

Abstract
Proton transfer (PT) through and across aqueous interfaces is a fundamental process in chemistry and biology. Notwithstanding its importance, it is not generally realized that interfacial PT is quite different from conventional PT in bulk water. Here we show that, in contrast with the behavior of strong nitric acid in aqueous solution, gas-phase HNO3 does not dissociate upon collision with the surface of water unless a few ions (> 1 per 106 H2O) are present. By applying online electrospray ionization mass spectrometry to monitor in situ the surface of aqueous jets exposed to HNO3(g) beams we found that 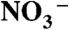 production increases dramatically on > 30-μM inert electrolyte solutions. We also performed quantum mechanical calculations confirming that the sizable barrier hindering HNO3 dissociation on the surface of small water clusters is drastically lowered in the presence of anions. Anions electrostatically assist in drawing the proton away from
production increases dramatically on > 30-μM inert electrolyte solutions. We also performed quantum mechanical calculations confirming that the sizable barrier hindering HNO3 dissociation on the surface of small water clusters is drastically lowered in the presence of anions. Anions electrostatically assist in drawing the proton away from 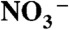 lingering outside the cluster, whose incorporation is hampered by the energetic cost of opening a cavity therein. Present results provide both direct experimental evidence and mechanistic insights on the counterintuitive slowness of PT at water-hydrophobe boundaries and its remarkable sensitivity to electrostatic effects.
lingering outside the cluster, whose incorporation is hampered by the energetic cost of opening a cavity therein. Present results provide both direct experimental evidence and mechanistic insights on the counterintuitive slowness of PT at water-hydrophobe boundaries and its remarkable sensitivity to electrostatic effects.
Keywords: air–water interface, acid-base, catalysis, nitric acid dissociation
Proton transfers (PTs) at water interfaces, such as water boundaries with air (1, 2) or lipid membranes (3), intervene in fundamental phenomena. Arguably the most important PTs are those that take place through and across water boundaries rather than in the bulk liquid. Interfacial PTs participate in the acidification of the ocean (4), the chemistry of atmospheric gases and aerosols (1, 5, 6), the generation of the electrochemical gradients that drive energy transduction across biomembranes (3, 7, 8), and in enzymatic function (9, 10) because the activation of neutral species is most generally accomplished via acid-base catalysis (11). Interfacial PT, in contrast with conventional PT in bulk water, depends sensitively on the extent of ion hydration because the density of water in interfacial layers vanishes within 1-nm (12). The acidity of hydronium at the interface, 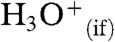 , is therefore expected to bridge that of
, is therefore expected to bridge that of 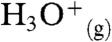 , which protonates most nonalkane species in the gas-phase (13), and
, which protonates most nonalkane species in the gas-phase (13), and 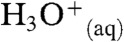 , which neutralizes only relatively strong bases in solution. Critically controlled by ion hydration in thin yet cohesive interfacial water layers that resist ion penetration, PT “on water” clearly confronts unique constraints. Species that behave as strong acids “in water” may become weak ones on water if dissociation were hindered by kinetic and/or thermodynamic factors in the interfacial region (14, 15).
, which neutralizes only relatively strong bases in solution. Critically controlled by ion hydration in thin yet cohesive interfacial water layers that resist ion penetration, PT “on water” clearly confronts unique constraints. Species that behave as strong acids “in water” may become weak ones on water if dissociation were hindered by kinetic and/or thermodynamic factors in the interfacial region (14, 15).
Herein we address these important issues and report the results of experiments in which we monitor the dissociation of gaseous nitric acid HNO3(g) molecules in collisions with interfacial water, H2O(if), reaction 1 (Eq. 1):
| [1] |
The Technique
Experiments were conducted by intersecting continuously refreshed surfaces of free-flowing aqueous jets with HNO3(g)/N2(g) beams at ambient temperature and pressure. The formation of interfacial nitrate, 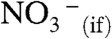 , was monitored in situ via surface-specific online electrospray mass spectrometry (ESMS) (16, 17) (SI Text and Figs. S1 and S2). ESMS is routinely used to analyze the composition of bulk liquids. However, we have demonstrated that by changing the instrumental configuration and operating parameters it is possible to sample the interfacial layers of the liquid jet. We have previously taken advantage of the high sensitivity, surface selectivity, and unequivocal identification capabilities of our modified electrospray mass spectrometer to investigate fast gas–liquid reactions on the surface of aqueous jets (5, 18). The claim that the mass spectra obtained in our instrument mostly reflect the ion composition of the outermost layers of the jet has been validated by showing that: (i) the relative anion abundances (i.e., the relative mass spectral signal intensities) measured on jets consisting of equimolar solutions of mixed salts are not identical but follow a normal Hofmeister series (as expected at the air–water interface and confirmed by other surface-sensitive techniques), and are specifically affected by surfactants (cationic or anionic) (19); and (ii) mass spectra of jets exposed to reactive gases reveal the presence of species necessarily produced at the interface rather than in the bulk liquid (20, 21).
, was monitored in situ via surface-specific online electrospray mass spectrometry (ESMS) (16, 17) (SI Text and Figs. S1 and S2). ESMS is routinely used to analyze the composition of bulk liquids. However, we have demonstrated that by changing the instrumental configuration and operating parameters it is possible to sample the interfacial layers of the liquid jet. We have previously taken advantage of the high sensitivity, surface selectivity, and unequivocal identification capabilities of our modified electrospray mass spectrometer to investigate fast gas–liquid reactions on the surface of aqueous jets (5, 18). The claim that the mass spectra obtained in our instrument mostly reflect the ion composition of the outermost layers of the jet has been validated by showing that: (i) the relative anion abundances (i.e., the relative mass spectral signal intensities) measured on jets consisting of equimolar solutions of mixed salts are not identical but follow a normal Hofmeister series (as expected at the air–water interface and confirmed by other surface-sensitive techniques), and are specifically affected by surfactants (cationic or anionic) (19); and (ii) mass spectra of jets exposed to reactive gases reveal the presence of species necessarily produced at the interface rather than in the bulk liquid (20, 21).
Mass spectrometers report the net charge that arrives at the detector per unit time. Therefore, the 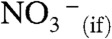 produced in reaction 1 on the surface of the electroneutral liquid jet can be detected after it has been separated from
produced in reaction 1 on the surface of the electroneutral liquid jet can be detected after it has been separated from 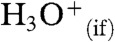 counterions. Separation is brought about during the pneumatic breakup of the liquid jet by a fast annular N2(g) nebulizer gas flow, which shears the outermost liquid layers into droplets carrying net charges of either sign. These droplets have size and net charge distributions, and carry more surface and electrostatic energies than the original jet, at the cost of the kinetic energy lost by the nebulizer gas. A key feature of our ESMS instrumental configuration is that the jet is orthogonal to the inlet to the mass spectrometer (Figs. S1 and S2). This geometry overwhelmingly favors the detection of ions emanating from the peripheral layers of the jet. Ions are ultimately ejected to the gas-phase because of severe charge crowding in the nanodroplets that result from extensive solvent evaporation (22). We have presented detailed data analysis (16), based on mass balances and the application of the kinetic theory of gases to fast gas–liquid reactions, which suggests that the thickness of the interfacial layers sampled in these experiments is certainly within a few nm, and most likely approximately 1 nm (see below and SI Text).
counterions. Separation is brought about during the pneumatic breakup of the liquid jet by a fast annular N2(g) nebulizer gas flow, which shears the outermost liquid layers into droplets carrying net charges of either sign. These droplets have size and net charge distributions, and carry more surface and electrostatic energies than the original jet, at the cost of the kinetic energy lost by the nebulizer gas. A key feature of our ESMS instrumental configuration is that the jet is orthogonal to the inlet to the mass spectrometer (Figs. S1 and S2). This geometry overwhelmingly favors the detection of ions emanating from the peripheral layers of the jet. Ions are ultimately ejected to the gas-phase because of severe charge crowding in the nanodroplets that result from extensive solvent evaporation (22). We have presented detailed data analysis (16), based on mass balances and the application of the kinetic theory of gases to fast gas–liquid reactions, which suggests that the thickness of the interfacial layers sampled in these experiments is certainly within a few nm, and most likely approximately 1 nm (see below and SI Text).
Results
Fig. 1 displays mass spectral 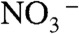 (m/z = 62) signal intensities, I62, as a function of pH (of the bulk aqueous solution) on liquid jets exposed to HNO3(g); I62 remains above detection limits on the surface of pH 4.5 to 9.5 jets, but sharply increases both on more basic and more acidic solutions to limiting values,
(m/z = 62) signal intensities, I62, as a function of pH (of the bulk aqueous solution) on liquid jets exposed to HNO3(g); I62 remains above detection limits on the surface of pH 4.5 to 9.5 jets, but sharply increases both on more basic and more acidic solutions to limiting values, 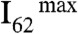 , above pH 11 and below pH 3. Notably, we found that
, above pH 11 and below pH 3. Notably, we found that 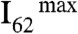 values are uniformly reached at all pH values on > 1-mM NaCl jets. The previously reported uptake coefficient of HNO3(g) on deionized water (γ > 0.1) (23), reveals that only a small fraction of the HNO3(g) molecules colliding with the surface of water are incorporated into the bulk liquid, where they fully dissociate [pKa(HNO3(aq)) = -1.4]. Therefore, the small
values are uniformly reached at all pH values on > 1-mM NaCl jets. The previously reported uptake coefficient of HNO3(g) on deionized water (γ > 0.1) (23), reveals that only a small fraction of the HNO3(g) molecules colliding with the surface of water are incorporated into the bulk liquid, where they fully dissociate [pKa(HNO3(aq)) = -1.4]. Therefore, the small 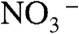 signals detected in our experiments on pure water jets indicate that we do sample their outermost interfacial layers (24, 25), and confirms that most of the mass-accommodated HNO3 diffuses in undissociated form through such layers. The fact that the production of
signals detected in our experiments on pure water jets indicate that we do sample their outermost interfacial layers (24, 25), and confirms that most of the mass-accommodated HNO3 diffuses in undissociated form through such layers. The fact that the production of 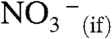 is dramatically enhanced by inert anions on water hints at the possibility that the barrier preventing HNO3 dissociation at the interface might be kinetic rather than thermodynamic (26, 27). In summary, the results of Fig. 1 and Fig. S3 provide evidence that HNO3(g) behaves as a weak acid on the surface of water, and extrinsic inert ions can significantly catalyze HNO3 dissociation therein (15, 28).
is dramatically enhanced by inert anions on water hints at the possibility that the barrier preventing HNO3 dissociation at the interface might be kinetic rather than thermodynamic (26, 27). In summary, the results of Fig. 1 and Fig. S3 provide evidence that HNO3(g) behaves as a weak acid on the surface of water, and extrinsic inert ions can significantly catalyze HNO3 dissociation therein (15, 28).
Fig. 1.

Electrospray mass spectral nitrate signal intensities (I62) detected on water or 1-mM NaCl microjets exposed to 3 × 1012 molecules cm-3 of gaseous nitric acid for approximately 10 μs as functions of pH. Solid, dashed lines are linear regression and 95% confidence limits, respectively, to the data on 1-mM NaCl. Error bars estimated from reproducibility tests. All experiments under 1 atm of N2 at 293 K.
The air–water interface of electrolyte solutions is preferentially populated by anions. This is borne out by the negative surface potential of most electrolyte solutions (29), by surface-specific spectroscopic studies (30–33), and by theoretical predictions. The adsorption of ions to the surface was surmised long ago from the surface tension minima observed in electrolyte solutions at approximately 1 mM. They were accounted for by electrostatic interactions among ions that saturate the surface of water at approximately 1 mM (34)—i.e., in the concentration range in which we observe an increase of HNO3 dissociation on water (30, 33). The saturation dependence of 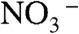 production on electrolyte concentration (Fig. S3A) can be ascribed to catalysis by anions A- adsorbed to identical, noninteracting sites of the air–water interface—i.e., I62 = I62,max[A-]/(K1/2 + [A-]) (30). We derive K1/2 = 128 μM (NaCl) and K1/2 = 77 μM (MgSO4) (i.e., the concentrations at which the interface would be half-saturated with catalyzing anions), which are commensurate with the values ([NaCl]max = 400 μM and [MgSO4]max = 200 μM) deduced from SHG experiments (30) (Fig. S3B). Although neither Cl- nor
production on electrolyte concentration (Fig. S3A) can be ascribed to catalysis by anions A- adsorbed to identical, noninteracting sites of the air–water interface—i.e., I62 = I62,max[A-]/(K1/2 + [A-]) (30). We derive K1/2 = 128 μM (NaCl) and K1/2 = 77 μM (MgSO4) (i.e., the concentrations at which the interface would be half-saturated with catalyzing anions), which are commensurate with the values ([NaCl]max = 400 μM and [MgSO4]max = 200 μM) deduced from SHG experiments (30) (Fig. S3B). Although neither Cl- nor 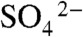 are as surface active as I- or
are as surface active as I- or 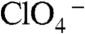 (32), they should approach the air–water interface far closer than the (Rion–ion) separations prevalent at the onset of catalytic effects (see below).
(32), they should approach the air–water interface far closer than the (Rion–ion) separations prevalent at the onset of catalytic effects (see below).
Hydronium, 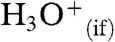 , the counterpart of
, the counterpart of 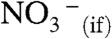 in reaction 1, was tracked by using hexanoic acid (PCOOH) as a proton scavenger. PCOOH is both a weak acid and a weak base in water: pKa(PCOOH)(aq) = 4.8,
in reaction 1, was tracked by using hexanoic acid (PCOOH) as a proton scavenger. PCOOH is both a weak acid and a weak base in water: pKa(PCOOH)(aq) = 4.8, 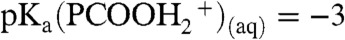 . However, we have shown that PCOOH is protonated on the surface of mildly acidic water, where it behaves as a stronger base:
. However, we have shown that PCOOH is protonated on the surface of mildly acidic water, where it behaves as a stronger base: 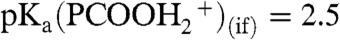 (17). Fig. 2 displays I117 (
(17). Fig. 2 displays I117 (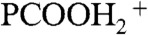 ), I118 (PCOOHD+), and I119 (
), I118 (PCOOHD+), and I119 (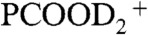 ) signal intensities from 1 mM PCOOH in 1∶1/D2O∶H2O jets (initially at pH 7) as functions of gas-phase HNO3(g) or DNO3(g) concentrations. Fig. 2 (Inset) shows the corresponding I62 and I115 (PCOO-) signal intensities versus HNO3(g) concentration. It is apparent that: (i) PCOO- is promptly neutralized upon exposure to the lowest HNO3(g)/DNO3(g) concentrations, whereas (ii) the protonation/deuteration (hydronation) of the weaker base PCOOH requires exposure to at least n > 2 × 1012 molecules cm-3. The fact that HNO3(g) dissociates on water containing the anions of either a stronger acid [pKa(HCl)(aq) = -7 versus pKa(HNO3)(aq) = -1.4] or a weaker one [pKa(PCOOH)(aq) = 4.8] supports the assertion that anions function as catalysts rather than proton acceptors. The appearance of hydronated species [(
) signal intensities from 1 mM PCOOH in 1∶1/D2O∶H2O jets (initially at pH 7) as functions of gas-phase HNO3(g) or DNO3(g) concentrations. Fig. 2 (Inset) shows the corresponding I62 and I115 (PCOO-) signal intensities versus HNO3(g) concentration. It is apparent that: (i) PCOO- is promptly neutralized upon exposure to the lowest HNO3(g)/DNO3(g) concentrations, whereas (ii) the protonation/deuteration (hydronation) of the weaker base PCOOH requires exposure to at least n > 2 × 1012 molecules cm-3. The fact that HNO3(g) dissociates on water containing the anions of either a stronger acid [pKa(HCl)(aq) = -7 versus pKa(HNO3)(aq) = -1.4] or a weaker one [pKa(PCOOH)(aq) = 4.8] supports the assertion that anions function as catalysts rather than proton acceptors. The appearance of hydronated species [(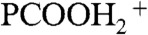 ), (PCOOHD+), and (
), (PCOOHD+), and (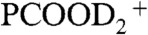 )] reveals that the surface of the jet has been acidified (from pH 7) to pH < 2.5. Because this is achieved under conditions in which the number of hydrons delivered by HNO3(g)/DNO3(g) on interfacial layers is much (approximately 103 times) smaller than those carried by the 50-μL min-1 1∶1/D2O∶H2O aqueous jet, the former must be confined to thin (Δ[cm]) interfacial layers during the lifetime of the jet. The relative abundances of the
)] reveals that the surface of the jet has been acidified (from pH 7) to pH < 2.5. Because this is achieved under conditions in which the number of hydrons delivered by HNO3(g)/DNO3(g) on interfacial layers is much (approximately 103 times) smaller than those carried by the 50-μL min-1 1∶1/D2O∶H2O aqueous jet, the former must be confined to thin (Δ[cm]) interfacial layers during the lifetime of the jet. The relative abundances of the 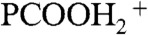 , PCOOHD+, and
, PCOOHD+, and 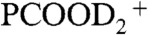 isotopologues are appreciably different under HNO3(g) or DNO3(g) (Fig. 2
A and B) and corroborate that the hydrons delivered by gaseous nitric acid remain (i.e., do not diffuse into and rapidly scramble their isotopic labels with the bulk solvent) in the interfacial layers sampled herein (SI Text and Fig. S4). The assumption that our experiments probe reactive events taking place in interfacial layers of molecular depth is therefore based on substantial evidence. From the frequency of HNO3(g) collisions with the jet given by the kinetic theory of gases, we estimate that Δ is approximately 1 × 10-7 cm (17) (SI Text).
isotopologues are appreciably different under HNO3(g) or DNO3(g) (Fig. 2
A and B) and corroborate that the hydrons delivered by gaseous nitric acid remain (i.e., do not diffuse into and rapidly scramble their isotopic labels with the bulk solvent) in the interfacial layers sampled herein (SI Text and Fig. S4). The assumption that our experiments probe reactive events taking place in interfacial layers of molecular depth is therefore based on substantial evidence. From the frequency of HNO3(g) collisions with the jet given by the kinetic theory of gases, we estimate that Δ is approximately 1 × 10-7 cm (17) (SI Text).
Fig. 2.

Electrospray mass spectral signal intensities of protonated isotopologues of hexanoic acid (PCOOH): 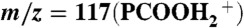 , m/z = 118(PCOOHD+), and
, m/z = 118(PCOOHD+), and 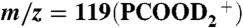 , detected on 1-mM PCOOH solutions in 1∶1/D2O∶H2O microjets (initially at pH 7) exposed to variable concentrations of gaseous HNO3 (A) or DNO3 (B). The inflection point corresponds to
, detected on 1-mM PCOOH solutions in 1∶1/D2O∶H2O microjets (initially at pH 7) exposed to variable concentrations of gaseous HNO3 (A) or DNO3 (B). The inflection point corresponds to 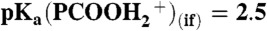 (17). Inset shows the evolution of the PCOO- (m/z = 115) and
(17). Inset shows the evolution of the PCOO- (m/z = 115) and 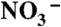 (m/z = 62) signals detected in negative ion mode. All experiments under 1 atm of N2 at 293 K.
(m/z = 62) signals detected in negative ion mode. All experiments under 1 atm of N2 at 293 K.
What is the minimum number of additional water molecules m that renders reaction 1 exoergic? The free energy required to produce a hydrated contact ion pair at the air–water interface, 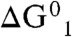 , can be estimated as the sum of the gas-phase process [
, can be estimated as the sum of the gas-phase process [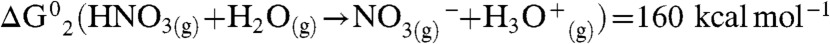 ] (13); plus the electrostatic energy released as the infinitely distant gas-phase point charges reach an approximately 3.3-Å separation in the contact ion pair (Eel = -100 kcal mol-1); plus the free energy of hydrating
] (13); plus the electrostatic energy released as the infinitely distant gas-phase point charges reach an approximately 3.3-Å separation in the contact ion pair (Eel = -100 kcal mol-1); plus the free energy of hydrating 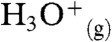 [
[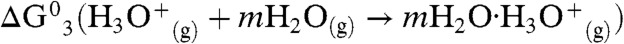 ]
]
Extant thermochemical data on (mH2O·H3O+) clusters (35) show that 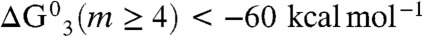 (i.e., reaction 1 is thermodynamically allowed for m≥4, even if
(i.e., reaction 1 is thermodynamically allowed for m≥4, even if 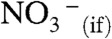 were not hydrated at all) (36). The hydration of
were not hydrated at all) (36). The hydration of 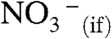 will, of course, contribute to the exoergicity of reaction 1. Because HNO3 is able to interact with at least four water molecules upon impact with the surface of water (28), the nature of the barrier-hindering reaction 1 remains to be elucidated. It has been proposed that acid-base equilibria at the air–water interface are shifted (relative to bulk water) toward neutral species by approximately ± 2 pKa units (37). In the case of nitric acid, pKa(HNO3(aq)) = -1.4, this proposal makes HNO3(if) a strong acid at the interface: pKa(HNO3(if)) of approximately 0, at variance with our observations. We wish to emphasize that in our experiments, in contrast with most other studies (38), HNO3 approaches the air–water interface from the vapor instead of the water side. Hence, gas-phase ion thermochemistry (13, 35) is a more appropriate framework for analyzing our results.
will, of course, contribute to the exoergicity of reaction 1. Because HNO3 is able to interact with at least four water molecules upon impact with the surface of water (28), the nature of the barrier-hindering reaction 1 remains to be elucidated. It has been proposed that acid-base equilibria at the air–water interface are shifted (relative to bulk water) toward neutral species by approximately ± 2 pKa units (37). In the case of nitric acid, pKa(HNO3(aq)) = -1.4, this proposal makes HNO3(if) a strong acid at the interface: pKa(HNO3(if)) of approximately 0, at variance with our observations. We wish to emphasize that in our experiments, in contrast with most other studies (38), HNO3 approaches the air–water interface from the vapor instead of the water side. Hence, gas-phase ion thermochemistry (13, 35) is a more appropriate framework for analyzing our results.
Against this background, we performed density functional theory calculations on HNO3 interacting with water decamers W10(W ≡ H2O) in the absence and presence of Cl- to ascertain the molecular basis of our experimental observations. Fig. 3
A and B display the calculated Gibbs free energy (ΔG0) and enthalpy (ΔH0) profiles at 300 K. We confirmed that HNO3 embedded in W10 clusters dissociates spontaneously, in accordance with common knowledge, thermodynamics, and Car–Parrinello molecular dynamics (CPMD) calculations (14, 15). In contrast, HNO3 binds as a molecule to the periphery of W10 via two hydrogen bonds with ΔH0 = -13.0 kcal mol-1 and (because of translational and rotational HNO3 entropy losses) ΔG0 = -1.2 kcal mol-1. The free energy barrier for transferring a proton from adsorbed HNO3 into the cluster while leaving a 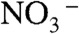 on its surface is quite large: ΔG‡ = 14.1 kcal mol-1, or 12.9 kcal mol-1 above the reactants. Weakly bound undissociated HNO3 is therefore rather stable toward dissociation and highly mobile on the surface of water. Remarkably, HNO3 not only binds more strongly (ΔH0 = -18.6 kcal mol-1, ΔG0 = -6.9 kcal mol-1) to clusters containing a Cl-, but the free energy barrier for transferring a proton to W10·Cl- is dramatically reduced: ΔG‡ = 1.2 kcal mol-1.
on its surface is quite large: ΔG‡ = 14.1 kcal mol-1, or 12.9 kcal mol-1 above the reactants. Weakly bound undissociated HNO3 is therefore rather stable toward dissociation and highly mobile on the surface of water. Remarkably, HNO3 not only binds more strongly (ΔH0 = -18.6 kcal mol-1, ΔG0 = -6.9 kcal mol-1) to clusters containing a Cl-, but the free energy barrier for transferring a proton to W10·Cl- is dramatically reduced: ΔG‡ = 1.2 kcal mol-1.
Fig. 3.

Calculated Gibbs free energies (ΔG0) and enthalpies (ΔH0) of reactants, adducts, transition states, and products of optimized water clusters in contact with nitric acid in the absence (A) and presence (B) of interfacial chloride. Proton wires highlighted. Energies in kcal mol-1.
Discussion
Calculations provide significant clues about the origin of the barrier to HNO3 dissociation on water. HNO3 binds to W10 both as H-bond donor and acceptor. However, the 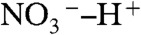 proton, an intrinsic water ion, cannot readily slip into cluster leaving
proton, an intrinsic water ion, cannot readily slip into cluster leaving 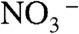 behind (Fig. 3A). The barrier to PT on the surface of water is therefore associated with the fact that (i) overcoming the electrostatic attraction in a disjoint (
behind (Fig. 3A). The barrier to PT on the surface of water is therefore associated with the fact that (i) overcoming the electrostatic attraction in a disjoint (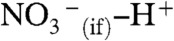 ) ion pair, or (ii) opening a cavity for
) ion pair, or (ii) opening a cavity for 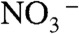 to follow after the proton into the cluster, entails significant energy costs. Calculations involving larger water clusters do not eliminate such barrier (28). Clearly, a chloride lets H3O+ advance further into the cluster primarily by countering the electrostatic bias imposed on H3O+ by laggardly
to follow after the proton into the cluster, entails significant energy costs. Calculations involving larger water clusters do not eliminate such barrier (28). Clearly, a chloride lets H3O+ advance further into the cluster primarily by countering the electrostatic bias imposed on H3O+ by laggardly 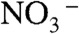 , rather than binding to it [recall that pKa(HCl)(aq) = -7]. We also noticed that the atomic rearrangements involved in binding HNO3 to W10 clusters are uncorrelated to those required for subsequent PT. In contrast, the stronger interaction between HNO3(g) and (Cl·W10)- clusters primes (Cl·W10⋯HNO3)- for PT. The reaction coordinate for PT on pure water is a combination of six internal modes involving displacements of heavy O atoms, whereas PT in the presence of chloride proceeds adiabatically along a three-link proton wire between quasi-degenerate solvent states (Fig. S5 A and B).
, rather than binding to it [recall that pKa(HCl)(aq) = -7]. We also noticed that the atomic rearrangements involved in binding HNO3 to W10 clusters are uncorrelated to those required for subsequent PT. In contrast, the stronger interaction between HNO3(g) and (Cl·W10)- clusters primes (Cl·W10⋯HNO3)- for PT. The reaction coordinate for PT on pure water is a combination of six internal modes involving displacements of heavy O atoms, whereas PT in the presence of chloride proceeds adiabatically along a three-link proton wire between quasi-degenerate solvent states (Fig. S5 A and B).
After establishing the role of electrostatics in the catalysis of HNO3 dissociation on small water clusters, we need to understand why catalytic effects are observed on > 30-μM electrolytes—i.e., at (Rion–ion) < 120-nm interfacial separations (SI Text) that vastly exceed the size of such clusters. On the basis of our calculations we envision that HNO3, after alighting on water, roams rather freely over its surface as HNO3(if) until it approaches an interfacial Cl-, whereupon it falls into a deeper potential well and undergoes prompt dissociation. In SI Text we estimate that average number of hops required by HNO3(if) to reach a Cl- on the surface of > 30-μM solutions would take a few nanoseconds (i.e., competitively with back desorption into the gas-phase) (16, 39). Recapitulating, present experimental results substantiate a key role for electrostatics in the mechanism of HNO3 dissociation at water-hydrophobe interfaces, and suggest that even sparse anions can effectively catalyze this process.
Implications
Our finding that PT across water-hydrophobic media interfaces is catalyzed by anions has important implications in many fields. Whether HNO3(g) dissociates on aqueous surfaces, for example, bears on various environmental issues. Whereas 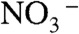 is a sink for active nitrogen in the atmosphere because it can be removed by dry and wet deposition, undissociated HNO3 may react via: 2HNO3(g) + NO(g) → 3NO2(g) + H2O(g), thereby sustaining the atmospheric impact of nitrogen oxides (40). Adsorption of HNO3(g) on ice also depends critically on whether HNO3 dissociates therein—i.e., whether coverage is a function of P or P1/2 (P ≡ HNO3(g) partial pressure) (41). Our results suggest that HNO3(g) will dissociate upon impact on most environmental aqueous surfaces, including premelted films on ice that contain electrolytes impurities at least at millimolar levels. Our demonstration that PT across internal water-hydrophobe interfaces is facilitated by electrostatics related to the concept of anion-mediated water bridges for PT in proteins (42) is at least consistent with the assumption that charge transfer events at water–protein interfaces are driven by electrostatic preorganization (43, 44). It also accounts for the fact that even weakly basic, mobile anions, such as chloride, may enhance proton motion along membrane surfaces without providing localized proton-binding sites (45, 46).
is a sink for active nitrogen in the atmosphere because it can be removed by dry and wet deposition, undissociated HNO3 may react via: 2HNO3(g) + NO(g) → 3NO2(g) + H2O(g), thereby sustaining the atmospheric impact of nitrogen oxides (40). Adsorption of HNO3(g) on ice also depends critically on whether HNO3 dissociates therein—i.e., whether coverage is a function of P or P1/2 (P ≡ HNO3(g) partial pressure) (41). Our results suggest that HNO3(g) will dissociate upon impact on most environmental aqueous surfaces, including premelted films on ice that contain electrolytes impurities at least at millimolar levels. Our demonstration that PT across internal water-hydrophobe interfaces is facilitated by electrostatics related to the concept of anion-mediated water bridges for PT in proteins (42) is at least consistent with the assumption that charge transfer events at water–protein interfaces are driven by electrostatic preorganization (43, 44). It also accounts for the fact that even weakly basic, mobile anions, such as chloride, may enhance proton motion along membrane surfaces without providing localized proton-binding sites (45, 46).
Experimental Methods
In our experiments, continuously refreshed, uncontaminated surfaces of free-flowing aqueous microjets exposed to < 8 × 1012 HNO3(g) molecules cm-3 for approximately 10 μs are monitored by online negative or positive ion ESMS. Fifty μL min-1 of deionized water or aqueous electrolyte solutions (pH-adjusted using concentrated NaOH or HCl) are injected as a microjet into the spraying chamber of an ES mass spectrometer held at 1 atm, 293 K via an electrically grounded, stainless steel pneumatic nozzle (100 μm) internal diameter (SI Text and Figs. S1 and S2) (47). A high-speed (approximately 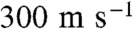 ) annular nebulizer N2(g) flow tears up the much slower (
) annular nebulizer N2(g) flow tears up the much slower (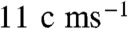 ) microjet into droplets charged with ion excesses of either sign. Ions are eventually ejected to the gas-phase, charge selected by a polarized inlet port orthogonal to the nozzle, and detected by mass spectrometry. We note that the velocity at which the liquid jet emerges from the nozzle is approximately 500 times slower than that required for observing electrokinetic effects in our experiments (48) (SI Text).
) microjet into droplets charged with ion excesses of either sign. Ions are eventually ejected to the gas-phase, charge selected by a polarized inlet port orthogonal to the nozzle, and detected by mass spectrometry. We note that the velocity at which the liquid jet emerges from the nozzle is approximately 500 times slower than that required for observing electrokinetic effects in our experiments (48) (SI Text).
Computational Methods
Gibbs free energies (ΔG) at 298 K were computed from calculated enthalpies (ΔH) and entropies (S) according to ΔG = Eelec + ZPE + Hvib - TSvib. Geometries of energy minima and transition states were optimized using the X3LYP functional (extended hybrid functional combined with Lee–Yang–Parr correlation functional) (49), the 6-31G** basis for light atoms (50), and 6-311G**++ for Cl- (51). Hessians at these geometries provided harmonic zero-point energies, vibrational enthalpies, and entropies. Neglect of anharmonicity effects (< 1 kcal mol-1) may not affect the main conclusions. After geometry optimization, the electronic energy Eelec was evaluated with the 6-311G**++ basis on all atoms. The free energies of nitric acid and nitrate at 1 atm were calculated using statistical mechanics for ideal gases (SI Text).
Supplementary Material
ACKNOWLEDGMENTS.
S.E. thanks the Japan Society for the Promotion of Sciences Postdoctoral Fellowship for Research Abroad. Research supported by National Science Foundation Grant AGS-964842 (to M.R.H.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1200949109/-/DCSupplemental.
References
- 1.Ravishankara AR. Heterogeneous and multiphase chemistry in the troposphere. Science. 1997;276:1058–1065. [Google Scholar]
- 2.Enami S, Mishra H, Hoffmann MR, Colussi AJ. Protonation and oligomerization of gaseous isoprene on mildly acidic surfaces: Implications for atmospheric chemistry. J Phys Chem A. 2012 doi: 10.1021/jp2110133. doi: 10.1021/jp2110133. [DOI] [PubMed] [Google Scholar]
- 3.Mitchell P. Coupling of phosphorylation to electron and hydrogen transfer by a chemi-osmotic type of mechanism. Nature. 1961;191:144–150. doi: 10.1038/191144a0. [DOI] [PubMed] [Google Scholar]
- 4.Hoegh-Guldberg O, et al. Coral reefs under rapid climate change and ocean acidification. Science. 2007;318:1737–1742. doi: 10.1126/science.1152509. [DOI] [PubMed] [Google Scholar]
- 5.Enami S, Hoffmann MR, Colussi AJ. Acidity enhances the formation of a persistent ozonide at aqueous ascorbate/ozone gas interfaces. Proc Natl Acad Sci USA. 2008;105:7365–7369. doi: 10.1073/pnas.0710791105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kinugawa T, et al. Conversion of gaseous nitrogen dioxide to nitrate and nitrite on aqueous surfactants. Phys Chem Chem Phys. 2011;13:5144–5149. doi: 10.1039/c0cp01497d. [DOI] [PubMed] [Google Scholar]
- 7.Sanden T, Salomonsson L, Brzezinski P, Widengren J. Surface-coupled proton exchange of a membrane-bound proton acceptor. Proc Natl Acad Sci USA. 2010;107:4129–4134. doi: 10.1073/pnas.0908671107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lane N, Allen JF, Martin W. How did LUCA make a living? Chemiosmosis in the origin of life. Bioessays. 2010;32:271–280. doi: 10.1002/bies.200900131. [DOI] [PubMed] [Google Scholar]
- 9.Villa J, Warshel A. Energetics and dynamics of enzymatic reactions. J Phys Chem B. 2001;105:7887–7907. [Google Scholar]
- 10.Benkovic SJ, Hammes-Schiffer S. A perspective on enzyme catalysis. Science. 2003;301:1196–1202. doi: 10.1126/science.1085515. [DOI] [PubMed] [Google Scholar]
- 11.Eigen M. Proton transfer acid-base catalysis and enzymatic hydrolysis. I. Elementary processes. Angew Chem Int Ed. 1964;3:1–15. [Google Scholar]
- 12.Arnett EM. Gas-phase proton transfer: A breakthrough for solution chemistry. Acc Chem Res. 1973;6:404–409. [Google Scholar]
- 13.Hunter EPL, Lias SG. Evaluated gas phase basicities and proton affinities of molecules: An update. J Phys Chem Ref Data. 1998;27:413–656. [Google Scholar]
- 14.Wang SZ, Bianco R, Hynes JT. Depth-dependent dissociation of nitric acid at an aqueous surface: Car–Parrinello molecular dynamics. J Phys Chem A. 2009;113:1295–1307. doi: 10.1021/jp808533y. [DOI] [PubMed] [Google Scholar]
- 15.Shamay ES, Buch V, Parrinello M, Richmond GL. At the water’s edge: Nitric acid as a weak acid. J Am Chem Soc. 2007;129:12910–12913. doi: 10.1021/ja074811f. [DOI] [PubMed] [Google Scholar]
- 16.Enami S, Hoffmann MR, Colussi AJ. Proton availability at the air/water interface. J Phys Chem Lett. 2010;1:1599–1604. [Google Scholar]
- 17.Enami S, Hoffmann MR, Colussi AJ. Superacid chemistry on mildly acidic water. J Phys Chem Lett. 2010;1:3488–3493. [Google Scholar]
- 18.Enami S, Hoffmann MR, Colussi AJ. Prompt Formation of organic acids in pulse ozonation of terpenes on aqueous surfaces. J Phys Chem Lett. 2010;1:2374–2379. [Google Scholar]
- 19.Cheng J, Vecitis C, Hoffmann MR, Colussi AJ. Experimental anions affinities for the air/water interface. J Phys Chem B. 2006;110:25598–25602. doi: 10.1021/jp066197k. [DOI] [PubMed] [Google Scholar]
- 20.Enami S, Hoffmann MR, Colussi AJ. Simultaneous detection of cysteine sulfenate, sulfinate, and sulfonate during cysteine interfacial ozonolysis. J Phys Chem B. 2009;113:9356–9358. doi: 10.1021/jp904316n. [DOI] [PubMed] [Google Scholar]
- 21.Yabushita A, et al. Anion-catalyzed dissolution of NO2 on aqueous microdroplets. J Phys Chem A. 2009;113:4844–4848. doi: 10.1021/jp900685f. [DOI] [PubMed] [Google Scholar]
- 22.Nguyen S, Fenn JB. Gas-phase ions of solute species from charged droplets of solutions. Proc Natl Acad Sci USA. 2007;104:1111–1117. doi: 10.1073/pnas.0609969104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vandoren JM, et al. Temperature dependence of the uptake coefficients of HNO3, HCl and N2O5 by water droplets. J Phys Chem. 1990;94:3265–3269. [Google Scholar]
- 24.Ardura D, Donaldson DJ. Where does acid hydrolysis take place? Phys Chem Chem Phys. 2009;11:857–863. doi: 10.1039/b812070f. [DOI] [PubMed] [Google Scholar]
- 25.Dempsey LP, Brastad SM, Nathanson GM. Interfacial acid dissociation and proton exchange following collisions of DCl with salty glycerol and salty water. J Phys Chem Lett. 2011;2:622–627. [Google Scholar]
- 26.Cox MJ, Siwick BJ, Bakker HJ. Influence of ions on aqueous acid-base reactions. ChemPhysChem. 2009;10:236–244. doi: 10.1002/cphc.200800406. [DOI] [PubMed] [Google Scholar]
- 27.Otten DE, Shaffer PR, Geissler PL, Saykally RJ. Elucidating the mechanism of selective ion adsorption to the liquid water surface. Proc Natl Acad Sci USA. 2012;109:701–705. doi: 10.1073/pnas.1116169109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bianco R, Wang SZ, Hynes JT. Theoretical study of the dissociation of nitric acid at a model aqueous surface. J Phys Chem A. 2007;111:11033–11042. doi: 10.1021/jp075054a. [DOI] [PubMed] [Google Scholar]
- 29.Jarvis NL, Scheiman MA. Surface potentials of aqueous electrolyte solutions. J Phys Chem. 1968;72:74–78. [Google Scholar]
- 30.Petersen PB, Saykally RJ. Adsorption of ions to the surface of dilute electrolyte solutions: The Jones–Ray effect revisited. J Am Chem Soc. 2005;127:15446–15452. doi: 10.1021/ja053224w. [DOI] [PubMed] [Google Scholar]
- 31.Lu SY, Jiang YJ, Zhou P, Zou JW, Wu TX. Geometric characteristics and energy landscapes of halogen–water–hydrogen bridges at protein-ligand interfaces. Chem Phys Lett. 2010;485:348–353. [Google Scholar]
- 32.Tian CS, Byrnes SJ, Han HL, Shen YR. Surface propensities of atmospherically relevant ions in salt solutions revealed by phase-sensitive sum frequency vibrational spectroscopy. J Phys Chem Lett. 2011;2:1946–1949. [Google Scholar]
- 33.Petersen PB, Saykally RJ. Probing the interfacial structure of aqueous electrolytes with femtosecond second harmonic generation spectroscopy. J Phys Chem B. 2006;110:14060–14073. doi: 10.1021/jp0601825. [DOI] [PubMed] [Google Scholar]
- 34.Dole M. A theory of surface tension of aqueous solutions. J Am Chem Soc. 1938;60:904–911. [Google Scholar]
- 35.Meot-Ner M. The ionic hydrogen bond. Chem Rev. 2005;105:213–284. doi: 10.1021/cr9411785. [DOI] [PubMed] [Google Scholar]
- 36.Leopold KR. Hydrated acid clusters. Annu Rev Phys Chem. 2011;62:327–349. doi: 10.1146/annurev-physchem-032210-103409. [DOI] [PubMed] [Google Scholar]
- 37.Zhao XL, Subrahmanyan S, Eisenthal KB. Determination of pKa at the air/water interface by second harmonic generation. Chem Phys Lett. 1990;171:558–562. [Google Scholar]
- 38.Lewis T, et al. Does nitric acid dissociate at the aqueous solution surface? J Phys Chem C. 2011;115:21183–21190. [Google Scholar]
- 39.Harper K, et al. Surface residence and uptake of methyl chloride and methyl alcohol at the air/water interface studied by vibrational sum frequency spectroscopy and molecular dynamics. J Phys Chem A. 2009;113:2015–2024. doi: 10.1021/jp808630v. [DOI] [PubMed] [Google Scholar]
- 40.Mochida M, Finlayson-Pitts BJ. FTIR studies of the reaction of gaseous NO with HNO3 on porous glass: Implications for conversion of HNO3 to photochemically active NOx in the atmosphere. J Phys Chem A. 2000;104:9705–9711. [Google Scholar]
- 41.Molina MJ, et al. Physical-chemistry of the H2SO4/HNO3/H2O system: Implications for polar stratospheric clouds. Science. 1993;261:1418–1423. doi: 10.1126/science.261.5127.1418. [DOI] [PubMed] [Google Scholar]
- 42.De Vivo M, et al. Proton shuttles and phosphatase activity in soluble epoxide hydrolase. J Am Chem Soc. 2007;129:387–394. doi: 10.1021/ja066150c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kamerlin SCL, Warshel A. At the dawn of the 21st century: Is dynamics the missing link for understanding enzyme catalysis? Proteins. 2010;78:1339–1375. doi: 10.1002/prot.22654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Warshel A, et al. Electrostatic basis for enzyme catalysis. Chem Rev. 2006;106:3210–3235. doi: 10.1021/cr0503106. [DOI] [PubMed] [Google Scholar]
- 45.Springer A, Hagen V, Cherepanov DA, Antonenko YN, Pohl P. Protons migrate along interfacial water without significant contributions from jumps between ionizable groups on the membrane surface. Proc Natl Acad Sci USA. 2011;108:14461–14466. doi: 10.1073/pnas.1107476108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ben-Amotz D. Unveiling electron promiscuity. J Phys Chem Lett. 2011;2:1216–1222. doi: 10.1021/jz2002875. [DOI] [PubMed] [Google Scholar]
- 47.Zilch LW, Maze JT, Smith JW, Ewing GE, Jarrold MF. Charge separation in the aerodynamic breakup of micrometer-sized water droplets. J Phys Chem A. 2008;112:13352–13363. doi: 10.1021/jp806995h. [DOI] [PubMed] [Google Scholar]
- 48.Duffin AM, Saykally RJ. Electrokinetic hydrogen generation from liquid water microjets. J Phys Chem C. 2007;111:12031–12037. [Google Scholar]
- 49.Xu X, Goddard WA. The X3LYP extended density functional for accurate descriptions of nonbond interactions, spin states, and thermochemical properties. Proc Natl Acad Sci USA. 2004;101:2673–2677. doi: 10.1073/pnas.0308730100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Clark T, Chandrasekhar J, Spitznagel GW, Schleyer PV. Efficient diffuse function-augmented basis sets for anion calculations. 3. The 3-21+G basis set for 1st-row elements, LI-F. J Comput Chem. 1983;4:294–301. [Google Scholar]
- 51.Francl MM, et al. Self-consistent molecular orbital methods. 23. A polarization type basis set for 2nd-row elements. J Chem Phys. 1982;77:3654–3665. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.