

Abstract
Cystinosis as a clinical entity is a progressive dysfunction of multiple organs caused by the accumulation of cystine in the tissues, leading, for example, to end-stage renal failure, diabetes, hypothyroidism, myopathy, and central nervous system deterioration. Brodin-Sartorius and colleagues present a long-term study on the impact of cysteamine therapy on these complications. The data show that cysteamine improves the outcome and complications of cystinosis but does not prevent them.
Cystinosis as a clinical entity is a progressive dysfunction of multiple organs caused by the accumulation of cystine in the lysosomes of all the cells in the body due to mutations in the ubiquitous gene CTNS, encoding the lysosomal cystine transporter protein cystinosin.1 The most severe and most frequent form of cystinosis is the infantile form, also called nephropathic cystinosis. Children consistently develop the syndrome of De Toni–Debré–Fanconi at 6–8 months of age, characterized by a severe fluid and electrolyte disturbance, growth retardation, and rickets.2 Corneal cystine crystals are also a constant symptom; they appear during the first decade of life, resulting in photophobia and visual impairment. The progressive cystine storage also leads to kidney failure, diabetes, myopathy, hypothyroidism, hypogonadism, and central nervous system deterioration. However, the extent and pace of these defects vary among patients.
Cysteamine, which reduces the intralysosomal cystine concentration, was first used in the treatment of cystinosis in 1976 and was approved by the US Food and Drug Administration in 1994. In cultured cells accumulating cystine and in patients’ white blood cells, it undisputedly clears the cystine very efficiently.3 However, cysteamine does not prevent the Fanconi syndrome even if started very early in life. Despite this, most publications on its impact on the late complications of cystinosis have been very optimistic and have suggested that cysteamine may prevent organ damage.4,5 Thus, some believed that this disease was cured. If complications were occurring in patients under treatment, either poor compliance with the treatment or the age at which the patients started the treatment was blamed. Yes, both of these have an impact on cysteamine therapy, as Brodin-Sartorius and colleagues (this issue) show.6 However, they also show that patients who started cysteamine therapy early in life and demonstrated good compliance still developed late complications. The new reality is that cysteamine delays the disease’s progression but does not cure it. End-stage renal failure, as well as the other complications, is still occurring. Thus, publications such as the one by Brodin-Sartorius and colleagues6 are important in demonstrating that cystinosis is still in need of new therapies.
The study presented by Brodin-Sartorius and colleagues 6 included 86 cystinosis patients and clearly showed that end-stage renal disease (ESRD) was delayed in some patients who started cysteamine before 5 years of age, but also that the majority of them still developed ESRD before the age of 15 years, the mean age at the start of ESRD being 13.4±4.8. According to the 2010 and 2011 annual reports of North American Pediatric Renal Trials and Collaborative Studies, 1.4% of children on dialysis and 2.1 % with kidney transplants have cystinosis. 7 Both dialysis and transplantation have significant negative health effects, and because of the severe shortage of donor organs, patients may wait three to six years for transplantation. Thus, the mortality and morbidity associated with renal failure are high enough to consider cystinosis uncured. Similarly, according to Brodin-Sartorius and colleagues’ study, 6 hypothyroidism appeared by 15 years of age even when cysteamine was started before 5 years of age. Neither the age of occurrence of neuromuscular disorders nor that of diabetes was delayed when cysteamine was taken before 5 years of age, but both occurred in fewer patients at the time of the study. Another comprehensive study on the long-term impact of cysteamine was performed by Gahl and colleagues on 100 individuals with nephropathic cystinosis, examined between 1985 and 2006.4 Their study showed a decrease in the frequency of symptoms with increased time on cysteamine. But still, 92% of the patients in their cohort had received a kidney transplant, 75% of patients had hypothyroidism, 69% had pulmonary dysfunction, 60% had a swallowing abnormality, and 50% had myopathy. Also, young people with cystinosis are still dying; the cause of death is not well characterized and would necessitate a comprehensive study. In the study by Brodin-Sartorius and colleagues, 6 27.9% of patients died, at a mean age of 26.8; the cause of death was infection or neuromuscular complications such as respiratory distress or swallowing impairment. In Gahl and colleagues’ study, 4 one-third of the patients died, at a mean age of 28.5 years. In addition to the causes of death already mentioned, this study also attributed death to renal complications and gastrointestinal damage.4
What is life like with cystinosis? In meeting families with cystinosis, we realized that this is an everyday fight, that cystinosis patients must take not only cysteamine every 6 h, but also many other drugs and supplements around the clock every day. Many of the children need a gastric tube to absorb and tolerate all these medications. And still they develop many complications and have frequent hospital visits. The question is, how we can gain a better idea of the status of health in patients taking cysteamine? Publications on a large cohort of patients such as Brodin-Sartorius and colleagues’, 6 recapitulating years of medical follow-up, are important. The major problem with this study is that these data are based on reviews of medical records, and the mean age at the start of the therapy was 9.9 years, much later than most cystinosis patients start cysteamine today. Thus, a historical review might influence negatively the data reported. How can current data be collected and reported? The new Cure Cystinosis International Registry (CCIR) could be a useful tool for this purpose. The CCIR uses a comprehensive questionnaire filled out online by patients or family members regarding the tolerance to cysteamine, their compliance, and the symptoms they are experiencing.8 The weekly statistical analyses are available for professionals. To date, 230 patients have registered, 221 of them have been treated with cysteamine, and 83.2% started cysteamine before 5 years of age, an age shown to be critical by Brodin-Sartorius and colleagues.6 Still, 29.9% of the patients who started cysteamine early had at least one kidney transplant, 78.2% of those before the age of 16 years. In addition, 47.8% of the patients who started cysteamine early experienced muscle weakness, and 23.9% hypothyroidism. These current data on a large cohort of patients show again that ESRD and complications are occurring even when cysteamine therapy is started early in life. In addition, cysteamine is challenging to take and difficult to tolerate. The need for frequent doses (oral every 6 h plus eye drops once an hour) necessitates that medications be carried for all activities, but the most significant impact of the every-6-h dosing schedule is that cystinosis patients never sleep through the night. Cysteamine also has a number of undesirable side effects, such as persistent odor and halitosis, along with more severe side effects of vomiting and diarrhea that render its administration difficult.9 Some patients stop taking cysteamine because they cannot tolerate it. Brodin-Sartorius and colleagues6 report that 28% of the patients in their study had extended periods without cysteamine. The CCIR reports that 7.8 % of the patients currently do not take cysteamine, 68.7 % of these because of the side effects and tolerance. The drug side effects and frequent dosing have a direct negative impact on the patients ’ and families’ quality of life. A comprehensive study in this area is necessary.
Why does cysteamine delay the complications of cystinosis but not prevent them? Cysteamine, by allowing cystine to exit the lysosomes, overcomes the major function of the missing cystinosin but does not replace cystinosin. Even if the level of cystine in white blood cells is low, what is the impact of cysteamine therapy on the tissues? Little is known about this, for obvious reasons, but data suggest that cystine storage in tissues is not completely cleared by cysteamine. 2,10 Moreover, the fact that cysteamine does not treat the proximal tubulopathy shows that the physiopathology of cystinosis is more complicated than expected, and cystinosin may have other functions still to be identified (Figure 1). Gahl et al.4 have mentioned that significantly more deaths and complications were observed in patients homozygous for the 57–kb CTNS deletion compared with patients with only mutations. Brodin-Sartorius and colleagues did not observe any significant difference between these two groups in survival or appearance of the renal and extrarenal complications. 6 A more comprehensive study on genotype– phenotype correlation would be necessary, but in regard to years of clinical reports, this correlation is not evident, which suggests the existence of modifier genes responsible for the broad range of phenotype in patients with or without cyste-amine (Figure 1). This could explain why, among patients who start cysteamine early and have good compliance, some will develop ESRD or other complications before 15 years of age and some will experience fewer symptoms, later in their lives. The existence of these genes that influence the phenotype of cystinosis is obvious in the different mouse models of cystinosis that Antignac’s group generated. The Ctns−/− mice on a mixed 129/Sv×C57BL/6 genetic background did not have any renal dysfunction, nor did the FVB/N pure-background mice, whereas the Ctns−/− mice on a pure C57BL/6 background developed renal dysfunction as early as 6 months of age and end-stage renal failure by 15 months.11 The identification of these other genes that influence the severity of the disease in the context of cystinosis should be of high interest and would help to better understand the physiopathology of cystinosis and the high variability of symptoms even with cysteamine therapy.
Figure 1. Physiopathology of cystinosis.
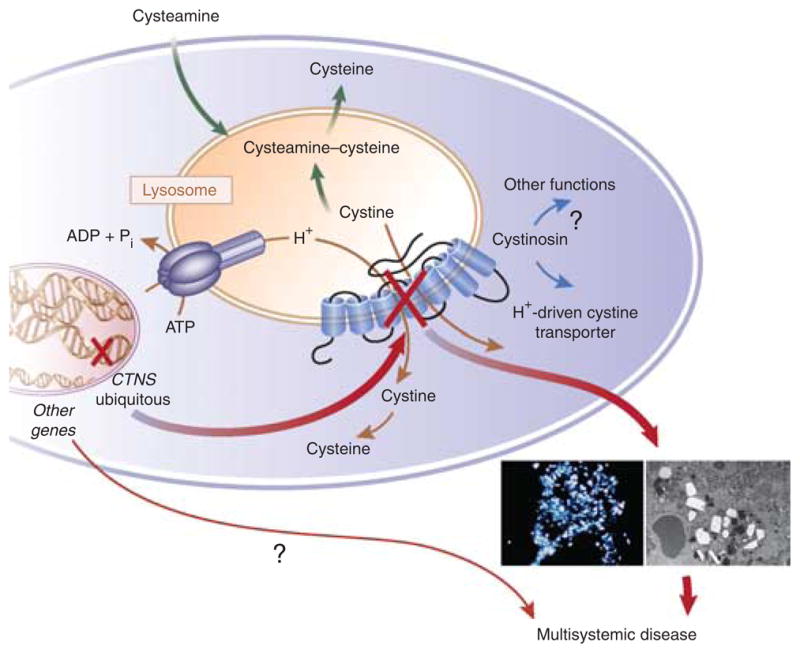
Cystinosis is a monogenic hereditary disease caused by mutations or deletions in the ubiquitous gene CTNS. This gene encodes a seven-transmembrane lysosomal protein, which is a proton-driven cystine transporter. However, the physiopathology of cystinosis suggests that cystinosin has other functions and/or other genes influence the pace and extent of the disease. This could explain the high variability in age of the appearance and severity of the complications observed in patients taking cysteamine, which allows cystine to exit the lysosomes. ADP, adenosine diphosphate; ATP, adenosine triphosphate; Pi, inorganic phosphate.
What is the outlook for patients with cystinosis? A new, delayed-release formulation of cysteamine (DR Cysteamine) is currently being developed by Raptor Pharmaceutical on the basis of a study conducted by Dohil et al.12 The company recently completed its pivotal, phase 3 clinical study. This new formulation needs to be taken twice daily, so the patients will be able to sleep through the night, and it will reduce the gastrointestinal side effects. Thus, the patients’ quality of life will be improved, but will the impact on the disease also be improved? Hematopoietic stem cell transplantation, which has a significant therapeutic benefit in the mouse model of cystinosis, 13,14 could be the next treatment for cystinosis. However, proof that cell therapy has the same benefits in patients as in mice still needs to be demonstrated.
Acknowledgments
I gratefully acknowledge Nancy Stack and Jerry Schneider for their review of and constructive comments on the manuscript, and Betty Cabrera, curator of the CCIR, for providing the latest data from the registry. I am funded by the Cystinosis Research Foundation and US National Institutes of Health grant RO1 DK090058-01, and R21 DK090548.
Footnotes
DISCLOSURE
The author declared no competing interests.
References
- 1.Kalatzis V, Cherqui S, Antignac C, et al. Cystinosin, the protein defective in cystinosis, is a H(+)-driven lysosomal cystine transporter. EMBO J. 2001;20:5940–5949. doi: 10.1093/emboj/20.21.5940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gahl WA, Thoene J, Schneider JA. Cystinosis: a disorder of lysosomal membrane transport. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The Metabolic and Molecular Bases of Inherited Disease. 8. Vol. 3. McGraw Hill; NewYork: 2001. pp. 5085–5108. [Google Scholar]
- 3.Thoene JG, Oshima RG, Crawhall JC, et al. Cystinosis. Intracellular cystine depletion by aminothiols in vitro and in vivo. J Clin Invest. 1976;58:180–189. doi: 10.1172/JCI108448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gahl WA, Balog JZ, Kleta R. Nephropathic cystinosis in adults: natural history and effects of oral cysteamine therapy. Ann Intern Med. 2007;147:242–250. doi: 10.7326/0003-4819-147-4-200708210-00006. [DOI] [PubMed] [Google Scholar]
- 5.Nesterova G, Gahl W. Nephropathic cystinosis: late complications of a multisystemic disease. Pediatr Nephrol. 2008;23:863–878. doi: 10.1007/s00467-007-0650-8. [DOI] [PubMed] [Google Scholar]
- 6.Brodin-Sartorius A, Tête M-J, Niaudet P, et al. Cysteamine therapy delays the progression of nephropathic cystinosis in late adolescents and adults. Kidney Int. 2012;81:179–189. doi: 10.1038/ki.2011.277. [DOI] [PubMed] [Google Scholar]
- 7.North American Pediatric RenalTrials and Collaborative Studies 2010 and 2011. [accessed June 2011];Annual Reports. 2010–2011 http://www.emmes.com/study/ped/annlrept/annlrept.html.
- 8.Cure Cystinosis International Registry. [accessed June 2011]; https://cystinosis.patientcrossroads.org.
- 9.Gahl WA, Reed GF, Thoene JG, et al. Cysteamine therapy for children with nephropathic cystinosis. N Engl J Med. 1987;316:971–977. doi: 10.1056/NEJM198704163161602. [DOI] [PubMed] [Google Scholar]
- 10.Gahl WA, Charnas L, Markello TC, et al. Parenchymal organ cystine depletion with long-term cysteamine therapy. Biochem Med Metab Biol. 1992;48:275–285. doi: 10.1016/0885-4505(92)90074-9. [DOI] [PubMed] [Google Scholar]
- 11.Nevo N, Chol M, Bailleux A, et al. Renal phenotype of the cystinosis mouse model is dependent upon genetic background. Nephrol Dial Transplant. 2010;25:1059–1066. doi: 10.1093/ndt/gfp553. [DOI] [PubMed] [Google Scholar]
- 12.Dohil R, Gangoiti JA, Cabrera BL, et al. Long-term treatment of cystinosis in children with twice-daily cysteamine. J Pediatr. 2010;156:823–827. doi: 10.1016/j.jpeds.2009.11.059. [DOI] [PubMed] [Google Scholar]
- 13.Syres K, Harrison F, Tadlock M, et al. Successful treatment of the murine model of cystinosis using bone marrow cell transplantation. Blood. 2009;114:2542–2552. doi: 10.1182/blood-2009-03-213934. [DOI] [PubMed] [Google Scholar]
- 14.Yeagy BA, Harrison F, Gubler MC, et al. Kidney preservation by bone marrow cell transplantation in hereditary nephropathy. Kidney Int. 2011;79:1198–1206. doi: 10.1038/ki.2010.537. [DOI] [PubMed] [Google Scholar]