

Abstract
More than 15% of cancer deaths worldwide are associated with underlying infections or inflammatory conditions, therefore understanding how inflammation contributes to cancer etiology is important for both cancer prevention and treatment. Inflamed tissues are known to harbor elevated etheno-base (ε-base) DNA lesions induced by the lipid peroxidation that is stimulated by reactive oxygen and nitrogen species (RONS) released from activated neutrophils and macrophages. Inflammation contributes to carcinogenesis in part via RONS-induced cytotoxic and mutagenic DNA lesions, including ε-base lesions. The mouse alkyl adenine DNA glycosylase (AAG, also known as MPG) recognizes such base lesions, thus protecting against inflammation-associated colon cancer. Two other DNA repair enzymes are known to repair ε-base lesions, namely ALKBH2 and ALKBH3; thus, we sought to determine whether these DNA dioxygenase enzymes could protect against chronic inflammation-mediated colon carcinogenesis. Using established chemically induced colitis and colon cancer models in mice, we show here that ALKBH2 and ALKBH3 provide cancer protection similar to that of the DNA glycosylase AAG. Moreover, Alkbh2 and Alkbh3 each display apparent epistasis with Aag. Surprisingly, deficiency in all 3 DNA repair enzymes confers a massively synergistic phenotype, such that animals lacking all 3 DNA repair enzymes cannot survive even a single bout of chemically induced colitis.
Introduction
Chronic inflammation is associated with an increased risk of cancer, and more than 15% of cancer deaths worldwide are associated with an underlying infection or inflammatory condition (1, 2). For example, patients with inflammatory bowel disease (IBD), encompassing both ulcerative colitis (UC) and Crohn’s disease, have an increased susceptibility to colorectal cancer; indeed, patients with extensive UC are at least 3- to 5-fold more likely to develop colorectal cancer than the general population (3). Further, the risk of colon cancer in patients with UC increases with the severity and duration of the inflammatory disease (4, 5). The association between cancer and intestinal inflammation is further strengthened by the fact that controlling or inhibiting inflammation in patients with IBD with long-term use of antiinflammatory drugs, such as nonsteroidal antiinflammatory drugs and selective cyclooxygenase-2 inhibitors, reduces cancer risk by 40%–50% (6, 7).
Increased levels of DNA base lesions have been documented in patients with chronic inflammatory conditions, including IBD (8–10). During an episodic inflammatory flare-up in patients with IBD, uncontrolled inflammation can cause tissue damage by the secretion of proinflammatory cytokines, chemokines, growth factors, and matrix-degrading enzymes. Importantly, such flare-ups also generate large quantities of reactive oxygen and nitrogen species (RONS) (11). RONS can directly induce DNA damage via oxidation and deamination of DNA bases, but, in addition, base damage occurs indirectly via lipid peroxidation; RONS react with polyunsaturated fatty acids to generate DNA-reactive aldehydes, including malondialdehyde and 4-hydroxy-2-nonenal (12). The terminal reaction of these aldehydes with DNA generates mutagenic and toxic etheno-base (ε-base) lesions and other exocyclic DNA adducts (13, 14). Moreover, RONS also induce the formation of nitrosamines that may methylate DNA (15, 16). Therefore, chronic inflammation, through multiple mechanisms, results in increased levels of 1,N6-ethenoadenine (εA), 1,N2-ethenoguanine (1,N2-εG), 3,N4-ethenocytosine (εC), 7,8-dihydro-8-oxoguanine (8oxoG), and possibly methylated base lesions in the DNA of affected tissues (8, 17, 18). In fact, patients with IBD exhibit 2- to 20-fold increases in εC adducts in colonic tissue compared with that in healthy controls (19). Evidence of systemic DNA damage is also suggested from the observation of increased DNA damage in peripheral leukocytes as well as the presence of DNA adducts in the urine during chronic inflammation (18, 20).
In a previous mouse study, we found that the mouse alkyladenine DNA glycosylase (AAG, also known as MPG and ANPG), which recognizes both ε-base and methylated base lesions, reduces colon damage and decreases tumor burden in response to chronic colonic inflammation (10). AAG initiates base excision repair and excises a wide range of structurally diverse damaged bases, including 3-methyladenine, 7-methylguanine, εA, 1,N2-εG, hypoxanthine, and 8oxoG, among others; in addition, AAG binds εC lesions with high affinity but cannot excise them from DNA (Figure 1 and refs. 21–30). In fact, we observed a greater accumulation of ε-adducted and oxidized DNA base lesions in the colonic epithelium of Aag–/– mice after an inflammation challenge when compared with those in WT mice (10). Moreover Aag–/– mice suffered increased colon tissue damage and elevated tumorigenesis compared with WT mice, indicating that the repair of inflammation-induced DNA lesions is important for colon cancer prevention. As mentioned, AAG tightly binds but is incapable of excising εC lesions (29, 31). Interestingly, we observed that following an intestinal inflammatory challenge, Aag–/– colons accumulate higher levels of εC, compared with that in WT colons, suggesting that AAG-εC binding somehow facilitates εC repair (10). It is possible that, although AAG does not directly excise εC, it may assist in its repair by recruiting other proteins to repair this RONS-induced εC DNA base lesion.
Figure 1. Published nucleic acid substrates for the AAG, ALKBH2, and ALKBH3 enzymes.
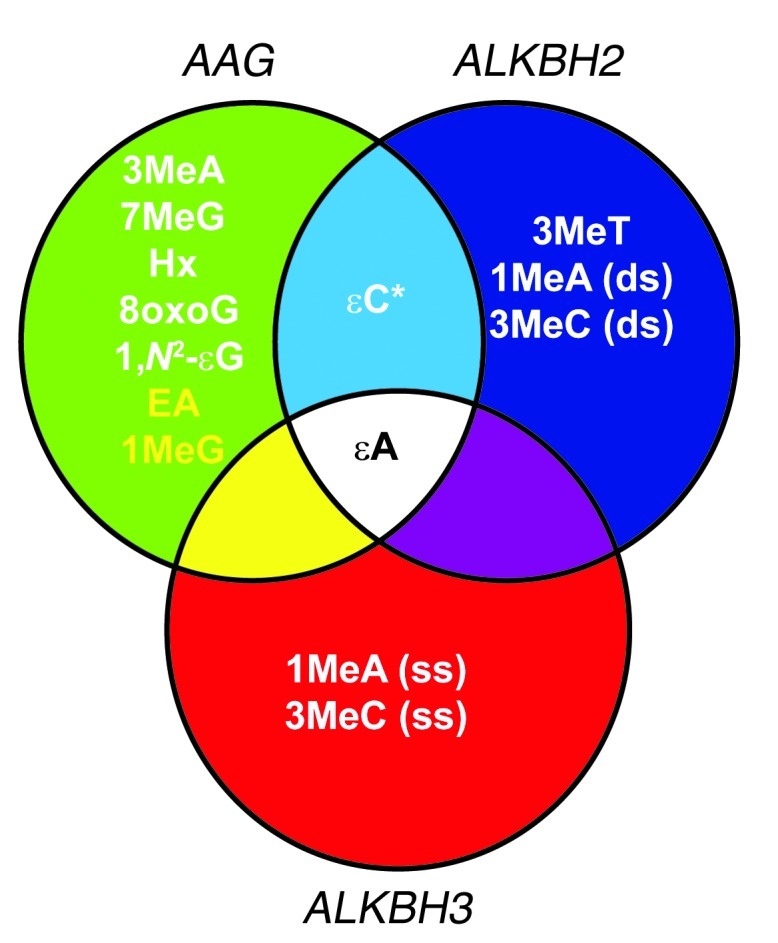
Substrates for the AAG, ALKBH2, and ALKBH3 enzymes have been illustrated either in vitro or in vivo, including 3-methyladenine (3MeA), 7-methylguanine (7MeG), hypoxanthine (Hx), 8oxoG, 1,N6-ethanoadenine (EA), 1,N2-εG, 1-methylguanine (1MeG), εC, εA, 3-methylthymine (3MeT), 1-methyladenine (1MeA), and 3-methylcytosine (3MeC). The asterisk indicates that AAG binds but is unable to excise εC base lesions. Lesions listed in yellow are known substrates of bacterial AlkB; ALKBH2 or ALKBH3 have yet to be tested for the ability to excise these lesions. ds, double strand; ss, single strand.
The E. coli AlkB DNA repair enzyme can repair ε-base lesions, such as εA and εC (32), in addition to simple alkylated adducts, such as 1-methyladenine and 3-methylcytosine (Figure 1 and refs. 33–36); repair is via a direct reversal oxidative demethylation reaction that is dependent on oxygen, iron, and α-ketoglutarate (33, 34). The preferred substrates for the human AlkB homologs (the ALKBH proteins) are still being elucidated, but it has been demonstrated that ALKBH2 repairs εA both in vivo and in vitro and that ALKBH3 repairs εA at least in vitro (Figure 1 and refs. 37, 38). Recently, ALKBH3-mediated DNA repair in vivo was shown to be coupled to DNA unwinding by the ASCC3 helicase (39). In addition, we recently showed that ALKBH2 efficiently repairs εC adducts in vitro (40). Since the mouse ALKBH2 and ALKBH3 enzymes are also likely to repair ε-base lesions, we sought to determine whether ALKBH2 and ALKBH3, like AAG, can protect against chronic inflammation-mediated colon carcinogenesis. It is important to note that in the absence of AAG, ε-base lesions continue to be removed from the genome, albeit very slowly (41, 42); we therefore hypothesized that ALKBH2 and ALKBH3 may be responsible for such redundant repair. To assess this, we induced chronic inflammatory colitis in Aag, Alkbh2, and Alkbh3 single-, double-, and triple-knockout mice, monitoring various aspects of colitis and colorectal cancer. Our results demonstrate that ALKBH2 and ALKBH3 modestly protect against inflammation-mediated colon carcinogenesis and that their effects are at least additive in the Alkbh2/Alkbh3 double-knockout mice. We also show that Aag appears to have an epistatic relationship with Alkbh2 and Alkbh3, because the addition of Alkbh2 or Alkbh3 mutations into the Aag–/– background did not exacerbate the phenotype. Most notably, we have uncovered a remarkable synergism among AAG, ALKBH2, and ALKBH3 in protecting against acute intestinal damage, as indicated by the inability of the Aag–/–Alkbh2–/–Alkbh3–/– triple mutant mice to recover from even a single bout of colitis induced by dextran sodium sulfate (DSS).
Results
ALKBH2 and ALKBH3 protect against azoxymethane/DSS-mediated colon carcinogenesis.
To determine whether the ALKBH2 or ALKBH3 enzymes play a protective role in chronic inflammation-associated carcinogenesis, we challenged Alkbh2–/–, Alkbh3–/–, and Alkbh2–/–Alkbh3–/– mice with the azoxymethane/DSS (AOM/DSS) treatment regimen. This 2-step carcinogenesis protocol has been extensively used to study inflammation-associated colon carcinogenesis (10, 43, 44); AOM acts as an initiator, and DSS, by perturbing the intestinal mucosal layer, enables enteric bacteria to elicit colonic inflammation. It is important to note that using our experimental conditions, neither AOM nor DSS alone significantly induce carcinogenesis in WT animals (10). Alkbh2–/–, Alkbh3–/–, and Alkbh2–/–Alkbh3–/– mice all exhibited greater sensitivity to AOM/DSS than WT mice, as indicated by decreased survival (P < 0.05) (Figure 2A). In fact, only 40% to 60% of Alkbh2–/–, Alkbh3–/–, and Alkbh2–/–Alkbh3–/– mice survived the entire treatment regimen compared with 80% of WT mice. Pathological data obtained from the mice euthanized during the experiment were combined with data obtained from mice that survived the entire experiment, because there were few statistically significant differences between the 2 groups in each of the following colon disease criteria: histopathological scores, spleen weight, colon length, and tumor multiplicity (Supplemental Table 1; supplemental material available online with this article; doi: 10.1172/JCI63338DS1).
Figure 2. ALKBH proteins are protective against AOM/DSS-mediated carcinogenesis.

(A) Kaplan-Meier survival curves show that during AOM/DSS treatment, compared with that for WT mice, there is a significant decrease in survival for Alkbh2–/– (P < 0.0001), Alkbh3–/– (P < 0.05), and Alkbh2–/–Alkbh3–/– (P < 0.0001). (B) Spleen weight as a percentage of body weight is plotted for WT (n = 36), Alkbh2–/– (n = 13), Alkbh3–/– (n = 26), and Alkbh2–/–Alkbh3–/– mice (n = 17). Data represent mean ± SEM. (C) Tumor multiplicity is presented as the number of tumors per mouse in WT (n = 31), Alkbh2–/– (n = 13), Alkbh3–/– (n = 25), and Alkbh2–/–Alkbh3–/– mice (n = 16). Data is presented as mean ± SEM.
All Alkbh-deficient mice (Alkbh2–/–, Alkbh3–/–, and Alkbh2–/–Alkbh3–/– mice) exhibited significant increases in spleen weight when compared with that of WT mice following AOM/DSS treatment (Figure 2B). Increased spleen weight is a common end point for colitis studies and reflects extramedullary hematopoiesis in the spleen plus lymphoid hyperplasia, a mechanism for coping with recurrent blood loss in the stools, as well as possible immune activation in the spleen. We also observed an additive increase in spleen weight in the Alkbh2–/–Alkbh3–/– mice, which exhibited significantly larger spleen weights compared with those of Alkbh3–/– mice (P = 0.0126) and trended toward an increase compared with Alkbh2–/– mice (P = 0.167) (Figure 2B).
Our previous studies illustrated that both the AOM and DSS challenges are required to induce significant colon tumorigenesis (10). Accordingly, AOM/DSS treatment resulted in colon tumors in the majority of mice for each genotype; 83% of WT, 93% of Alkbh3–/–, and 100% of Alkbh2–/– and Alkbh2–/–Alkbh3–/– mice developed tumors in the distal colon (Figure 2C). Mice lacking ALKBH2 are more susceptible than WT mice to colon tumorigenesis, as illustrated by increased tumor multiplicity (P = 0.041) (Figure 2C). In contrast, Alkbh3–/– mice did not develop significantly more tumors than WT mice (P = 0.269). Despite this, an ALKBH3 deficiency did contribute to disease progression in the absence of ALKBH2, since we observed a synergistic increase in tumor induction in Alkbh2–/–Alkbh3–/– mice, when compared with that in either Alkbh2–/– (P = 0.005) or Alkbh3–/– mice (P < 0.001). Taken together, these results show that both the ALKBH2 and ALKBH3 DNA repair proteins protect against inflammation-associated colon carcinogenesis.
Histopathological analysis of colon tissue revealed an additive increase in hyperplasia in Alkbh2–/–Alkbh3–/– mice compared with that in the single mutants and an increase in the criteria of dysplasia/neoplasia and area affected compared with that in WT and Alkbh3–/– mice following the AOM/DSS treatment (Supplemental Figure 1A). Alkbh2–/– mice also exhibited increased scores for dysplasia/neoplasia and area affected compared with those of WT mice. Although Alkbh3–/– mice exhibited an increase in the inflammation score, compared with that of all other genotypes, as well as increased spleen weight and decreased colon length, this single mutant did not exhibit significant increases in tumor multiplicity or other histopathological criteria compared with WT mice.
AAG and ALKBH enzymes exhibit overlap in their protection against AOM/DSS-mediated colon carcinogenesis.
Given the overlap in substrate specificity between the ALKBH enzymes and AAG (Figure 1), we investigated whether the absence of Alkbh2 or Alkbh3 in an Aag-deficient background might result in further susceptibility to AOM/DSS treatment compared with Aag–/– mice (10). We treated WT, Aag–/–, Aag–/–Alkbh2–/–, and Aag–/–Alkbh3–/– mice with AOM/DSS. Compared with WT mice, all 3 repair-deficient strains exhibited greater sensitivity to the AOM/DSS treatment, as shown by decreased survival that is similar for all 3 mutant strains (P < 0.0001) (Figure 3A). Again, since the histopathological and other disease end points did not differ significantly with length of survival, we combined data from mice euthanized mid-treatment with data from mice euthanized after the end of treatment (Supplemental Table 2). The spleen weights were similar for all of the genotypes (data not shown). However, as demonstrated in Figure 3B, all 3 genotypes (Aag–/–, Aag–/–Alkbh2–/–, and Aag–/–Alkbh3–/–) exhibited significant decreases in colon length when compared with that in WT mice; repeated DSS-induced colon injury and healing is known to cause colon shortening. Surprisingly, there were no additional decreases in the colon length of Aag–/–Alkbh2–/– or Aag–/–Alkbh3–/– mice as compared with that of Aag–/– mice, suggesting an epistatic relationship between Aag and Alkbh2 and between Aag and Alkbh3. In this experimental cohort, all of the mice developed colon tumors, and Aag–/– and Aag–/–Alkbh2–/– mice developed significantly more colon tumors than WT mice (Figure 3C). The Aag–/–Alkbh3–/– mice also trended toward increased tumor multiplicity, but this did not quite reach statistical significance (P = 0.078). Again, these results suggest an epistatic relationship between Aag and the Alkbh genes, which will be considered further in the Discussion. Histopathological criteria indicate no significant increases in disease severity for Aag–/–Alkbh2–/– mice compared with that for WT mice (Supplemental Figure 1B). However, we did observe a significant increase in the criteria of dysplasia and area affected for Aag–/–Alkbh3–/– mice compared with that for all other genotypes; interestingly, as mentioned above, this did not translate into increased tumor formation in the Aag–/–Alkbh3–/– mice but does suggest that these mice may be prone to develop tumors that are broad based, with a higher grade of dysplasia.
Figure 3. Aag–/–, Aag–/–Alkbh2–/–, and Aag–/–Alkbh3–/– mice all exhibit increased susceptibility to AOM/DSS treatment.

(A) Kaplan-Meier survival curves show decreased survival for Aag–/–, Aag–/–Alkbh2–/–, and Aag–/–Alkbh3–/– mice compared with that for WT mice during the AOM/DSS treatment. (B) The change in colon length as compared with that in untreated controls is plotted for WT (n = 18), Aag–/– (n = 9), Aag–/–Alkbh2–/– (n = 20), and Aag–/–Alkbh3–/– mice (n = 9). Data represent mean ± SEM. (C) Tumor multiplicity is presented as the number of tumors per mouse in WT (n = 18), Aag–/– (n = 9), Aag–/–Alkbh2–/– (n = 19), and Aag–/–Alkbh3–/– mice (n = 9). Data is presented as mean ± SEM.
Aag–/–Alkbh2–/–Alkbh3–/– triple-knockout mice exhibit extreme sensitivity to AOM/DSS treatment.
Since the addition of an ALKBH2 or ALKBH3 deficiency had little effect in the Aag–/– mice, it was necessary to construct the triple mutant strain to further explore the interaction of AAG-initiated base excision repair and ALKBH-mediated direct reversal of DNA base damage in protecting against inflammation in the colon. We therefore tested the susceptibility of Aag–/–Alkbh2–/–Alkbh3–/– triple mutant mice to AOM/DSS treatment. To our surprise, all of the triple mutant mice (n = 29) became moribund between 2 and 3 days after the first 5-day DSS cycle (Figure 4A). Histopathological analysis revealed that Aag–/–Alkbh2–/–Alkbh3–/– mice exhibited massive intestinal injury, with a pathological diagnosis of severe acute necrotizing colitis. The lesions were observed predominantly in the transverse and distal colon, occasionally to a milder degree in the proximal colon, and never in the small intestine. The colonic lesions in the Aag–/–Alkbh2–/–Alkbh3–/– mice exhibited mucosal (surface and crypt epithelial) degeneration, necrosis with edema, and extensive areas of loss of tissue architecture and substantial neutrophil infiltration (Figure 4B). The corresponding WT and Aag–/– controls completed the 5 cycles of the AOM/DSS treatment regimen, and, accordingly, these mice exhibited colon hyperplasia, epithelial dysfunction, and increased inflammation in the distal colon (Figure 4B).
Figure 4. Aag–/–Alkbh2–/–Alkbh3–/– mice do not survive AOM/DSS treatment.

(A) Kaplan-Meier survival curves illustrate complete lethality of Aag–/–Alkbh2–/–Alkbh3–/– mice following the first DSS cycle. (B) H&E-stained sections of colons harvested 2 days following DSS withdrawal (12 days after AOM dose) in Aag–/–Alkbh2–/–Alkbh3–/– mice and at the completion of the AOM/DSS treatment (100 days after AOM dose) in WT and Aag–/– mice. Consistent with the completion of the AOM/DSS treatment in the WT and Aag–/– cohorts, their colons exhibited regions of hyperplasia, inflammation, and minimal dysplasia. Scale bar: 100 μm. The asterisk indicates submucosal edema and associated scattered inflammatory cells; the arrow indicates epithelial erosion and necrosis.
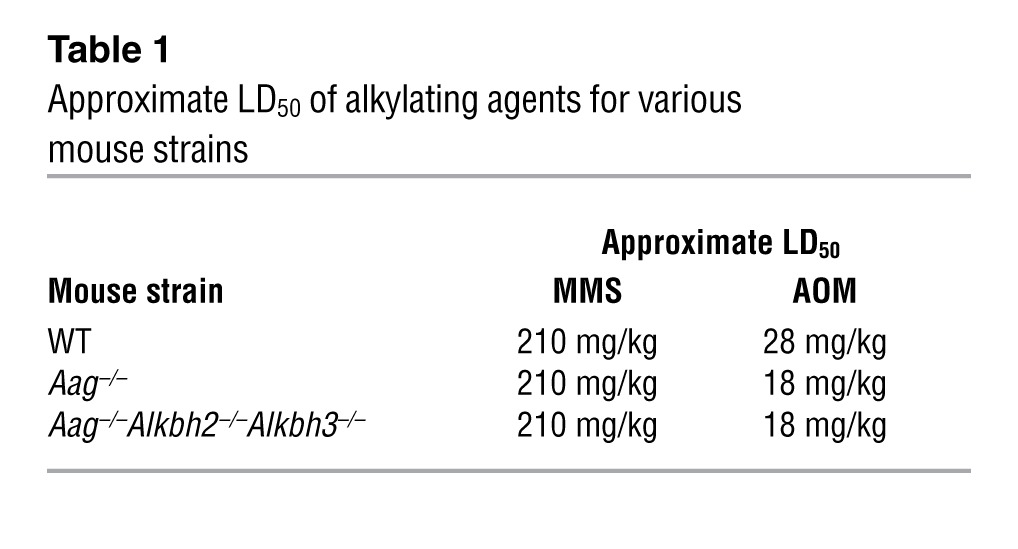
AOM, a SN1 colonotropic alkylating agent, and DSS, an intestinal epithelial irritant, each induce colon-specific damage (43, 45). To determine which agent was responsible for the extreme sensitivity observed in the Aag–/–Alkbh2–/–Alkbh3–/– triple mutant mice, we examined whole-body sensitivity to AOM in the Aag–/–Alkbh2–/–Alkbh3–/– mice by determining their approximate LD50. As illustrated in Table 1, Aag–/–Alkbh2–/–Alkbh3–/– mice did not exhibit increased whole-body sensitivity to AOM when compared with Aag–/– mice; however, both Aag–/– and Aag–/–Alkbh2–/–Alkbh3–/– mice did appear to be slightly more sensitive to AOM when compared with WT mice. This slight increase in whole-body sensitivity to AOM cannot explain the surprising lethality of the Aag–/–Alkbh2–/–Alkbh3–/– mice during the AOM/DSS experiment, in which AOM was administered at a dose lower than this LD50. We also examined whole-body sensitivity to the prototypical SN2 alkylating agent methylmethane sulfonate (MMS) and found that WT, Aag–/–, and Aag–/–Alkbh2–/–Alkbh3–/– mice all displayed the same approximate LD50 (Table 1). Together, these data indicate that Aag–/–Alkbh2–/–Alkbh3–/– mice did not exhibit significantly increased susceptibility to SN1 or SN2 alkylating agents, indicating that the extreme pathology observed in the triple mutant mice following AOM/DSS treatment is not due to AOM sensitivity. We next explored whether Aag–/–Alkbh2–/–Alkbh3–/– mice are specifically sensitive to DSS-induced colitis.
Table 1 .
Approximate LD50 of alkylating agents for various mouse strains
AAG, ALKBH2, and ALKBH3 repair enzymes are essential to survive DSS-induced colitis.
We challenged WT, Aag–/–, and Aag–/–Alkbh2–/–Alkbh3–/– mice with a single 5-day cycle of DSS (2.5%) to determine whether DSS-induced disruption of the colonic mucosa on its own is sufficient to induce lethality in Aag–/–Alkbh2–/–Alkbh3–/– mice. Between 2 and 3 days following withdrawal of DSS, all the Aag–/–Alkbh2–/–Alkbh3–/– mice (n = 11) became moribund and required euthanasia, whereas the vast majority of WT and Aag–/– mice fully recovered from the DSS treatment but were euthanized at day 13 for histological comparison with the Aag–/–Alkbh2–/–Alkbh3–/– mice (Figure 5A). As seen with AOM/DSS, the Aag–/–Alkbh2–/–Alkbh3–/– mice exposed to DSS alone exhibited severe intestinal (colonic) mucosal erosions/ulcerations, degeneration, necrosis, and edema. Since WT and Aag–/– mice survived 8 days after DSS withdrawal, their intestines, while exhibiting some regions of ulceration, also exhibited regions that had recovered from the intestinal injury, and these regions included regenerative crypts and intact mucosa; presumably, the regenerated colonic architecture was derived from surviving mesenchymal cells, stem cells, and any other cells that are required for such tissue repair (Figure 5B and ref. 46).
Figure 5. DSS treatment is lethal to Aag–/–Alkbh2–/–Alkbh3–/– mice.

(A) Kaplan-Meier survival curves of WT (n = 11), Aag–/– (n = 9), and Aag–/–Alkbh2–/–Alkbh3–/– (n = 11) mice following 1 5-day cycle of DSS (2.5%). (B) Examples of colon pathology when the mice became moribund (2 days after DSS withdrawal, 7 days following start of DSS treatment) in Aag–/–Alkbh2–/–Alkbh3–/– mice. WT and Aag–/– mice are shown at completion of study (8 days after DSS withdrawal, 13 days after start of DSS treatment). (C and D) Levels of (C) εA base lesions and (D) 1,N2-εG base lesions are quantitated from colonic epithelium for WT, Aag–/–, and Aag–/–Alkbh2–/–Alkbh3–/– mice under untreated conditions (day 0), immediately following DSS (1.75%) withdrawal (day 5), and 4 days following DSS withdrawal (day 9). Statistical significance was determined by 2-way ANOVA with Bonferroni post test. Data are shown as the mean ± SD. (E) H&E-stained colon sections are shown from all mice 7 days following start of DSS treatment. (F) Histopathological scores of epithelial defects, mucosal degeneration, edema, inflammation, and hyperplasia are shown for WT (n = 10), Aag–/– (n = 10), and Aag–/–Alkbh2–/–Alkbh3–/– (n = 5) mice harvested 7 days following initiation of DSS (2.5%) treatment. Data represent mean ± SEM. (B and E) Asterisk indicate edema; arrows indicate epithelial ulceration. Scale bar: 100 μm.
We next measured endogenous levels of εA (Figure 5C) and 1,N2-εG base lesions (Figure 5D) in colonic epithelial cells of untreated and DSS-treated (1.75%) mice to determine whether the accumulation of unrepaired DNA base lesions may contribute to increased DSS toxicity in Aag–/–Alkbh2–/–Alkbh3–/– mice. Two-way ANOVA analysis illustrates that there was an accumulation of εA (P = 0.0012) and 1,N2-εG (P < 0.0001) DNA lesions in the genome of colonic epithelial cells following DSS treatment. At day 9 (4 days after DSS withdrawal), Aag–/–Alkbh2–/–Alkbh3–/– mice exhibited significantly increased levels of εA compared with those of WT mice (Figure 5C) and significantly increased levels of 1,N2-εG base lesions compared with those of both WT and Aag–/– mice (Figure 5D). This finding suggests that, like AAG, ALKBH2 and/or ALKBH3 repairs 1,N2-εG base lesions; this substrate has not previously been illustrated for ALKBH2 or ALKBH3 (30, 47, 48). As these ε-base lesions can be transcription-blocking, replication-blocking, mutagenic, and cytotoxic lesions, their accumulation likely explains, at least in part, the extreme DSS toxicity seen in the Aag–/–Alkbh2–/–Alkbh3–/– mice (49–51).
The reasons why Aag–/–Alkbh2–/–Alkbh3–/– mice do not survive DSS treatment may include the following: they develop more severe intestinal lesions than WT mice; they are unable to regenerate functional tissue after injury; or both. We evaluated the first possibility by examining the intestinal lesions in all genotypes 2 days following DSS withdrawal, when the Aag–/–Alkbh2–/–Alkbh3–/– mice became moribund. Surprisingly, all genotypes exhibited equivalently severe intestinal lesions with extensive edema, epithelial necrosis, and inflammation infiltrates (Figure 5E). Histopathological analysis illustrated similar levels of epithelial defects, mucosal degeneration, and edema in all genotypes (Figure 5F). Interestingly, the Aag–/–Alkbh2–/–Alkbh3–/– colons actually exhibited decreased scores for inflammation when compared with those of WT colons (P < 0.05); thus, even in the face of an apparently less severe inflammatory response, the Aag–/–Alkbh2–/–Alkbh3–/– mice went on to a much worse phenotype (Figure 5F). The Aag–/–Alkbh2–/–Alkbh3–/– mice also displayed less severe hyperplasia than WT mice, perhaps indicating an inability to respond appropriately to mucosal degeneration and epithelial defects or perhaps because the inflammatory response is muted (Figure 5F). Full-body gross examination and detailed histopathological analysis of most organs was also performed on WT, Aag–/–, and Aag–/–Alkbh2–/–Alkbh3–/– mice in untreated conditions as well as 2 days following DSS (2.5%) withdrawal. There were no obvious phenotypic differences among any of the genotypes in untreated conditions, and no significant pathological differences were found in DSS-treated mice, with the sole exception of the severe lesions observed in the transverse to distal colon. As severe colitis can result in intestinal bleeding and eventually anemia, we performed a complete blood count to determine whether the increased lethality in DSS-treated Aag–/–Alkbh2–/–Alkbh3–/– mice could be attributed to an increased penetrance of anemia. However, all genotypes exhibited equivalent hematocrit, hemoglobin, white blood cell counts, and red blood cell counts that were within normal limits, indicating that the DSS-treated mice did not differentially succumb to anemia (Supplemental Figure 2).
In summary, although all mice exhibited severe intestinal lesions 2 days after DSS withdrawal, the WT and Aag–/– mice survived, exhibiting visible tissue repair of the intestinal epithelium 6 days after DSS withdrawal, whereas Aag–/–Alkbh2–/–Alkbh3–/– mice were totally unable to recover from the same initial lesions (Figure 5B). The histopathological analysis of the colon suggested that Aag–/–Alkbh2–/–Alkbh3–/– mice are unable to mount an appropriate tissue repair response following the DSS-mediated epithelial damage, resulting in a total inability to withstand DSS-induced colitis.
AAG, ALKBH2, and ALKBH3 repair enzymes protect against some but not all inflammatory challenges.
Having established that Aag–/–Alkbh2–/–Alkbh3–/– DNA repair-deficient mice are unable to survive the induction of colonic inflammation by DSS, we set out to determine the generality of this dramatic phenotype. We chose 2 different kinds of inflammatory challenge: one that, like DSS exposure, is stimulated by bacterial cell constituents and a second that is not stimulated by bacterial cell constituents. We thus challenged WT and Aag–/–Alkbh2–/–Alkbh3–/– mice with either LPS to induce bacterial endotoxemia or with the hormone mimetic cerulein to induce acute inflammation in the pancreas (pancreatitis). LPS, derived from the outer membranes of gram-negative bacteria, induces systemic inflammation that is mediated through TLR signaling (52–54), and cerulein induces pancreatitis that is mediated through cholecystokinin receptor signaling (55).
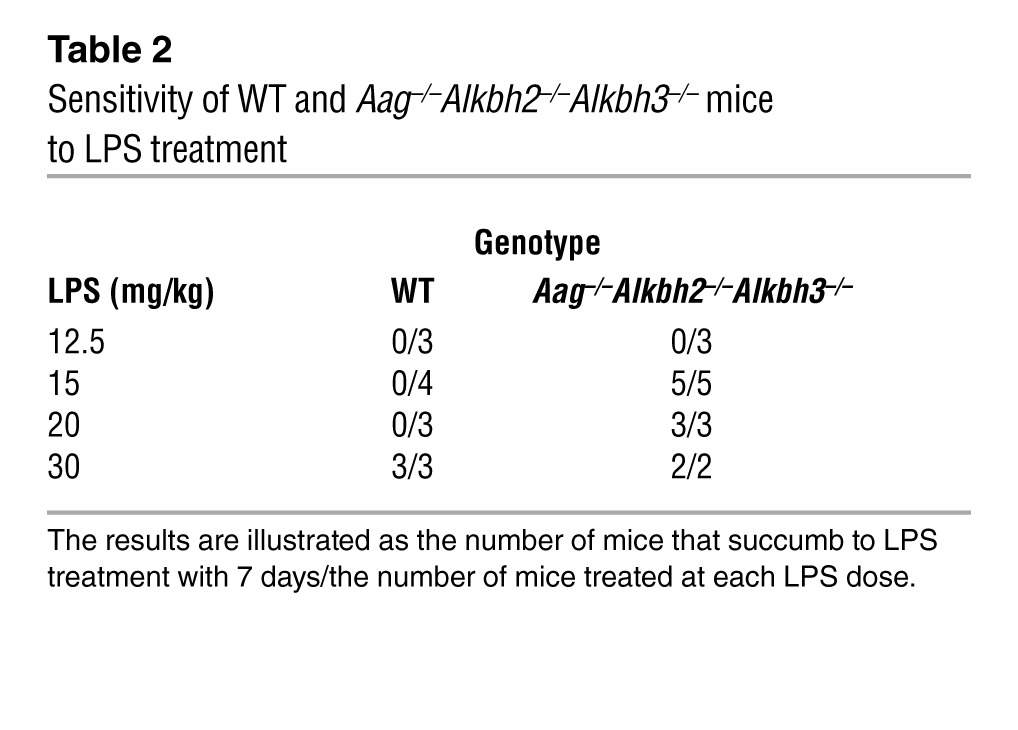
Twenty-four hours following cerulein treatment, all mice exhibited evidence of pancreatic damage, with edema and acinar cell degeneration and/or necrosis; however, there was no difference in disease severity among the genotypes (Figure 6A), just as there was no difference in initial colonic damage for DSS-treated mice. To determine whether recovery from such cerulein-induced damage is impaired in the Aag–/–Alkbh2–/–Alkbh3–/– mice, we examined recovery from this pancreatic challenge. Surprisingly, both WT and Aag–/–Alkbh2–/–Alkbh3–/– mice exhibited comparable recovery after the cerulein treatment, as indicated by pancreatic pathology 7 days following the acute pancreatic challenge (Figure 6). Thus, the induction of and recovery from cerulein-induced inflammatory injury was equivalent between WT and Aag–/–Alkbh2–/–Alkbh3–/– mice. The results upon exposing mice to bacterial endotoxemia induced by LPS were very different. WT mice survived all LPS doses up to 30 mg/kg and only required euthanasia following treatment with 30 mg/kg LPS (Table 2). In contrast, Aag–/–Alkbh2–/–Alkbh3–/– mice were much more sensitive than WT mice to LPS; 100% of Aag–/–Alkbh2–/–Alkbh3–/– mice succumbed to treatment at doses of 15 mg/kg LPS and above, although all survived at 12.5 mg/kg LPS (Table 2). Together these experiments illustrate that Aag–/–Alkbh2–/–Alkbh3–/– mice are more susceptible than WT mice to some, but not all, inflammatory challenges and further suggest that this sensitivity may be specific to TLR-mediated inflammation that is induced upon exposure to bacterial constituents.
Figure 6. Aag–/–Alkbh2–/–Alkbh3–/– mice are capable of recovering from cerulein treatment.

(A) H&E-stained sections of pancreata are shown from untreated WT and Aag–/–Alkbh2–/–Alkbh3–/– mice as well as following cerulein treatment. Mice recovered for either 24 hours or 7 days following the 6 hourly injections of cerulein (50 μg/kg). Scale bar: 100 μm. (B) Pathology scores of cerulein-treated WT and Aag–/–Alkbh2–/–Alkbh3–/– mice, showing the sum of the scores for edema, inflammation, and acinar cell degeneration. Individual data points represent total pathological scores of individual mice, and horizontal bars denote the mean score of mice.
Table 2 .
Sensitivity of WT and Aag–/–Alkbh2–/–Alkbh3–/– mice to LPS treatment
Discussion
Inflammation and cancer were linked together as early as 1863 when Virchow noted that cancers often occur at sites of chronic inflammation (2). Bouts of chronic inflammation can result in cycles of tissue damage, cellular proliferation, and eventually tissue repair (56). Following tissue injury, inflammatory cells initiate a complex response of chemical signals to repair the damaged tissue. However, activated inflammatory cells rapidly release RONS as a mechanism to prevent bacterial infections and as a result cause deleterious DNA damage. Inflammatory cells also release chemical mediators, such as cytokines and chemokines that can directly (or indirectly through the induction of NF-κB and STAT-3) promote the proliferation of surrounding tissues, invasion and metastasis of tumor cells, and generation of a proangiogenic microenvironment (57, 58). This strong relationship linking inflammation and cancer has resulted in the inclusion of “an inflammatory environment” as an emerging hallmark in Hanahan and Weinberg’s hallmarks of carcinogenesis (59).
Pattern recognition receptors (PRR) are important mediators of IBD, with evidence coming from both genetic association studies and animal models (60). The recognition of commensal bacteria by TLRs and the resulting MYD88-dependent signaling cascade are essential for regaining intestinal homeostasis following DSS treatment (60–62). Further, NOD-like receptors, the inflammasome, and the downstream mediator IL-18 are also required for proper host responses to DSS treatment (63). Accordingly, numerous proteins in these pathways (notably MYD88, TLR2, TLR4, IL-1R, IL-18R, among others) are essential in preventing DSS-induced lethality; mice deficient in these proteins exhibit decreased survival and increased morbidity following DSS treatment (64–66). Since the majority of mouse models hitherto reported to display dramatically increased DSS sensitivity have defects in PRR signaling, it was surprising to find that Aag–/–Alkbh2–/–Alkbh3–/– mice exhibit DSS sensitivity comparable to that observed in Myd88–/– mice (65). In fact, 2 recent studies have demonstrated connections between DNA damage responses and pattern recognition receptors (PRR) responses mediated by MYD88. Salcedo et al. (67) reported that Myd88–/– colonic tissue shows altered regulation of numerous DNA-damage genes (i.e., Parp1, Msh2, Msh3, Atm, and Atr) following AOM/DSS treatment, and Menendez et al. (68) reported that the human TLR gene family is integrated into the p53/DNA damage response network. These observations, together with the data presented here, indicate an important role for DNA repair in regulating intestinal homeostasis following DSS treatment.
IBD is a well-established example of chronic inflammation resulting in enhanced tumor risk, with patients with IBD exhibiting a 3- to 5-fold greater risk of cancer (3). Here, the episodic inflammation that occurs with repeated flare-ups in patients with IBD was simulated by the colonic irritant DSS in WT and numerous DNA repair-deficient mice. We determine that deficiencies in the direct DNA repair proteins ALKBH2 and ALKBH3 resulted in greater susceptibility to inflammation-associated colon cancer. As we have shown previously, the absence of AAG activity also increases susceptibility in this carcinogenesis model, likely due to the wide variety of DNA lesions it repairs (10, 69–71). Examining AOM/DSS-mediated tumorigenesis in numerous knockout mouse models allowed us to examine functional overlap and possible redundancy between AAG, ALKBH2, and ALKBH3 in vivo. The increased tumor multiplicity observed in Aag–/– and Alkbh2–/– single-knockout mice illustrates the protective roles of these proteins in inflammation-associated carcinogenesis and suggests that other repair proteins are unable to fully compensate in their absence. Additionally, we observed a synergistic increase in tumorigenesis in the Alkbh2–/–Alkbh3–/– mice, compared with that in Alkbh2–/– and Alkbh3–/– mice. Despite their overlap in DNA base substrates (Figure 1), AAG is unable to fully compensate in the absence of the 2 ALKBH proteins. We were surprised to find no additivity in tumorigenesis in the Aag–/–Alkbh2–/– or Aag–/–Alkbh3–/– mice compared with that in the relevant single mutants, indicating that AAG may be dominant to any additional phenotypic effect caused by absence of ALKBH2 or ALKBH3 and suggesting a possible epistatic relationship. Since AAG can bind to a number of ALKBH enzyme substrates, it is not hard to imagine that under certain circumstances AAG may facilitate ALKBH enzyme binding. Alternatively, it is possible that we failed to observe additivity in Aag–/–Alkbh2–/– or Aag–/–Alkbh3–/– mice because we reached an upper threshold in colon shortening and the number of tumors that develop following AOM/DSS treatment. This could prevent us from observing additional sensitivity/tumors in the Aag–/–Alkbh2–/– or Aag–/–Alkbh3–/– mice. Interestingly, Aag–/–, Aag–/–Alkbh2–/–, Aag–/–Alkbh3–/–, and Alkbh2–/–Alkbh3–/– mice all exhibited tumor multiplicity of approximately 20 tumors per mouse, which is similar to the maximum number of tumors typically reported for mice exhibiting increased susceptibility to AOM/DSS-induced carcinogenesis (43, 66, 72).
Our most surprising finding was that a single cycle of DSS-induced colitis resulted in absolute lethality in Aag–/–Alkbh2–/–Alkbh3–/– triple-knockout mice. This striking phenotype differs dramatically from the various double mutant combinations, which can survive up to 5 DSS cycles (during AOM/DSS treatment). During the AOM/DSS treatment, all of the Aag–/–Alkbh2–/–Alkbh3–/– mice succumbed to severe necrotizing colitis following the first DSS cycle, indicating that DNA repair mediated by AAG, ALKBH2, and ALKBH3 is absolutely essential for colon homeostasis, colon tissue repair, and whole-animal survival during acute inflammatory conditions. It is important to note that following 1 cycle of 2.5% DSS, all genotypes, including WT, developed equivalently severe intestinal ulcerations, and the extent of epithelial defects was the same for all the genotypes. Interestingly, the Aag–/–Alkbh2–/–Alkbh3–/– mice exhibited decreased histopathological scores for inflammation and hyperplasia compared with those of WT mice, indicative of an impaired response in these animals. Thus, Aag–/–Alkbh2–/–Alkbh3–/– mice are simply unable to recover from the DSS-induced epithelial lesions. Importantly, Aag–/–Alkbh2–/–Alkbh3–/– mice exhibited increased sensitivity to LPS-mediated endotoxemia, although they were no more sensitive than WT mice to cerulein-induced pancreatitis. These differences perhaps reflect the fact that cerulein induces inflammation via cholecystokinin receptors, whereas DSS and LPS both induce inflammation via TLR-mediated signaling (54, 55). We speculate that TLR-mediated signaling may involve the induction of Aag, Alkbh2, and Alkbh3 in cells that is crucial for colonic tissue repair and that in the absence of functional AAG, ALKBH2, and ALKBH3 repair enzymes, these cells simply do not survive exposure to RONS.
Remarkably, although the Aag–/–Alkbh2–/–Alkbh3–/– mice exhibited increased sensitivity to certain inflammatory challenges, a deficiency in these 3 DNA repair proteins did not significantly affect their ability to survive exposure to 2 different genotoxic DNA methylating agents (AOM and MMS). This suggests that the DNA base lesions induced during an inflammatory response to DSS or LPS are different than those induced by AOM and MMS, presumably reflect the generation of ε-base lesions but not methylated-base lesions. Consistent with this, Aag–/–Alkbh2–/–Alkbh3–/– mice exhibited increased levels of toxic and mutagenic εA and 1,N2-εG in colonic epithelial cells following DSS treatment. The in vivo studies reported here suggest that there is substantial redundancy among the 3 repair proteins, AAG, ALKBH2, and ALKBH3, and that together they provide enough DNA repair to prevent the lethality induced by the inflammation associated with DSS or LPS exposure. Although substrate overlap among these proteins was previously reported (37, 69), we provide in vivo evidence that no other DNA repair activity is able to adequately compensate in the absence of AAG, ALKBH2, and ALKBH3 during TLR-mediated acute inflammatory stress. Further, the redundancy among these 3 repair proteins underscores the importance of DNA repair for coping with inflammatory stresses that include both acute and chronic inflammation. We propose that AAG, ALKBH2, and ALKBH3 may be potential targets for genetic studies of IBD and human colorectal cancer. Indeed, AAG levels have already been shown to be elevated in inflamed regions, compared with those in uninflamed regions, of the colon in patients with UC (73). Similar monitoring of ALKBH2 and ALKBH3 levels in patients with IBD may also be warranted.
Methods
Animals.
Aag–/–, Alkbh2–/–, and Alkbh3–/– mice were described previously (25, 74). The Alkbh2–/–Alkbh3–/– mice (on a mixed C57BL/6 129Sv/Ev background) were rederived upon arrival at Massachusetts Institute of Technology. All mice used for experiments were on a mixed B6/129 background. Age-matched mice of both genders were 6–10 weeks old at the start of the treatment regimen.
Treatments.
We used the combination of AOM and DSS to induce chronic colitis and colorectal cancer (10, 43). This treatment consists of a single dose of 12.5 mg/kg AOM (Midwest Research Institute, NCI Chemical Repository) injected i.p., followed by 5 cycles of DSS. One DSS cycle consisted of 5 days of 2.5% DSS (MW 36,000–50,000, MP Biomedicals) in the drinking water, followed by 16 days of standard drinking water.
Whole-animal sensitivity to AOM and MMS (Sigma-Aldrich) was performed as in Deichmann and LeBlanc (75). In brief, 5–6 animals were i.p. injected with a series of doses, each being 50% more than the previous dose. The lowest dose that induces signs of lethality within 30 days is the approximate LD50.
To induce acute pancreatitis, mice were i.p. injected hourly with cerulein (Sigma-Aldrich) (50 μg/kg) for 6 injections. The mice were euthanized 24 hours or 7 days following the first injection; the pancreata were harvested and fixed in formalin for histological analysis.
Sensitivity to LPS (from E. coli 026:B6, Sigma-Aldrich) was assessed by i.p. injecting a range of LPS doses (5 mg/kg to 30 mg/kg) and evaluating survival over a 7-day period.
Tissue collection.
During the AOM/DSS experiment, mice were weighed and monitored regularly and euthanized if moribund or if they exhibited greater than 20% loss in body weight. Five days following the conclusion of the final DSS cycle, the cohorts were euthanized by CO2 asphyxiation. Subsequently, each mouse was weighed, and the colon and spleen were carefully dissected from each mouse. The colon (from cecum to anus) was removed from each mouse, washed with 1X PBS, and cut open lengthwise; the colon length was measured, and the number of polyps was counted. The spleen was weighed, and both the spleen and colon were fixed in 10% neutral buffered formalin for 18 hours. Blood obtained from a cardiac puncture was collected in EDTA-containing blood collection tubes (Sardstedt) to perform a complete blood count (Hemavet 950FS, Drew Scientific Inc.).
Tissue processing and histopathology.
Tissues were embedded in paraffin, sectioned at 5 μm, and stained with H&E at The David H. Koch Institute for Integrative Cancer Research at MIT Histology Core Facility. All H&E-stained slides were then blindly scored by a board-certified veterinary pathologist (S. Muthupalani). AOM/DSS colons were scored as described previously (10). Colons from mice treated with a single DSS cycle were scored for the additional criteria of edema and mucosal necrosis/degeneration. Cerulein-treated pancreata samples were scored for the levels of inflammation, edema, and acinar cell degeneration/necrosis/loss on a 0–4 scale; the sum of these criteria was the overall histopathological score.
Measurement of modified bases.
Six- to eight-week-old male mice were administered 1.75% DSS for 5 days. To prolong survival, mice were supplemented subcutaneously with 1 ml sterile 0.9% sodium chloride on day 8 and 9. Colons were harvested from untreated mice (day 0), immediately following DSS treatment (day 5), and 4 days following DSS withdrawal (day 9). Colonic epithelial cells were harvested as previously described (10). εA and 1,N2-εG lesions were quantified as described previously (10, 76, 77). Measurements were carried out on DNA prepared from 3 or more animals per group, with the following exceptions: Aag–/– (untreated), Aag–/–Alkbh2–/–Alkbh3–/– (untreated), and Aag–/– (day 5), all of which were prepared from 2 animals.
Statistics.
Statistical analyses were performed using GraphPad Prism software. Data are represented as mean ± SEM or mean ± SD, as indicated in figure legends. Statistical significance was determined by performing Mann-Whitney test or 2-way ANOVA with Bonferroni post-test analysis. Kaplan-Meier survival curves were generated, and difference in survival was determined using the log-rank test. A P value was considered significant if P < 0.05.
Study approval.
Mice were housed in an Association of Assessment and Accreditation of Laboratory Animal Care–accredited facility. All procedures were approved by the Massachusetts Institute of Technology Committee on Animal Care.
Supplementary Material
Acknowledgments
We thank Yusuf Erkul for contributions during the initiation of this project. We also thank Alicia Caron, Weijia Zhang, and Ruben Chevere at The David H. Koch Institute for Integrative Cancer Research Histology Core for technical help. This work was supported by NIH grants R01-CA075576, R01-CA055042, R01-CA149261, and P30-ES02109 (to L.D. Samson). C.-Y.I. Lee was supported by NIH grant T32-ES007020, and L.D. Samson is an American Cancer Society Research Professor.
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Citation for this article: J Clin Invest. 2012;122(7):2680–2689. doi:10.1172/JCI63338.
Lisiane B. Meira’s present address is: Faculty of Health and Medical Sciences, University of Surrey, Guildford, Surrey, United Kingdom.
Chun-Yue I. Lee’s present address is: Reata Pharmaceuticals Inc., Irving, Texas, USA.
References
- 1.Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature. 2008;454(7203):436–444. doi: 10.1038/nature07205. [DOI] [PubMed] [Google Scholar]
- 2.Balkwill F, Mantovani A. Inflammation and cancer: back to Virchow? Lancet. 2001;357(9255):539–545. doi: 10.1016/S0140-6736(00)04046-0. [DOI] [PubMed] [Google Scholar]
- 3.Harpaz N, Polydorides AD. Colorectal dysplasia in chronic inflammatory bowel disease: pathology, clinical implications, and pathogenesis. Arch Pathol Lab Med. 2010;134(6):876–895. doi: 10.5858/134.6.876. [DOI] [PubMed] [Google Scholar]
- 4.Hardy RG, Meltzer SJ, Jankowski JA. ABC of colorectal cancer. Molecular basis for risk factors. BMJ. 2000;321(7265):886–889. doi: 10.1136/bmj.321.7265.886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Eaden JA, Abrams KR, Mayberry JF. The risk of colorectal cancer in ulcerative colitis: a meta-analysis. Gut. 2001;48(4):526–535. doi: 10.1136/gut.48.4.526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dannenberg AJ, Subbaramaiah K. Targeting cyclooxygenase-2 in human neoplasia: rationale and promise. Cancer Cell. 2003;4(6):431–436. doi: 10.1016/S1535-6108(03)00310-6. [DOI] [PubMed] [Google Scholar]
- 7.Smalley W, DuBois R. Colorectal cancer and nonsteroidal anti-inflammatory drugs. Adv Pharmacol. 1997;39:1–20. doi: 10.1016/s1054-3589(08)60067-8. [DOI] [PubMed] [Google Scholar]
- 8.Tardieu D, Jaeg JP, Deloly A, Corpet DE, Cadet J, Petit CR. The COX-2 inhibitor nimesulide suppresses superoxide and 8-hydroxy-deoxyguanosine formation, and stimulates apoptosis in mucosa during early colonic inflammation in rats. Carcinogenesis. 2000;21(5):973–97. doi: 10.1093/carcin/21.5.973. [DOI] [PubMed] [Google Scholar]
- 9.Bartsch H. Hunting for electrophiles that harm human DNA: Frits Sobels Award Lecture. Mutagenesis. 2002;17(4):281–287. doi: 10.1093/mutage/17.4.281. [DOI] [PubMed] [Google Scholar]
- 10.Meira LB, et al. DNA damage induced by chronic inflammation contributes to colon carcinogenesis in mice. J Clin Invest. 2008;118(7):2516–2525. doi: 10.1172/JCI35073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420(6917):860–867. doi: 10.1038/nature01322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wiseman H, Halliwell B. Damage to DNA by reactive oxygen and nitrogen species: role in inflammatory disease and progression to cancer. Biochem J. 1996;313(pt 1):17–29. doi: 10.1042/bj3130017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chung FL, Chen HJ, Nath RG. Lipid peroxidation as a potential endogenous source for the formation of exocyclic DNA adducts. Carcinogenesis. 1996;17(10):2105–2111. doi: 10.1093/carcin/17.10.2105. [DOI] [PubMed] [Google Scholar]
- 14.Chung FL, Nath RG, Ocando J, Nishikawa A, Zhang L. Deoxyguanosine adducts of t-4-hydroxy-2-nonenal are endogenous DNA lesions in rodents and humans: detection and potential sources. Cancer Res. 2000;60(6):1507–1511. [PubMed] [Google Scholar]
- 15.Bartsch H, Ohshima H, Pignatelli B, Calmels S. Endogenously formed N-nitroso compounds and nitrosating agents in human cancer etiology. Pharmacogenetics. 1992;2(6):272–277. doi: 10.1097/00008571-199212000-00005. [DOI] [PubMed] [Google Scholar]
- 16.Wu Y, Brouet I, Calmels S, Bartsch H, Ohshima H. Increased endogenous N-nitrosamine and nitrate formation by induction of nitric oxide synthase in rats with acute hepatic injury caused by Propionibacterium acnes and lipopolysaccharide administration. Carcinogenesis. 1993;14(1):7–10. doi: 10.1093/carcin/14.1.7. [DOI] [PubMed] [Google Scholar]
- 17.Bartsch H, Nair J. Potential role of lipid peroxidation derived DNA damage in human colon carcinogenesis: studies on exocyclic base adducts as stable oxidative stress markers. Cancer Detect Prev. 2002;26(4):308–312. doi: 10.1016/S0361-090X(02)00093-4. [DOI] [PubMed] [Google Scholar]
- 18.Nair U, Bartsch H, Nair J. Lipid peroxidation-induced DNA damage in cancer-prone inflammatory diseases: a review of published adduct types and levels in humans. Free Radic Biol Med. 2007;43(8):1109–1120. doi: 10.1016/j.freeradbiomed.2007.07.012. [DOI] [PubMed] [Google Scholar]
- 19.Nair J, Gansauge F, Beger H, Dolara P, Winde G, Bartsch H. Increased etheno-DNA adducts in affected tissues of patients suffering from Crohn’s disease, ulcerative colitis, and chronic pancreatitis. Antioxid Redox Signal. 2006;8(5–6):1003–1010. doi: 10.1089/ars.2006.8.1003. [DOI] [PubMed] [Google Scholar]
- 20.Westbrook AM, Wei B, Braun J, Schiestl RH. Intestinal mucosal inflammation leads to systemic genotoxicity in mice. Cancer Res. 2009;69(11):4827–4834. doi: 10.1158/0008-5472.CAN-08-4416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Saparbaev M, Laval J. Excision of hypoxanthine from DNA containing dIMP residues by the Escherichia coli, yeast, rat, and human alkylpurine DNA glycosylases. Proc Natl Acad Sci U S A. 1994;91(13):5873–5877. doi: 10.1073/pnas.91.13.5873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gallagher P, Brent T. Partial purification and characterization of 3-methyladenine-DNA glycosylase from human placenta. Biochemistry. 1982;21(25):6404–6409. doi: 10.1021/bi00268a013. [DOI] [PubMed] [Google Scholar]
- 23.Singer B, et al. Both purified human 1,N6-ethenoadenine-binding protein and purified human 3-methyladenine-DNA glycosylase act on 1,N6-ethenoadenine and 3-methyladenine. Proc Natl Acad Sci U S A. 1992;89(20):9386–9390. doi: 10.1073/pnas.89.20.9386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.O’Connor TR. Purification and characterization of human 3-methyladenine-DNA glycosylase. Nucleic Acids Res. 1993;21(24):5561–5569. doi: 10.1093/nar/21.24.5561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Engelward BP, et al. Base excision repair deficient mice lacking the Aag alkyladenine DNA glycosylase. Proc Natl Acad Sci U S A. 1997;94(24):13087–13092. doi: 10.1073/pnas.94.24.13087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hang B, Singer B, Margison GP, Elder RH. Targeted deletion of alkylpurine-DNA-N-glycosylase in mice eliminates repair of 1,N6-ethenoadenine and hypoxanthine but not of 3,N4-ethenocytosine or 8-oxoguanine. Proc Natl Acad Sci U S A. 1997;94(24):12869–12874. doi: 10.1073/pnas.94.24.12869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Miao F, Bouziane M, O’Connor TR. Interaction of the recombinant human methylpurine-DNA glycosylase (MPG protein) with oligodeoxyribonucleotides containing either hypoxanthine or abasic sites. Nucleic Acids Res. 1998;26(17):4034–4041. doi: 10.1093/nar/26.17.4034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bessho T, et al. Repair of 8-hydroxyguanine in DNA by mammalian N-methylpurine-DNA glycosylase. Proc Natl Acad Sci U S A. 1993;90(19):8901–8904. doi: 10.1073/pnas.90.19.8901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gros L, Maksimenko AV, Privezentzev CV, Laval J, Saparbaev MK. Hijacking of the human alkyl-N-purine-DNA glycosylase by 3,N4-ethenocytosine, a lipid peroxidation-induced DNA adduct. J Biol Chem. 2004;279(17):17723–17730. doi: 10.1074/jbc.M314010200. [DOI] [PubMed] [Google Scholar]
- 30.Lee C, et al. Recognition and processing of a new repertoire of DNA substrates by human 3-methyladenine DNA glycosylase (AAG). Biochemistry. 2009;48(9):1850–1861. doi: 10.1021/bi8018898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lingaraju GM, Davis CA, Setser JW, Samson LD, Drennan CL. Structural basis for the inhibition of human alkyladenine DNA glycosylase (AAG) by 3,N4-ethenocytosine containing DNA. J Biol Chem. 2011;286(15):13205–13213. doi: 10.1074/jbc.M110.192435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Delaney JC, et al. AlkB reverses etheno DNA lesions caused by lipid oxidation in vitro and in vivo. Nat Struct Mol Biol. 2005;12(10):855–860. doi: 10.1038/nsmb996. [DOI] [PubMed] [Google Scholar]
- 33.Trewick SC, Henshaw TF, Hausinger RP, Lindahl T, Sedgwick B. Oxidative demethylation by Escherichia coli AlkB directly reverts DNA base damage. Nature. 2002;419(6903):174–178. doi: 10.1038/nature00908. [DOI] [PubMed] [Google Scholar]
- 34.Falnes PO, Johansen RF, Seeberg E. AlkB-mediated oxidative demethylation reverses DNA damage in Escherichia coli. Nature. 2002;419(6903):178–182. doi: 10.1038/nature01048. [DOI] [PubMed] [Google Scholar]
- 35.Delaney JC, Essigmann JM. Mutagenesis, genotoxicity, and repair of 1-methyladenine, 3-alkylcytosines, 1-methylguanine, and 3-methylthymine in alkB Escherichia coli. Proc Natl Acad Sci U S A. 2004;101(39):14051–14056. doi: 10.1073/pnas.0403489101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Koivisto P, Robins P, Lindahl T, Sedgwick B. Demethylation of 3-methylthymine in DNA by bacterial and human DNA dioxygenases. J Biol Chem. 2004;279(39):40470–40474. doi: 10.1074/jbc.M407960200. [DOI] [PubMed] [Google Scholar]
- 37.Ringvoll J, et al. AlkB homologue 2-mediated repair of ethenoadenine lesions in mammalian DNA. Cancer Res. 2008;68(11):4142–4149. doi: 10.1158/0008-5472.CAN-08-0796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mishina Y, Yang CG, He C. Direct repair of the exocyclic DNA adduct 1,N6-ethenoadenine by the DNA repair AlkB proteins. J Am Chem Soc. 2005;127(42):14594–14595. doi: 10.1021/ja055957m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dango S, et al. DNA unwinding by ASCC3 helicase is coupled to ALKBH3-dependent DNA alkylation repair and cancer cell proliferation. Mol Cell. 2011;44(3):373–384. doi: 10.1016/j.molcel.2011.08.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fu D, Samson LD. Direct repair of 3,N(4)-ethenocytosine by the human ALKBH2 dioxygenase is blocked by the AAG/MPG glycosylase. DNA Repair (Amst). 2012;11(1):46–52. doi: 10.1016/j.dnarep.2011.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Barbin A, Wang R, O’Connor PJ, Elder RH. Increased formation and persistence of 1,N6-Ethenoadenine in DNA is not associated with higher susceptibility to carcinogenesis in alkylpurine- DNA-N-Glycosylase knockout mice treated with vinyl carbamate. Cancer Res. 2003;63(22):7699–7703. [PubMed] [Google Scholar]
- 42.Ham A-JL, et al. New immunoaffinity-LC-MS/MS methodology reveals that Aag null mice are deficient in their ability to clear 1,N6-etheno-deoxyadenosine DNA lesions from lung and liver in vivo. DNA Repair. 2004;3(3):257–265. doi: 10.1016/j.dnarep.2003.11.003. [DOI] [PubMed] [Google Scholar]
- 43.Neufert C, Becker C, Neurath MF. An inducible mouse model of colon carcinogenesis for the analysis of sporadic and inflammation-driven tumor progression. Nat Protoc. 2007;2(8):1998–2004. doi: 10.1038/nprot.2007.279. [DOI] [PubMed] [Google Scholar]
- 44.Grivennikov S, et al. IL-6 and Stat3 are required for survival of intestinal epithelial cells and development of colitis-associated cancer. Cancer Cell. 2009;15(2):103–113. doi: 10.1016/j.ccr.2009.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kitajima S, Takuma S, Morimoto M. Changes in colonic mucosal permeability in mouse colitis induced with dextran sulfate sodium. Exp Anim. 1999;48(3):137–143. doi: 10.1538/expanim.48.137. [DOI] [PubMed] [Google Scholar]
- 46.Pull SL, Doherty JM, Mills JC, Gordon JI, Stappenbeck TS. Activated macrophages are an adaptive element of the colonic epithelial progenitor niche necessary for regenerative responses to injury. Proc Natl Acad Sci U S A. 2005;102(1):99–104. doi: 10.1073/pnas.0405979102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dosanjh MK, Chenna A, Kim E, Fraenkel-Conrat H, Samson L, Singer B. All four known cyclic adducts formed in DNA by the vinyl chloride metabolite chloroacetaldehyde are released by a human DNA glycosylase. Proc Natl Acad Sci U S A. 1994;91(3):1024–1028. doi: 10.1073/pnas.91.3.1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Saparbaev M, et al. 1,N(2)-ethenoguanine, a mutagenic DNA adduct, is a primary substrate of Escherichia coli mismatch-specific uracil-DNA glycosylase and human alkylpurine-DNA-N-glycosylase. . J Biol Chem. 2002;277(30):26987–26993. doi: 10.1074/jbc.M111100200. [DOI] [PubMed] [Google Scholar]
- 49.Barbin A. Etheno-adduct-forming chemicals: from mutagenicity testing to tumor mutation spectra. Mutat Res. 2000;462(2–3):55–69. doi: 10.1016/s1383-5742(00)00014-4. [DOI] [PubMed] [Google Scholar]
- 50.Dimitri A, Goodenough AK, Guengerich FP, Broyde S, Scicchitano DA. Transcription processing at 1,N2-ethenoguanine by human RNA polymerase II and bacteriophage T7 RNA polymerase. J Mol Biol. 2008;375(2):353–366. doi: 10.1016/j.jmb.2007.10.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Basu AK, Wood ML, Niedernhofer LJ, Ramos LA, Essigmann JM. Mutagenic and genotoxic effects of three vinyl chloride-induced DNA lesions:1,N6-ethenoadenine, 3,N4-ethenocytosine, and 4-amino-5-(imidazol-2-yl)imidazole. Biochemistry. 1993;32(47):12793–12801. doi: 10.1021/bi00210a031. [DOI] [PubMed] [Google Scholar]
- 52.Hoshino K, et al. Cutting edge: Toll-like receptor 4 (TLR4)-deficient mice are hyporesponsive to lipopolysaccharide: evidence for TLR4 as the Lps gene product. J Immunol. 1999;162(7):3749–3752. [PubMed] [Google Scholar]
- 53.Chow JC, Young DW, Golenbock DT, Christ WJ, Gusovsky F. Toll-like receptor-4 mediates lipopolysaccharide-induced signal transduction. J Biol Chem. 1999;274(16):10689–10692. doi: 10.1074/jbc.274.16.10689. [DOI] [PubMed] [Google Scholar]
- 54.Poltorak A, et al. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science. 1998;282(5396):2085–2088. doi: 10.1126/science.282.5396.2085. [DOI] [PubMed] [Google Scholar]
- 55.Kim H. Cerulein pancreatitis: oxidative stress, inflammation, and apoptosis. Gut and Liver. 2008;2(2):74–80. doi: 10.5009/gnl.2008.2.2.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cordon-Cardo C, Prives C. At the crossroads of inflammation and tumorigenesis. J Exp Med. 1999;190(10):1367–1370. doi: 10.1084/jem.190.10.1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Balkwill F, Charles KA, Mantovani A. Smoldering and polarized inflammation in the initiation and promotion of malignant disease. Cancer Cell. 2005;7(3):211–217. doi: 10.1016/j.ccr.2005.02.013. [DOI] [PubMed] [Google Scholar]
- 58.Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. 2010;140(6):883–899. doi: 10.1016/j.cell.2010.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 60.Asquith M, Powrie F. An innately dangerous balancing act: intestinal homeostasis, inflammation, and colitis-associated cancer. J Exp Med. 2010;207(8):1573–1577. doi: 10.1084/jem.20101330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rakoff-Nahoum S, Hao L, Medzhitov R. Role of toll-like receptors in spontaneous commensal-dependent colitis. Immunity. 2006;25(2):319–329. doi: 10.1016/j.immuni.2006.06.010. [DOI] [PubMed] [Google Scholar]
- 62.Cario E. Toll-like receptors in inflammatory bowel diseases: a decade later. Inflamm Bowel Dis. 2010;16(9):1583–1597. doi: 10.1002/ibd.21282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Schroder K, Tschopp J. The inflammasomes. Cell. 2010;140(6):821–832. doi: 10.1016/j.cell.2010.01.040. [DOI] [PubMed] [Google Scholar]
- 64. Zaki MH, Boyd KL, Vogel P, Kastan MB, Lamkanfi M, Kanneganti T-D. The NLRP3 inflammasome protects against loss of epithelial integrity and mortality during experimental colitis. Immunity. 2010. 32 3 379 391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rakoff-Nahoum S, Paglino J, Eslami-Varzaneh F, Edberg S, Medzhitov R. Recognition of commensal microflora by toll-like receptors is required for intestinal homeostasis. Cell. 2004;118(2):229–241. doi: 10.1016/j.cell.2004.07.002. [DOI] [PubMed] [Google Scholar]
- 66.Dupaul-Chicoine J, et al. Control of intestinal homeostasis, colitis, and colitis-associated colorectal cancer by the inflammatory caspases. Immunity. 2010;32(3):367–378. doi: 10.1016/j.immuni.2010.02.012. [DOI] [PubMed] [Google Scholar]
- 67.Salcedo R, et al. MyD88-mediated signaling prevents development of adenocarcinomas of the colon: role of interleukin 18. J Exp Med. 2010;207(8):1625–1636. doi: 10.1084/jem.20100199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Menendez D, Shatz M, Azzam K, Garantziotis S, Fessler MB, Resnick MA. The Toll-like receptor gene family is integrated into human DNA damage and p53 networks. PLoS Genet. 2011;7(3):e1001360. doi: 10.1371/journal.pgen.1001360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lee CY, et al. Recognition and processing of a new repertoire of DNA substrates by human 3-methyladenine DNA glycosylase (AAG). Biochemistry. 2009;48(9):1850–1861. doi: 10.1021/bi8018898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wyatt MD, Allan JM, Lau AY, Ellenberger TE, Samson LD. 3-methyladenine DNA glycosylases: structure, function, and biological importance. Bioessays. 1999;21(8):668–676. doi: 10.1002/(SICI)1521-1878(199908)21:8<668::AID-BIES6>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 71.Wirtz S, Nagel G, Eshkind L, Neurath MF, Samson LD, Kaina B. Both base excision repair and O6-methylguanine-DNA methyltransferase protect against methylation-induced colon carcinogenesis. Carcinogenesis. 2010;31(12):2111–2117. doi: 10.1093/carcin/bgq174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Xiao H, et al. The Toll-interleukin-1 receptor member SIGIRR regulates colonic epithelial homeostasis, inflammation, and tumorigenesis. Immunity. 2007;26(4):461–475. doi: 10.1016/j.immuni.2007.02.012. [DOI] [PubMed] [Google Scholar]
- 73.Hofseth LJ, et al. The adaptive imbalance in base excision-repair enzymes generates microsatellite instability in chronic inflammation. J Clin Invest. 2003;112(12):1887–1894. doi: 10.1172/JCI19757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ringvoll J, et al. Repair deficient mice reveal mABH2 as the primary oxidative demethylase for repairing 1meA and 3meC lesions in DNA. EMBO J. 2006;25(10):2189–2189. doi: 10.1038/sj.emboj.7601109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Deichmann WB, LeBlanc TJ. Determination of the approximate lethal dose with about six animals. . J Ind Hyg Toxicol. 1943;25:415–417. [Google Scholar]
- 76.Pang B, et al. Lipid peroxidation dominates the chemistry of DNA adduct formation in a mouse model of inflammation. Carcinogenesis. 2007;28(8):1807–1813. doi: 10.1093/carcin/bgm037. [DOI] [PubMed] [Google Scholar]
- 77.Taghizadeh K, et al. Quantification of DNA damage products resulting from deamination, oxidation and reaction with products of lipid peroxidation by liquid chromatography isotope dilution tandem mass spectrometry. Nat Protoc. 2008;3(8):1287–1298. doi: 10.1038/nprot.2008.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.