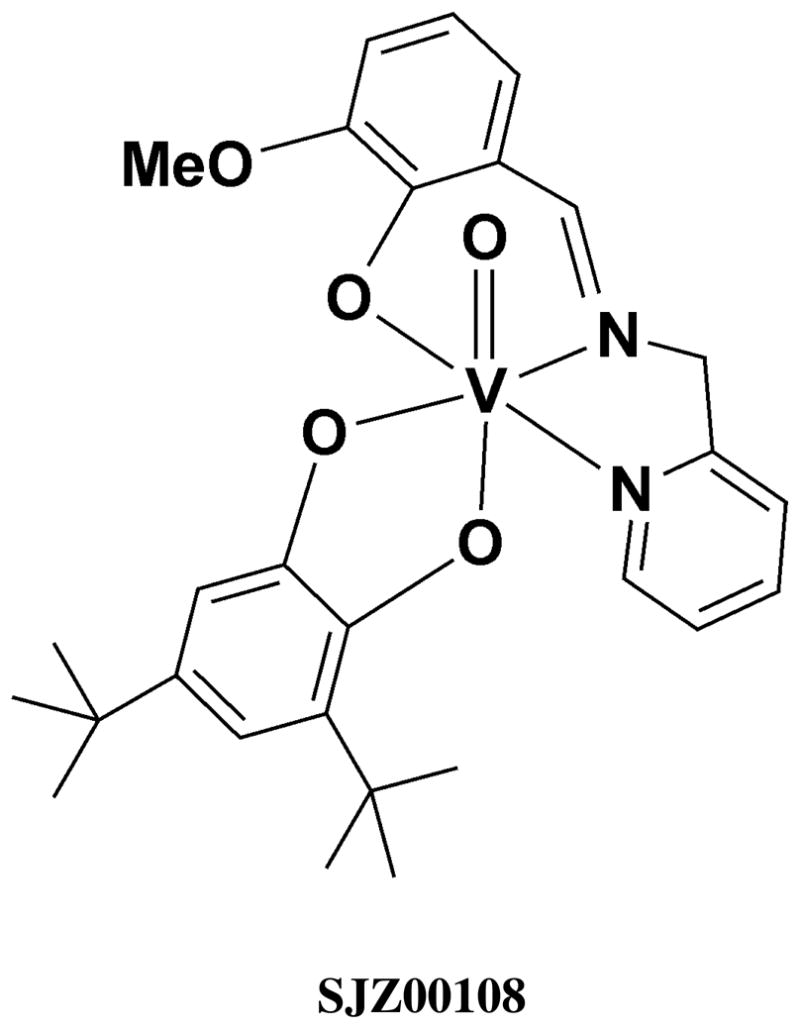
Redox non-innocent vanadium(V)-catechol complex (3,5-di-tertiarybutylcatecholato- -{N-(2-methylpyridine)-3-methoxysalicylideneaminato} oxovanadium(V) (SJZ00108).36
Figure 2.
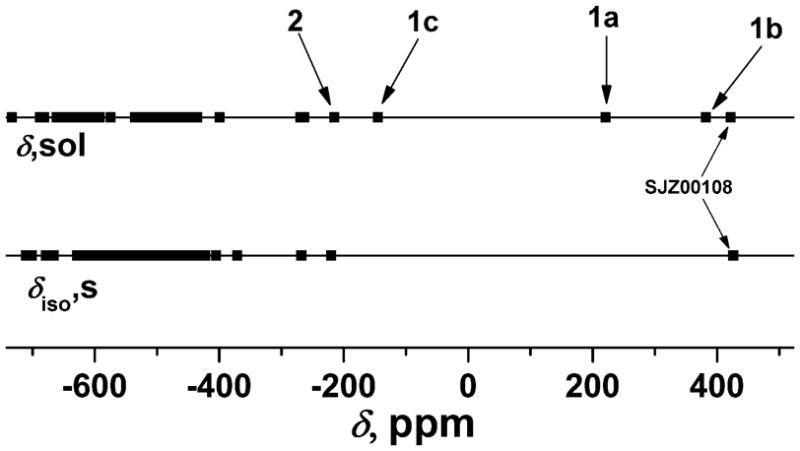
Comparison between 51V isotropic chemical shifts obtained from solid-state and solution NMR for all the compounds reported thus far in the literature demonstrating a missing domain in the solid-state NMR investigation.
Normal and redox inactive (innocent) ligands are generally described as having a large energy gap between the HOMO and LUMO. Non-innocent ligands have a smaller energy gap as illustrated for comparison with the normal ligands in Figure 3. As shown recently for titanium (Ti) this energy gap for the non innocent ligands decrease upon complexation61,63 and this is also illustrated in Figure 3. These considerations are not exclusive to simple solution Ti-complexes but extend to other metal-catechol complexes and surface systems. For example, changes in HOMO and LUMO energies were observed for catechol complexes formed on the interface of rutile TiO2(110); but in this case the shifts in orbital energies resulted in the orbitals are no longer at the HOMO-LUMO gap.64 Changes in the electronic properties of these systems by substitution on the catechol moiety in catechol complexes with mononuclear Ti(IV) or TiO2 nanoparticles were not found to be reflected in the reduction potential in these complexes.65 V-catechol complexes in solution were, however, found to be sensitive to the subtle changes in electronic structure16 and therefore excellent candidates to show the decrease previously reported in the HOMO-LUMO gap compared to the free catechol66 also included in Figure 3. Substitution of the catechol-ligand with electron donating groups raises both the HOMO and LUMO compared to the free ligand. However, in the case of the t-Bu group the HOMO is raised more than the LUMO, and the HOMO-LUMO gap is decreased in the complex and this is illustrated in Figure 3. In contrast, substitution by bromo-substituents lowers the HOMO and the LUMO.66 However, since the HOMO is lowered more than the LUMO the result is a net increase in the HOMO-LUMO gap66 as shown in Figure 3. We propose that the electronic modifications that take place in the vanadium catechol complexes upon substitution can be observed directly in their 51V NMR chemical shifts since the vanadium is sensitive to its electronic environment.
Figure 3.
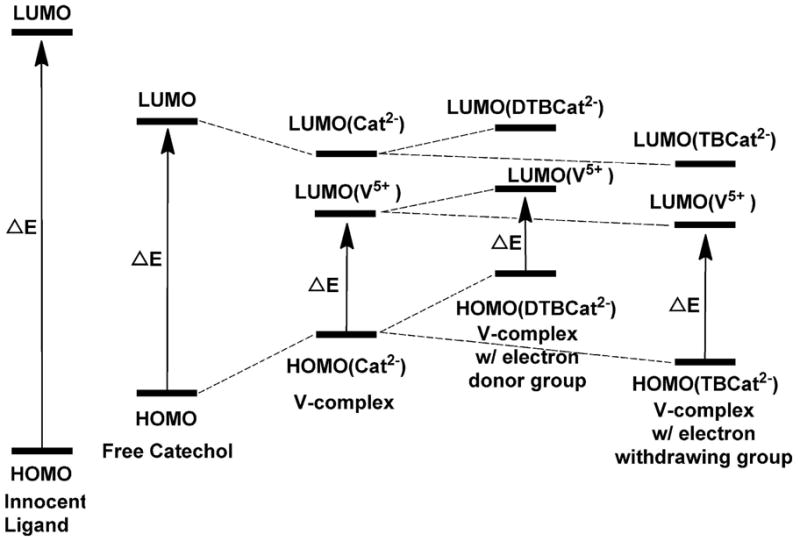
Qualitative presentation of molecular orbital energies and electronic excitation in free and vanadium-coordinated o-dioxolenes. For comparison separation between HOMO-LUMO for redox innocent ligands is also shown (left). The solid arrows correspond to the lowest energy excitation in each system.
Quantum chemical calculations have also been used to predict the 51V NMR parameters and/or the electronic structure of a wide range of vanadium-containing bioinorganic compounds26–29,33,34,36,66,67 and proteins,37,68–70 including vanadium complexes containing non-innocent ligands.66 DFT calculations generally predict the NMR parameters accurately. However, when a ligand is non-innocent with large 51V isotropic chemical shifts, the experimentally observed de-shielding is less well described.66
Herein, we present 51V SSNMR studies of a series of vanadium(V)-catechol complexes (Figure 4) to test the hypothesis that SSNMR observables (i.e. quadrupolar and chemical shift anisotropy tensors) can effectively describe the electronic properties of the complexes with redox non-innocent ligands. We also performed quantum chemical characterization of one of these complexes and found limited agreement between values calculated and experimentally measured. Our experimental results were used to provide information needed for application of 51V SSNMR parameters in characterizing the electronic properties of systems containing non-innocent ligands. Our findings also demonstrate that the observed quadrupolar couplings are relatively small and cover a narrow range of values reflecting symmetric electronic charge distribution. Therefore, these quadrupolar interaction parameters alone are not sufficient to define the electronic structure of these complexes. On the contrary, the chemical shift anisotropy tensor elements accessible from 51V SSNMR measurements are a highly sensitive probe of the subtle differences in the electronic distribution and orbital occupancy in these compounds. The vanadium catechol complexes were used in this because the 51V is a half-integer quadrupolar nucleus whose NMR observables are highly sensitive to the local environment. However, similar experiments with other NMR active metal nuclei could be used for characterization of other redox active complexes.
Figure 4.
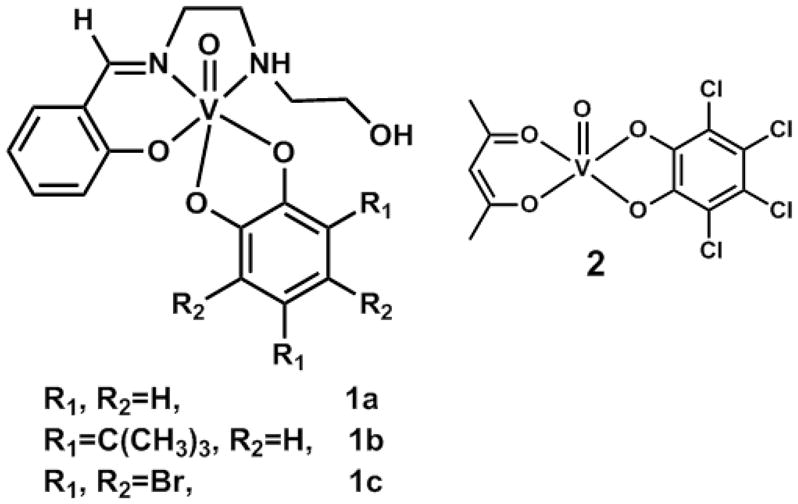
Molecular structures of the four vanadium(V)-o-dioxolene compounds under investigation. [VO(hshed)(Cat)] (1a), [VO(hshed)(DTBCat)] (1b), [VO(hshed)(TBCat)] (1c), and [VO(acac)(TCCat)] (2).
Experimental Section
Materials
Vanadyl acetylacetonate, [VO(acac)2] (99.99 %), vanadyl sulfate hydrate, [VO(SO4).H2O], salicyldehyde (98 %), N-(2-hydroxyethyl)ethylenediamine (99 %), catechol (≥ 99 %), tetrabromocatechol (96 %), 3,5-di-tert-butylcatechol (99 %), tetrachloro-o-quinone (97 %) were purchased from Sigma-Aldrich. Sodium hydroxide was purchased from Fisher Scientific. All reagents were commercially available and used as received. Solvents were ACS grade and used as received. Figure 4 shows the structures of the compounds selected for this study. Compounds 1a-1c are from the VVO-hshed series while compound 2 (acetylacetonatotetrachlorocatecholatooxovanadium(V)) represents a VVO-acac compound. By going from the VVO-hshed series to compound 2 we are changing the type and also the denticity of the ancillary ligand.
Abbreviations
Acac = 2,4-pentanedionate; hshed = N-salicylidenyl-N′-(2-hydroxyethyl)ethylenediamine; Cat = catechol; DTBCat = 3,5-di-tert-butylcatechol; TBCat = tetrabromocatechol; and TCCat = tetrachlorocatechol.
Syntheses of Compounds
The compounds of the VVO-hshed series (1a-1c) were prepared as described previously16 by first preparing the [VO2(hshed)]2 precursor. The pentacoordinate acetylacetonatotetrachlorocatecholatooxovanadium(V) compound (2) was prepared using the method described by Galeffi et al.57 Sample purities were checked by solution 1H NMR spectroscopy and were in agreement with previous reports.16,57
Solution 51V NMR Spectroscopy
The solution 51V NMR spectra were acquired on a Varian INOVA-300 spectrometer (7.0 T) at 78.9 MHz. The 51V NMR spectra were generally acquired using single pulse excitation with a pulse angle of 60°, a spectral width of 83.6 kHz, and an acquisition time of 0.096 s. The 51V chemical shifts were obtained using an external reference of VOCl3 (δiso = 0.0 ppm).
51V SSNMR Spectroscopy
Solid-state 51V NMR spectra were recorded on a 9.4 T Tecmag Discovery spectrometer (51V Larmor frequency of 105.23 MHz). A 3.2 mm Varian T3 MAS probe was employed for all solid-state NMR experiments. Neat VOCl3 was used as an external reference (δiso = 0.0 ppm).71 This sample was also used to determine the 90° pulse width which was set to 4.0 μs (γB1/2π ≈ 62 kHz). Between 8.0 and 16.0 mg of sample was packed into a 3.2 mm thick-wall rotor. For each compound, 51V MAS SSNMR spectra were acquired at ωr = 13, 17, and 20 kHz. The MAS frequency was controlled to within ±5 Hz by a Tecmag MAS controller. The temperature was calibrated for this probe at different MAS frequencies using a PbNO3 temperature sensor,34 and the actual temperature at the sample was 25 °C maintained to within ±1 °C throughout the experiments using the Tecmag temperature controller.
The magic angle was set by maximizing the number of rotational echoes observed in the 23Na NMR free-induction decay of solid NaNO3. All spectra were recorded using a single pulse excitation experiment with a pulse width of 1.0 μs and a spectral width of 1.0 MHz. A recycle delay of 1 s was used for all experiments. A total of 8192 scans were acquired for each compound. The spectra were processed by MestReNova NMR data processing software with a Gaussian line broadening function of 300 Hz and baseline correction. The isotropic chemical shifts were determined by the analyses of the spectra collected at the different MAS frequencies. 51V chemical shift anisotropy (δσ, ησ) and quadrupolar (CQ, ηQ) parameters as well as the relative tensor orientations described by the Euler angles (α, β, γ) were extracted by numerical simulations of the spinning sideband patterns using SIMPSON72 software package. The best-fit values are shown in Table 1.
Table 1.
Experimental 51V NMR parameters for six and five coordinated vanadium(V)-o-dioxolene compounds (1a - 2)a,b
| Compound | Solid state
|
Solution | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CQ/MHz ± 0.1 | ηQ ± 0.05 | δiso/ppm ± 5 | δσ/ppm ± 30 | ησ ± 0.05 | δ11/ppm ± 30 | δ22/ppm ± 30 | δ33/ppm ± 30 | α/degree ± 10 | β/degree ±15 | γ/degree ±30 | δiso/ppm | |
|
| ||||||||||||
| 1a | 4.0 | 1.00 | 58 | −243 | 0.93 | −185 | 66 | 292 | 81 | 70 | 87 | 22116 |
| 1b | 3.4 | 0.60 | 531 | 437 | 0.90 | 968 | 509 | 116 | 10 | 40 | 120 | 38216 |
| 1c | 4.2 | 1.0 | 31 | −314 | 0.65 | −314 | 54 | 258 | 45 | 45 | 90 | −14516 |
| 2 | 4.1 | 0.77 | 3219 | −302 | 0.70 | −521 | −145 | 9 | 40 | 40 | 75 | 215 (This work) |
| SJZ0010816 | 6.0±0.4 | 0.7±0.05 | 426.3± 3 | 570±19 | 0.6±0.1 | - | - | - | 0±60 | 0±10 | 30±30 | 422 & 37516 |
The chemical shift parameters are defined such that |δxx – δiso |≤ |δyy – δiso |≤ |δzz – δiso | and δiso = (δxx + δyy + δzz)/3, δσ = δzz − δiso, ησ = (δyy − δxx)/( δzz − δiso) according to the Haeberlen-Mehring-Spiess convention.16 Here δii denotes the principal components of the chemical shift tensor.
The EFG parameters are CQ = eQVZZ/h and ηQ = (VXX − VYY)/VZZ where |VZZ| ≥ |VYY| ≥ |VXX|, e is the electron charge, and h is Planck’s constant.
In this work, the chemical shift parameters are defined such that |δxx – δiso |≤ |δyy – δiso |≤ |δzz – δiso | and δiso = (δxx + δyy + δzz)/3, δσ = δzz − δiso, ησ = (δyy − δxx)/(δzz − δiso) according to the Haeberlen-Mehring-Spiess convention.37 δii denotes the principal components of the chemical shift tensor. The EFG parameters are CQ = eQVZZ/h and ηQ = (VXX − VYY)/VZZ where |VZZ| ≥ |VYY| ≥ |VXX|, e is the electron charge and h is Planck’s constant.
Density Functional Theory calculations
Quantum chemical calculations for the structurally characterized VO(hshed)(Cat) complex (compound 1a) were performed with Density Functional Theory in Gaussian03.73 The 51V magnetic shielding and EFG tensors were computed using B3LYP functional and several basis sets (6-311+G, 6-311++G, TZV, TZVP, Wachters+f). Calculations were carried out using either the non-optimized X-ray geometry or geometry optimized structures at the same level of theory as the NMR parameter calculations, as specified in Table 2. The 51V chemical shifts are referenced with respect to VOCl3 (0 ppm) whose absolute magnetic shielding tensor was calculated at the same level of theory with prior geometry optimization.
Table 2.
| Method | Solid state
|
Solution | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CQ/MHz | ηQ | δiso/ppm | δσ/ppm | ησ | δ11/ppm | δ22/ppm | δ33/ppm | α/degree | β/degree | γ/degree | δiso/ppm | |
|
| ||||||||||||
| Experiment | 4.0 ±0.1 | 1.00 ±0.05 | 58 ±5 | −243 ±30 | 0.93 ±0.05 | 291 ±30 | 68 ±30 | −185 ±30 | 81 ±10 | 70 ±15 | 87 ± 30 | 22116 |
| b3lyp/6-311+G (non-optimized) | 7.76 | 0.38 | −16.3 | −472.0 | 0.82 | 413.2 | 26.2 | −488.3 | 89 | 109 | 69 | |
| b3lyp/TZV (non-optimized) | 7.00 | 0.39 | −51.4 | −477.3 | 0.84 | 387.7 | −13.2 | −528.7 | 87 | 115 | 71 | |
| b3lyp/6-311++G (proton geometries optimized) | 7.97 | 0.32 | −32.0 | −461.8 | 0.77 | 376.7 | 21.1 | −493.8 | 149 | 21 | 16 | |
| b3lyp/TZVP (proton geometries optimized) | 7.79 | 0.32 | 30.1 | −434.0 | 0.92 | 446.7 | 47.5 | −403.9 | 150 | 24 | 23 | |
| b3lyp/6-311++G (all atom geometries optimized) | 10.66 | 0.45 | −233.3 | −484.3 | 0.62 | 159.0 | −141.3 | −717.6 | 92 | 4 | 148 | |
| b3lyp/Wachters+f on V, 6-31*G on other atoms | 9.92 | 0.50 | 61.7 | −333.9 | 0.89 | 377.2 | 80.1 | −272.2 | 82 | 107 | 64 | |
The chemical shift parameters are defined such that |δxx – δiso |≤ |δyy – δiso |≤ |δzz – δiso | and δiso = (δxx + δyy + δzz)/3, δσ = δzz − δiso, ησ = (δyy − δxx)/( δzz − δiso) according to the Haeberlen-Mehring-Spiess convention.16 Here δii denotes the principal components of the chemical shift tensor.
The EFG parameters are CQ = eQVZZ/h and ηQ = (VXX − VYY)/VZZ where |VZZ| ≥ |VYY| ≥ |VXX|, e is the electron charge, and h is Planck’s constant.
Results
51V Solution NMR Spectroscopy
A number of oxovanadium(V)catecholato complexes with chemical shifts ranging from −200 to +400 ppm have been prepared (1a – 1c, 2), Figure 4. Compound 1a contains a redox-active catechol ligand in a bidentate dianionic mode to the oxovanadium(V) center resulting in a downfield chemical shift of 221 ppm for this non-innocent complex, Table 1. Upon introducing tert-butyl groups to the catechol ring (1b), further downfield chemical shift (at 382 ppm) is observed (Table 1). Introducing electron withdrawing bromo-substituents on the catechol moiety (1c) results in an upfield shift (−145 ppm). The pentacoordinate complex 2 has a chemical shift of −215 ppm. From Figure 2 it is evident that these four compounds represent a chemical shift region not observed previously for the majority of vanadium(V) compounds.
51V Solid-State NMR (SSNMR) Spectroscopy
To further explore the electronic impact of these noninnocent ligands on the vanadium nucleus in compounds 1a-1c and 2, we performed 51V solid-state MAS NMR investigations. The 51V MAS NMR spectra of the four oxovanadium(V) -catecholato compounds (1a-1c, 2) were acquired at 9.4 T using three spinning frequencies (13, 17, and 20 kHz). Figure 5 shows 51V NMR spectra of the four vanadium(V) catechol complexes (1a-1c, 2) recorded at the MAS frequency of 17 kHz. The chemical shift anisotropy and quadrupolar NMR parameters, as well as the relative orientation, were extracted by fitting the 51V MAS spectra. The experimental and best-fit simulated spectra for compound 1a acquired at MAS frequencies of 7, 13, 17, and 20 kHz are illustrated in Figure 6. The experimental and best-fit simulated spectra for compounds 1b, 1c, and 2 are presented in the Supporting Information. A summary of the SSNMR parameters obtained from these 51V MAS NMR spectra is presented in Table 1.
Figure 5.
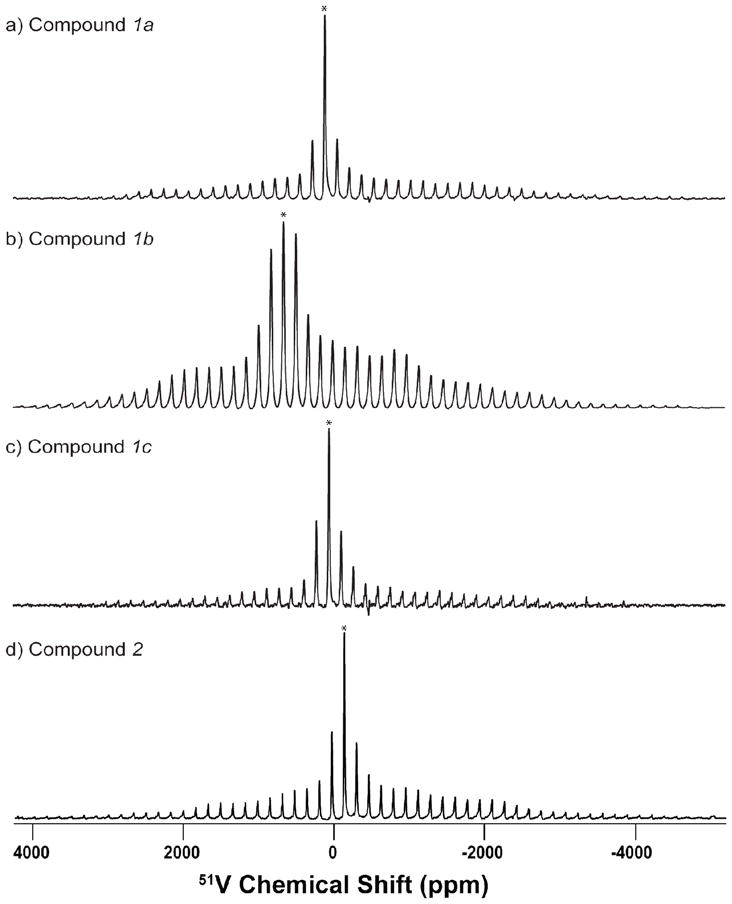
51V solid-state NMR spectra of the four vanadium(V)-o-dioxolene compounds of the series VVO-hshed (1a – 1c) (a–c) and [VO(acac)(TCCat)] (2) (d) obtained at a magnetic field of 9.4 T with MAS frequency of 17 kHz. 8192 scans were accumulated for each spectrum, and the pulse delay was 1.0 s. Experimental spectra are shown in black and best-fit simulated spectra are shown in red. The simulated spectra were obtained from SIMPSON using the NMR parameters listed in Table 1.
Figure 6.
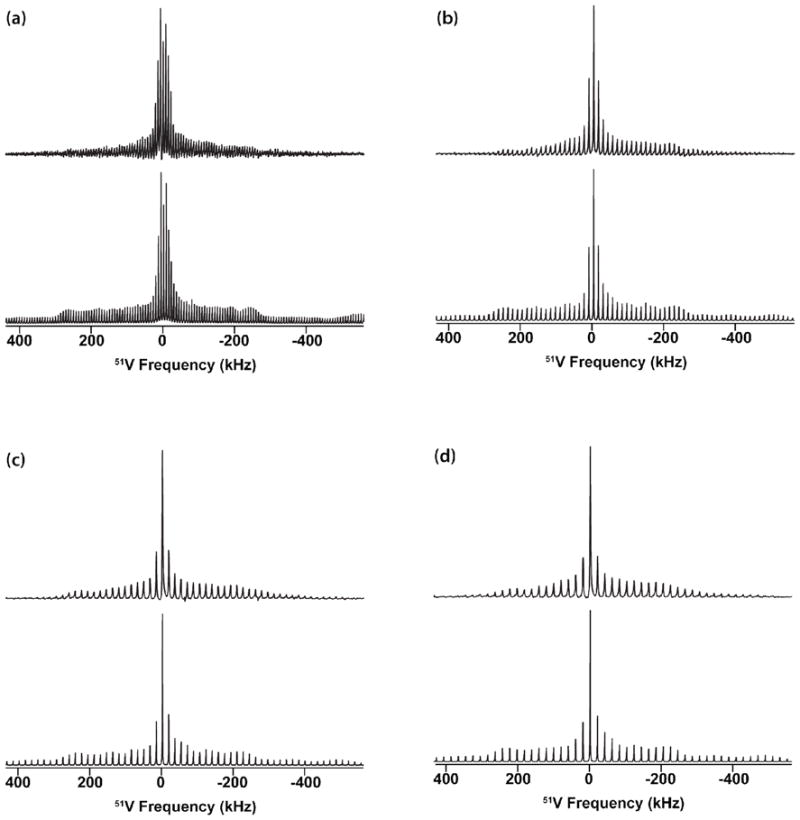
Experimental (top) and simulated (bottom) 51V solid-state NMR spectra of [VO(hshed)(Cat)] acquired at the MAS frequencies of (a) 7 kHz, (b) 13 kHz, (c) 17 kHz and (d) 20 kHz. The spectra were simulated with the following parameters: CQ = 4.0 ± 0.1 MHz; δσ= −243 ± 30 ppm; ηQ = 1.0 ± 0.05; ησ = 0.93 ± 0.05; α = 81 ± 10; β= 70 ± 15; γ = 87 ± 15.
Analysis of the results presented in Table 1 indicates that, quite surprisingly, the quadrupolar coupling constants for the four complexes are relatively small and cover a very narrow range of 3.4 – 4.2 MHz. On the other hand, a very wide 51V isotropic chemical shift range is observed in these compounds and is similar to the solution NMR findings. Complex 1a exhibits an isotropic chemical shift value at 58 ppm, complex 1b at 531 ppm, 1c at −1 ppm, and 2 at −219 ppm. These results show that the non-innocent characteristic of these complexes is reflected in their isotropic solid-state chemical shifts lying outside of the more common region of −300 to −700 ppm. Interestingly, the chemical shift anisotropy, δσ, does not vary to as large extent as the isotropic shifts and is in the range of 243 to 437 ppm for these complexes. Analysis of the individual CSA tensor components, δ11, δ22, and δ33, indicates that all three tensor components vary dramatically with the series of complexes under investigation warranting further analysis of the individual orbital contributions to the corresponding magnetic shielding anisotropy tensors.
The isotropic chemical shifts for compounds 1a - 1c, 2 and other known vanadium(V) complexes were plotted as a function of solution chemical shifts (Figure 7).26–38,49 The linear relationship shows that the expected correlation between solid state (δiso) and solution isotropic chemical shifts (δ) generally holds although significantly more scatter in the current data is observed for these non-innocent complexes.
Figure 7.
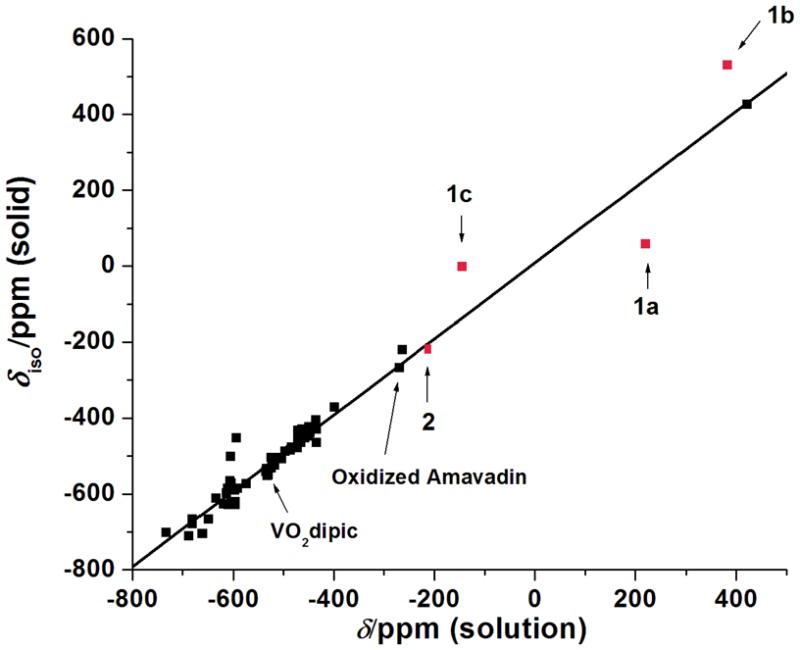
Plot of the solution 51V NMR isotropic chemical shifts vs. experimentally obtained solid-state NMR chemical shifts for all the compounds reported hitherto in the literature along with the results reported in this study establishing a linear correlation between the different methods of investigations; solid state versus solution.
51V NMR Parameters of VO(hshed)(Cat) Complex (1a): Density Functional Theory
To understand the experimental 51V solid-state NMR parameters we have conducted DFT calculations of the EFG and CSA tensors for the only structurally characterized compound from the current series under study, VO(hshed)(Cat) (compound 1a). DFT calculations of the 51V chemical shifts have already been reported for this compound.66 However, the EFG tensor has not been computed and, furthermore, prior to our work only the isotropic solution chemical shift was determined. In Table 2 the results of the DFT calculations conducted at different levels of theory are summarized. Similar to the previous report,66 the agreement between the experimental and calculated isotropic chemical shifts is rather poor for most of the methods used with the exception of the calculations performed with the Wachters+f basis set on vanadium atoms. In the latter case, the computed and experimental isotropic shifts agree remarkably well (within 3.7 ppm). Moreover, in this calculation, the principal components of the computed CSA tensor are also in reasonable agreement with the experiment (δσ agrees to within 90 ppm, and ησ–to within 0.04) and the computed values are similar to the previously reported calculations conducted at a comparable level of theory.66
On the other hand, the calculated quadrupolar coupling constant deviates significantly from the experimental value of 4.0 MHz and for the different basis sets the CQ ranges between 7.0 and 10.7 MHz. When the Wachters+f basis set is used on vanadium atoms (this level of theory yields the best agreement between experiment and theory for the CSA tensor), CQ is 9.9 MHz. In our previous studies of multiple V(V) complexes with various mainly innocent ligands,27–29,36,67 DFT calculations accurately predicted the experimentally observed quadrupolar coupling constants with the experimental/calculated CQ ratios (never exceeding 30% and typically within 5–15%). Therefore, the large discrepancies in CQ between the experiment and calculation observed in this study are surprising.
Discussion
The potential of using magic angle spinning (MAS) 51V SSNMR spectroscopy to evaluate the electronic structure of metal complexes with redox active ligands was investigated. Assessment of the interactions between vanadium(V) and a large number of ligands has been done using 51V SSNMR spectroscopy.27–29,36,37,67 51V SSNMR analysis has provided detailed electronic information regarding the active site of haloperoxidases and its catalytic mechanism.37 Recently we found two nonoxovanadium(V) compounds with unusual solution chemical shifts (ca. −260 ppm) for which we gained additional insight from solid-state NMR analysis of 51V EFG and CSA tensors.67 Despite multiple reports, many important and ubiquitous classes of vanadium(V) compounds have not yet been examined by 51V SSNMR spectroscopy. As demonstrated in this work, analysis of the 51V SSNMR observables such as quadrupolar and chemical shift anisotropy provides fundamental information about the electronic structure of these systems as well as documenting a less traditional method for gaining information on these challenging systems.
The ternary vanadium(V) catechol complexes (1a – 1c, 2) examined in this work contain one coligand in addition to one catechol moiety and, as a result, have unusual electronic properties. Solution NMR spectroscopy of non-innocent vanadium complexes has been reported;16 however, no generalized understanding on the relationships between different NMR parameters and the electronic structure in such compounds emerged to date. Bryliakov et al.74 have shown how one can utilize 51V NMR spectroscopy for characterizing the intermediates in vanadium-catalyzed oxidation reactions. Furthermore, a linear correlation has been demonstrated between isotropic chemical shifts in the solid state (δiso) and in solution (δ) of vanadium complexes formed from innocent ligands. The 51V NMR chemical shifts for these complexes are in the range of −300 ppm to −700 ppm.18,38,67 However, unusual electronic properties have been found in complexes formed from redox non-innocent ligands such as catechols16,75 and hydroxylamines.67 In these systems, strong de-shielding of the metal nucleus is observed with the chemical shift range dramatically extending outside the −300 ppm to −700 ppm region which is a manifestation of the significant changes in the electronic properties of these complexes.
According to the experimental results, the vanadium(V) catechol complexes (1a – 1c, 2) under investigation exhibit relatively small quadrupolar coupling constants in the range between 3.4 and 4.2 MHz. Similarly small quadrupolar coupling constants (ranging between 3.0 and 3.9 MHz) were observed in complexes of heptacoordinate geometry with dipicolinic acid ligands.28 In comparison, hexacoordinate compounds typically show somewhat larger quadrupolar coupling constants, in the range of 3.0–6.3 MHz.27,33,34,36,38 Pentacoordinate and octacoordinate complexes typically exhibit even greater quadrupolar coupling constants, 4.3–8.3 MHz.27,33,34,36,38,67 In vanadium haloperoxidases, quadrupolar coupling constants of the vanadium site are even larger. In the resting state of vanadium chloroperoxidase (VCPO) at pH 8.0, CQ = 10.5 MHz37 while CQ ranges between 13 and 17 MHz in the VCPO resting state at pH 6.3 and in its two mutants.(S. Bolte, K. Ooms, R. Renirie, R. Wever, and T. Polenova, unpublished results) Therefore, the experimental results documenting small similar quadrupolar coupling constants in catechol complexes are somewhat surprising in light of the fact that the coordination geometries between 1a-1c and 2 differ; 1a-1c are hexacoordinate while 2 is a pentacoordinate complex. We also note that, surprisingly, the DFT calculations predict the quadrupolar coupling constant for compound 1a to be much greater than the experimental value and ranging between 7.0 and 10.7 MHz depending on the basis set employed (see the Results section). The current experimental results in conjunction with the reports cited above indicate that in the oxovanadium(V)-catecholato complexes under investigation the electronic charge distribution is unexpectedly similar and relatively symmetric across the series. The electronic charge distribution in oxovanadium(V)hydroxylamido complexes examined previously were similar28 where quadrupolar interaction parameters were small but measurable by changing functionality on the ligands. On the contrary, the chemical shift anisotropy tensor elements as well as the isotropic chemical shifts in solution and in the solid state span a very broad range between most systems investigated and compound SJZ0010836 (Figure 1). The unusual electronic properties in the vanadium(V) catechol complexes are therefore appropriately reflected in the observed chemical shifts (Figure 7).
Isotropic 51V NMR chemical shifts for the selected compounds cover a very broad range from −200 to 400 ppm in solution and from −219 to 530 ppm in the solid state. These experimental results provide the information needed for the application of 51V SSNMR parameters in characterizing the electronic properties of systems containing non-innocent ligands and that have chemical shifts outside the range of −300 ppm to −700 ppm where no data were available before this work (Figure 2). Solution and solid-state isotropic chemical shifts exhibit linear correlation for the compounds under study thereby extending the known linear correlation for vanadium complexes (Figure 7) and thus documenting that the solution and solid-state structure are the same. On the other hand, there is no relationship between isotropic chemical shift (either solution or solid state) and the chemical shift anisotropy, which is due to the fact that the individual CSA tensor values (δ11, δ22, and δ33) vary widely within this series of complexes (see Table 1). All of the CSA tensors are rhombic and it is also noteworthy that Euler angles describing the relative orientations of the CSA tensors with respect to the EFG tensors also vary within the series (except in 1c and 2, where both tensors are collinear).
The above variations in the 51V CSA parameters are not surprising because there are significant differences in the electronic properties within the series of complexes under study. To understand the nature of the contributions of the individual MOs to the CSA tensor and to the electronic charge distribution that defines the EFG tensor in these compounds, quantum chemical calculations of the NMR parameters were done of the structurally characterized complex is VO(hshed)(Cat) (compound 1a). Our results discussed above and summarized in Table 2 indicate that while the experimental and calculated isotropic chemical shifts are in excellent agreement, the reduced anisotropy δσ is only predicted to within 90 ppm using Wachters+f basis set on vanadium. These findings are in line with the previous report where DFT calculations evaluated the chemical shifts of this and other vanadium catechol complexes.66 This limited agreement between the experimental and theoretical chemical shifts in 51V(V) complexes with non-innocent ligands has been attributed to several possible factors:66 i) that the paramagnetic contributions in the closed-shell wavefunctions are severely underestimated; ii) that a temperature-dependent paramagnetic term arises from non-zero spin density at the metal due to the mixing of V(IV)-semiquinone configurations into the electronic ground state. In addition, the contributions of the frontier molecular orbitals (HOMO and LUMO) in VO(hshed)(Cat) to the 51V magnetic shieldings were analyzed and it was demonstrated, that the “non-innocent” ligand character resulting in strong deshielding is due not only to the bidentate catechol ligand but also to the coligand tridentate salicylideneaminato Schiff base,66 which is consistent with an earlier work by Pecoraro.16 The X-ray crystal structures for the other compounds are needed before detailed quantum chemical analyses of compounds 1b, 1c, and 2 other complexes based on their NMR parameters can be carried out.
Substitution of the catechol moiety induces small electronic changes in the complex and for vanadium catechol complexes should result in measurable differences in the HOMO-LUMO gap as illustrated in Figure 3. How the electronic properties of the complexes are represented in their NMR spectra can be understood in a qualitative manner by an empirical analysis of the orbital contributions to chemical shifts. According to Ramsey’s equation,76 the shielding, σ, at the NMR active nucleus is defined as the sum of the generally positive diamagnetic (σdia) component and a paramagnetic (σpara) component (Equation 1):
| (1) |
For the majority of metal-containing complexes, the σdia term is controlled by the core electrons and thus constant for a given metal nucleus.16,28 The donor atoms of the ligand(s) and the valence molecular orbital(s) of the metal therefore do not contribute significantly to σdia, but instead tune the σpara term and thus the shielding.77–80 The σpara term is dependent on the average mixing between the symmetry matched excited and ground states of the molecule in the presence of an applied magnetic field,16,28 which in turn is determined by the relative energy separation (ΔE) between the HOMO and the LUMO in these complexes. In fact, σpara varies inversely with ΔE and it also contains a <r−3> term indicating that only the orbitals near the metal center need to be considered.16,28 The overlap of the ligand’s valence π-orbitals and the metal’s d-orbitals controls the degree of mixing between them, which eventually directs the electronic delocalization.79–80 Indeed, the extent of ligand-to-metal charge transfer (LMCT) controls the electron density redistribution surrounding the central nucleus16 and is readily modified using substitutions on the catechol.66 The oxovanadium(V)catecholato complexes 1a – 1c and 2 under study possess variable electronic properties of the nucleus, which can be probed by changes in Equation (1)’s σpara term.
The small energy gap between the HOMO and the LUMO in vanadium catechol complexes makes these systems ideally suited for the analysis of electronic effects using 51V SSNMR spectroscopy. Electronic excitation in free catechol is attributed to the transition from π HOMO to π* LUMO61,66 but this is not observed in its vanadium complexes (Figure 3). In complexes, the predominantly vanadium-based LUMO is lower in energy than the ligand-based LUMO for 1a-1c. The non-innocent nature of these ligands results in little separation between the HOMO and the LUMO in these complexes as well as in a few vanadium(V)-sulfur complexes.26 This is in contrast to other vanadium(V) compounds without any redox-active ligand(s) which exhibit high excitation energy (Figure 3). Empirical molecular orbital methods were used for several heavy metals e.g. 51V, 95Mo and 183W to examine the experimental observations made in solution state.16,81 These calculations revealed that the small HOMO-LUMO gap and the effect on σpara could explain the downfield chemical shifts.16,81 Complexes investigated previously have upfield chemical shifts (from −300 to −700 ppm) with the exception of SJZ0010836 (Figure 1). Since insufficient information is available for compounds with chemical shifts outside the range −300 to −700 ppm and since the coordination of catechol gives rise to the 51V NMR isotropic chemical shifts in the range between −145 and +382 ppm, we took the opportunity to explore the moderate-to-large deshielding at the vanadium center in the o-dioxolene complexes (1a–1c).
51V NMR isotropic chemical shifts as well as the principal components of the CSA tensor in compounds 1a–1c changed drastically upon modification of the o-dioxolene ligand indicating considerable change of the HOMO-LUMO energy gap. Compound 1a, δ(solution) = 221 ppm/δiso(solid) = 58 ppm, is halfway between that in the V(V) complexes examined previously and complex SJZ0010836 (Figure 1). Introduction of tertiary butyl groups to the catechol ring (1b) results in electron redistribution within the complex increasing the energies of both HOMO and LUMO in 1b. Since the HOMO increases more in energy than the LUMO, the HOMO-LUMO gap is decreased (Figure 3) which is directly observed as a downfield shift for 1b (δ(solution) = 382 ppm/δiso(solid) = 531 ppm). In contrast, as illustrated in Figure 3, bromo substitution on the catechol ring increases the HOMO-LUMO gap leading to an upfield shift in 1c (δ(solution) = −145 ppm/δiso(solid) = −1 ppm). These three compounds are the first example of complexes with 51V chemical shifts in this range. Furthermore, the dramatic variation in chemical shifts with subtle substitutions on the ligand can be rationalized as the tuning of the HOMO-LUMO gap. In contrast, for other series of known V(V) complexes, little change in isotropic chemical shifts both in solution and in the solid state was observed upon substitution on the ligand.27,28 In those cases local electronic environments surrounding the vanadium site are similar.
An alternative approach to obtain compounds with similar electronic properties is to combine a non-innocent ligand with another redox-inactive oxygen-rich co-ligand. By changing the supporting ligand donor sites from tridentate (ONN) (as in 1a-1c) to a bidentate (OO) acetylacetonate (2), we are introducing an additional oxygen donor to the vanadium metal center. Changing the nitrogen atom from the ternary ligand (as in 1a–1c) by a harder donor atom, oxygen, stabilizes the energy level of HOMO. This results in a larger HOMO and LUMO gap in compound (2), which is reflected in the observed upfield chemical shift (−215 ppm in solution and −219 ppm in the solid state, respectively). In contrast, replacing bromo- substituents by chloro- on catechol moiety has very little influence on 51V NMR chemical shifts as documented earlier by Pecoraro et al.16 and consistent with minor changes in the HOMO-LUMO gap.
Conclusions
We have examined a series of four vanadium(V) catechol complexes by 51V solid-state NMR spectroscopy. The objective with this work was to address whether SSNMR observables, quadrupolar and chemical shift anisotropy tensors, can effectively describe the electronic properties of the metal complexes containing redox non-innocent ligands and those isotropic chemical shifts are in the ranges not explored previously. Because 51V is a sensitive NMR-active nucleus, we used vanadium(V)-catechol complexes for these studies. In addition to the SSNMR studies, one complex that was characterized structurally by X-ray crystallography was also examined used DFT calculations of the NMR parameters.
We discovered that the 51V quadrupolar coupling constants are surprisingly small and similar, indicating a relatively symmetric electronic charge distribution across the series. To the contrary, the isotropic chemical shifts and the chemical shift anisotropy parameters cover a broad range and extend the scale of chemical shifts beyond the commonly observed values for vanadium complexes with normal ligands. The observed effects can be attributed to changes in the HOMO-LUMO energy gaps, which are tuned by the nature of the substituent on the catechol ligand. After complexation, the HOMO-LUMO gap of the ligand decreases. When the catechol is substituted with electron-donating groups, the HOMO-LUMO gap decreases, and the corresponding chemical shift for the complex is downfield from the vanadium-catechol complex. When the catechol is substituted with an electron-withdrawing group, the HOMO –LUMO gap increases, which is reflected in an upfield shift. These findings contribute to the generalized understanding of the relationships between the NMR parameters and the electronic structure in metal complexes.
Supplementary Material
Acknowledgments
D. C. C. acknowledges financial support of the National Science Foundation (CHE-0533189 and CHE-0750079). T. P. acknowledges financial support of the National Science Foundation (CHE-0750079) and the National Institutes of Health (P30RR031160 from NCRR).
Footnotes
51V solid-state experimental and simulated solid-state NMR spectra (9.4 T) of the three vanadium(V)-o-dioxolene compounds of the series VVO-hshed (1b – 1c) and [VO(acac)(TCCat)] (2) acquired at the MAS rates of 13, 17 and 20 kHz. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Jorgensen CK. Coord Chem Rev. 1966;1:15. [Google Scholar]
- 2.Costas M, Mehn MP, Jensen MP, Que L. Chem Rev. 2004;104:939. doi: 10.1021/cr020628n. [DOI] [PubMed] [Google Scholar]
- 3.Thorstensen K, Romslo I. Biochem J. 1990;271:1. doi: 10.1042/bj2710001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Klabunde T, Krebs B. Struct Bond. 1997;89:177. [Google Scholar]
- 5.Wang Y, DuBois JL, Hedman B, Hodgson KO, Stack TD. Science. 1998;279:537. doi: 10.1126/science.279.5350.537. [DOI] [PubMed] [Google Scholar]
- 6.Mukhopadhyay S, Mandal SK, Bhaduri S, Armstrong WH. Chem Rev. 2004;104:3981. doi: 10.1021/cr0206014. [DOI] [PubMed] [Google Scholar]
- 7.Kaim W, Schwederski B. Coord Chem Rev. 2010;254:1580. [Google Scholar]
- 8.Ward MD, McCleverty JA. J Chem Soc Dalton Trans. 2002:275. [Google Scholar]
- 9.Pierpont CG. Coord Chem Rev. 2001;216:99. [Google Scholar]
- 10.Pierpont CG, Lange CW. Prog Inorg Chem. 1994;41:331. [Google Scholar]
- 11.Wang PG, Xian M, Tang XP, Wu XJ, Wen Z, Cai TW, Janczuk AJ. Chem Rev. 2002;102:1091. doi: 10.1021/cr000040l. [DOI] [PubMed] [Google Scholar]
- 12.Cass ME, Gordon NR, Pierpont CG. Inorg Chem. 1986;25:3962. [Google Scholar]
- 13.Cass ME, Greene DL, Buchanan RM, Pierpont CG. J Am Chem Soc. 1983;105:2680. [Google Scholar]
- 14.Chang HC, Kitagawa S. Angew Chem Int Ed. 2002;41:130. doi: 10.1002/1521-3773(20020104)41:1<130::aid-anie130>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 15.Chatterjee PB, Bhattacharya K, Kundu N, Choi KY, Clerac R, Chaudhury M. Inorg Chem. 2009;48:804. doi: 10.1021/ic801985g. [DOI] [PubMed] [Google Scholar]
- 16.Cornman CR, Colpas GJ, Hoeschele JD, Kampf J, Pecoraro VL. J Am Chem Soc. 1992;114:9925. [Google Scholar]
- 17.Attia AS, Pierpont CG. Inorg Chem. 1998;37:3051. [Google Scholar]
- 18.Lapina OB, Khabibulin DF, Shubin AA, Terskikh VV. Prog Nucl Magn Res Spec. 2008;53:128. [Google Scholar]
- 19.Plevin MJ, Bryce DL, Boisbouvier J. Nat Chem. 2010;2:466. doi: 10.1038/nchem.650. [DOI] [PubMed] [Google Scholar]
- 20.Tran TT, Herfort D, Jakobsen HJ, Skibsted J. J Am Chem Soc. 2009;131:14170. doi: 10.1021/ja905223d. [DOI] [PubMed] [Google Scholar]
- 21.Ellis PD, Lipton AS. Ann Rep NMR Spec. 2007;60:1. [Google Scholar]
- 22.Rovnyak D, Baldus M, Wu G, Hud NV, Feigon J, Griffin RG. J Am Chem Soc. 2000;122:11423. [Google Scholar]
- 23.Weiss JWE, Bryce DL. J Phys Chem A. 2010;114:5119. doi: 10.1021/jp101416k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bryce DL. Dalton Trans. 2010;39:8593. doi: 10.1039/c0dt00416b. [DOI] [PubMed] [Google Scholar]
- 25.Brown C, Achey R, Fu RQ, Gedris T, Stiegman AE. J Am Chem Soc. 2005;127:11590. doi: 10.1021/ja052579c. [DOI] [PubMed] [Google Scholar]
- 26.Rehder D, Polenova T, Buhl M. Ann Rep NMR Spec. 2007;62:49. [Google Scholar]
- 27.Bolte SE, Ooms KJ, Polenova T, Baruah B, Crans DC, Smee JJ. J Chem Phys. 2008:128. doi: 10.1063/1.2830239. [DOI] [PubMed] [Google Scholar]
- 28.Ooms KJ, Bolte SE, Smee JJ, Baruah B, Crans DC, Polenova T. Inorg Chem. 2007;46:9285. doi: 10.1021/ic7012667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Smee JJ, Epps JA, Ooms K, Bolte SE, Polenova T, Baruah B, Yang LQ, Ding WJ, Li M, Willsky GR, la Cour A, Anderson OP, Crans DC. J Inorg Biochem. 2009;103:575. doi: 10.1016/j.jinorgbio.2008.12.015. [DOI] [PubMed] [Google Scholar]
- 30.Huang WL, Todaro L, Francesconi LC, Polenova T. J Am Chem Soc. 2003;125:5928. doi: 10.1021/ja029246p. [DOI] [PubMed] [Google Scholar]
- 31.Huang WL, Todaro L, Yap GPA, Beer R, Francesconi LC, Polenova T. J Am Chem Soc. 2004;126:11564. doi: 10.1021/ja0475499. [DOI] [PubMed] [Google Scholar]
- 32.Chen L, Kaiser JM, Polenova T, Yang J, Rienstra CM, Mueller LJ. J Am Chem Soc. 2007;129:10650. doi: 10.1021/ja073498e. [DOI] [PubMed] [Google Scholar]
- 33.Nica S, Buchholz A, Rudolph M, Schweitzer A, Waechtler M, Breitzke H, Buntkowsky G, Plass W. Eur J Inorg Chem. 2008:2350. [Google Scholar]
- 34.Gutmann T, Schweitzer A, Wachtler M, Breitzke H, Blichholz A, Plass W, Buntkowsky G. Phys Chem Chem Phys. 2008;222:1389. [Google Scholar]
- 35.Schweitzer A, Gutmann T, Wachtler M, Breitzke H, Buchholz A, Plass W, Buntkowsky G. Solid State Nucl Mag Resn. 2008;34:52. doi: 10.1016/j.ssnmr.2008.02.003. [DOI] [PubMed] [Google Scholar]
- 36.Pooransingh N, Pomerantseva E, Ebel M, Jantzen S, Rehder D, Polenova T. Inorg Chem. 2003;42:1256. doi: 10.1021/ic026141e. [DOI] [PubMed] [Google Scholar]
- 37.Pooransingh-Margolis N, Renirie R, Hasan Z, Wever R, Vega AJ, Polenova T. J Am Chem Soc. 2006;128:5190. doi: 10.1021/ja060480f. [DOI] [PubMed] [Google Scholar]
- 38.Fenn A, Wachtler M, Gutmann T, Breitzke H, Buchholz A, Lippold I, Plass W, Buntkowsky G. Solid State Nucl Mag Resn. 2009;36:192. doi: 10.1016/j.ssnmr.2009.11.003. [DOI] [PubMed] [Google Scholar]
- 39.Skibsted J, Nielsen NC, Bildsoe H, Jakobsen HJ. J Am Chem Soc. 1993;115:7351. [Google Scholar]
- 40.Lapina OB, Khabibulin DF, Shubin AA, Bondareva VM. J Mol Cat Chem. 2000;162:381. [Google Scholar]
- 41.Skibsted J, Jacobsen CJH, Jakobsen HJ. Inorg Chem. 1998;37:3083. [Google Scholar]
- 42.Buhl M. Angew Chem Int Ed. 1998;37:142. [Google Scholar]
- 43.Spengler J, Anderle F, Bosch E, Grasselli RK, Pillep B, Behrens P, Lapina OB, Shubin AA, Eberle HJ, Knozinger H. J Phys Chem B. 2001;105:10772. [Google Scholar]
- 44.Jenkins JE, Creager MS, Lewis RV, Holland GP, Yarger JL. Biomacromolecules. 2010;11:192. doi: 10.1021/bm9010672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Holland GP, Cherry BR, Jenkins JE, Yarger JL. J Mag Resn. 2010;202:64. doi: 10.1016/j.jmr.2009.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Creager MS, Jenkins JE, Thagard-Yeaman LA, Brooks AE, Jones JA, Lewis RV, Holland GP, Yarger JL. Biomacromolecules. 2010;11:2039. doi: 10.1021/bm100399x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Butler A, Danzitz MJ, Eckert H. J Am Chem Soc. 1987;109:1864. [Google Scholar]
- 48.Vilter H, Rehder D. Inorg Chim Acta Bioinorg Chem. 1987;136:L7. [Google Scholar]
- 49.Crans DC, Smee JJ, Gaidamauskas E, Yang LQ. Chem Rev. 2004;104:849. doi: 10.1021/cr020607t. [DOI] [PubMed] [Google Scholar]
- 50.Butler A, Walker JV. Chem Rev. 1993;93:1937. [Google Scholar]
- 51.Rehder D, Holst H, Priebsch W, Vilter H. J Inorg Biochem. 1991;41:171. [Google Scholar]
- 52.Cornman CR, Kampf J, Pecoraro VL. Inorg Chem. 1992;31:1981. [Google Scholar]
- 53.Pierpont CG, Attia AS. Collect Cze Chem Commun. 2001;66:33. [Google Scholar]
- 54.Chatterjee PB, Abtab SMT, Bhattacharya K, Endo A, Shotton EJ, Teat SJ, Chaudhury M. Inorg Chem. 2008;47:8830. doi: 10.1021/ic800815p. [DOI] [PubMed] [Google Scholar]
- 55.Cooper SR, Koh YB, Raymond KN. J Am Chem Soc. 1982;104:5092. [Google Scholar]
- 56.Cornman CR, Kampf J, Lah MS, Pecoraro VL. Inorg Chem. 1992;31:2035. [Google Scholar]
- 57.Galeffi B, Postel M, Grand A, Rey P. Inorg Chim Acta. 1987;129:1. [Google Scholar]
- 58.Kabanos TA, White AJP, Williams DJ, Woollins JD. J Chem Soc Chem Commun. 1992:17. [Google Scholar]
- 59.Kabanos TA, Slawin AMZ, Williams DJ, Woollins JD. J Chem Soc Dalton Trans. 1992:1423. [Google Scholar]
- 60.Persson P, Bergstrom R, Lunell S. J Phys Chem B. 2000;104:10348. [Google Scholar]
- 61.Duncan WR, Prezhdo OV. J Phys Chem B. 2005;109:365. doi: 10.1021/jp046342z. [DOI] [PubMed] [Google Scholar]
- 62.Yin CX, Finke RG. J Am Chem Soc. 2005;127:13988. doi: 10.1021/ja052998+. [DOI] [PubMed] [Google Scholar]
- 63.Sanchez-de-Armas R, San-Miguel MA, Oviedo J, Marquez A, Sanz JF. Phys Chem Chem Phys. 2011;13:1506. doi: 10.1039/c0cp00906g. [DOI] [PubMed] [Google Scholar]
- 64.Li SC, Wang JG, Jacobson P, Gong XQ, Selloni A, Diebold U. J Am Chem Soc. 2009;131:980. doi: 10.1021/ja803595u. [DOI] [PubMed] [Google Scholar]
- 65.Creutz C, Chou MH. Inorg Chem. 2008;47:3509. doi: 10.1021/ic701687k. [DOI] [PubMed] [Google Scholar]
- 66.Geethalakshmi KR, Waller MP, Buhl M. Inorg Chem. 2007;46:11297. doi: 10.1021/ic701650y. [DOI] [PubMed] [Google Scholar]
- 67.Ooms KJ, Bolte SE, Baruah B, Choudhary MA, Crans DC, Polenova T. Dalton Trans. 2009:3262. doi: 10.1039/b820383k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhang Y, Gascon JA. J Inorg Biochem. 2008;102:1684. doi: 10.1016/j.jinorgbio.2008.04.006. [DOI] [PubMed] [Google Scholar]
- 69.Schneider CJ, Zampella G, Greco C, Pecoraro VL, De Gioia L. Eur J Inorg Chem. 2007:515. [Google Scholar]
- 70.Zampella G, Fantucci P, Pecoraro VL, De Gioia L. Inorg Chem. 2006;45:7133. doi: 10.1021/ic060555g. [DOI] [PubMed] [Google Scholar]
- 71.Harris RK, Becker ED, De Menezes SMC, Goodfellow R, Granger P. Pure Appl Chem. 2001;73:1795. [Google Scholar]
- 72.Bak M, Rasmussen JT, Nielsen TENC. J Magn Reson. 2000;147:296. doi: 10.1006/jmre.2000.2179. [DOI] [PubMed] [Google Scholar]
- 73.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Montgomery JA, Jr, Vreven T, Kudin KN, Burant JC, Millam JM, Iyengar SS, Tomasi J, Barone V, Mennucci B, Cossi M, Scalmani G, Rega N, Petersson GA, Nakatsuji H, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Klene M, Li X, Knox JE, Hratchian HP, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Ayala PY, Morokuma K, Voth GA, Salvador P, Dannenberg JJ, Zakrzewski VG, Dapprich S, Daniels AD, Strain MC, Farkas O, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Ortiz JV, Cui Q, Baboul AG, Clifford S, Cioslowski J, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Martin RL, Fox DJ, Keith T, Al-Laham MA, Peng CY, Nanayakkara A, Challacombe M, Gill PMW, Johnson B, Chen W, Wong MW, Gonzalez C, Pople JA. Gaussian 03, revision C02. Gaussian, Inc; Wallingford, CT: 2004. [Google Scholar]
- 74.Bryliakov KP, Karpyshev NN, Fominsky SA, Tolstikov AG, Talsi EP. J Mol Cat Chem. 2001;171:73. [Google Scholar]
- 75.Frank P, Robinson WE, Kustin K, Hodgson KO. J Inorg Biochem. 2001;86:635. doi: 10.1016/s0162-0134(01)00231-8. [DOI] [PubMed] [Google Scholar]
- 76.Ramsey NF. Phys Rev. 1950;78:699. [Google Scholar]
- 77.Kanda K, Nakatsuji H, Yonezawa T. J Am Chem Soc. 1984;106:5888. [Google Scholar]
- 78.Nakatsuji H, Kanda K, Endo K, Yonezawa T. J Am Chem Soc. 1984;106:4653. [Google Scholar]
- 79.Schreckenbach G. J Chem Phys. 1999;110:11936. [Google Scholar]
- 80.Wilson PJ, Amos RD, Handy NC. Phys Chem Chem Phys. 2000;2:187. [Google Scholar]
- 81.Mondal JU, Schultz FA, Brennan TD, Scheidt WR. Inorg Chem. 1988;27:3950. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.