

Abstract
T cells spend the majority of their time perusing lymphoid organs in search of cognate antigen presented by antigen presenting cells (APCs) and then quickly recirculate through the bloodstream to another lymph node. Therefore, regulation of a T-cell response is dependent upon the ability of cells to arrive in the correct location following chemokine gradients (“go” signal) as well as to receive appropriate T-cell receptor (TCR) activation signals upon cognate antigen recognition (“stop” signal). However, the mechanisms by which T cells regulate these go and stop signals remain unclear. We found that overexpression of the hematopoietic-specific RhoH protein in the presence of chemokine signals resulted in decreased Rap1–GTP and LFA-1 adhesiveness to ICAM-1, thus impairing T-cell chemotaxis; while in the presence of TCR signals, there were enhanced and sustained Rap1–GTP and LFA-1 activation as well as prolonged T:APC conjugates. RT-PCR analyses of activated CD4+ T cells and live images of T-cell migration and immunological synapse (IS) formation revealed that functions of RhoH took place primarily at the levels of transcription and intracellular distribution. Thus, we conclude that RhoH expression provides a key molecular determinant that allows T cells to switch between sensing chemokine-mediated go signals and TCR-dependent stop signals.
Regulation of a T-cell response is dependent upon the ability of cells to migrate within a lymphoid organ following chemokine signals as well as to form stable and prolonged T:antigen presenting cell (APC) contacts to receive appropriate activation signals upon cognate antigen recognition (1). Integrin-mediated adhesion is needed for extravasation through high endothelial venules (HEVs) and intranodal migration during which APC scanning occurs (2). The fibroblastic reticular cell network serves as a matrix upon which T cells migrate and expresses ICAM-1 as well as CCL21, CCL19, and CXCL12 (3). Adhesive force generated by LFA-1 ligation is also essential for the maintenance of the immunological synapse (IS) and the signal integration necessary for complete T-cell activation (4).
Although enhanced LFA-1 adhesiveness is equally important for both migration and T-cell activation, the biological outcomes of chemokine and T-cell-receptor (TCR)–mediated LFA-1 activation in T cells are quite different. Intracellular cues in response to cognate interactions lead to cells deciding to stop despite continued chemokine-driven migratory signals. Conversely, following a period of stable contact coincident with T-cell activation, cells again become responsive to chemokine-induced migratory signals and ignore the continued presence of ligands for the TCR. Rap1, a member of the Ras family of small GTPases, modulates the affinity and avidity of LFA-1 during TCR and chemokine triggered LFA-1 adhesion to ICAM-1 (5). Calcium and diacylglycerol-regulated guanine nucleotide exchange factor I (CalDAG–GEFI) and C3G are key GEFs that activate Rap1 (6) and regulator of adhesion and polarization enriched in lymphocytes (RAPL), through its association with Rap1, is a crucial effector molecule involved in Rap1-mediated LFA-1 activation during TCR and chemokine-triggered LFA-1 adhesion to ICAM-1 (7). Several negative regulators of LFA-1 activation have been described, including RhoH, a member of the Rho family of small GTPases (8). Overexpression of RhoH is inhibitory to chemokine-induced migration, whereas RhoH is involved in proximal TCR signaling (9–13).
In this study, we have addressed a longstanding question of how T cells differentially regulate LFA-1 adhesiveness during chemokine-mediated active migration (go) and TCR-mediated stable IS formation (stop). Here, we provide evidence that RhoH performs distinct functions by inhibiting chemokine-mediated migration and enhancing TCR-induced adhesiveness, suggesting that RhoH may serve as a rheostat that modulates the CD4 T-cell response.
Results
Opposing Roles for RhoH in Chemokine-Receptor and TCR-Induced Rap1 Activation.
Chemokine and TCR signals induce the open conformation of LFA-1, via inside-out signaling (2). A significant body of primary literature supports the presence of common intermediates between the inside-out signaling pathways downstream of chemokine and TCR-induced activation of LFA-1, including CAS and Rap1 activation (Fig. S1). As CAS is reported to impact Rap1 activation by modulation of C3G activity, we focused on Rap1 activation in this study as a major point of overlap between TCR and chemokine-receptor–induced LFA-1 adhesiveness.
The conversion of LFA-1 from a bent to open conformation is mediated by Rap1 and its GEFs. It has been speculated that RhoH may interfere with Rap1 activation making this a probable target for modulating both chemokine and TCR-induced activation of LFA-1 (8). Importantly, RhoH, like Cbl-b, has a negative impact on LFA-1–mediated adhesion (2). RhoH also inhibits several effector functions of Rho family small GTPases, such as NF-kB activation induced by Rac1 and cdc42 and RhoA and p38MAPK activation by Rac1 and cdc42 (14), yet these would not directly impact inside-out signaling to induce LFA-1 activation. Therefore, these observations had led us to hypothesize that RhoH negatively regulates Rap1 activation, thus preventing LFA-1 adhesion in T cells. To investigate the effects of RhoH on Rap1 activation in T cells, we assessed the stimulation-dependent activation of Rap1 in control and cells overexpressing RhoH. To overexpress RhoH, we generated a fusion protein in which monomeric red fluorescence protein (mRFP) was fused to the C terminus of RhoH (RhoH–mRFP). The Jurkat cell line was transiently transfected with control vector (mRFP) or RhoH–mRFP, then stimulated with chemokine CXCL12 (Fig. 1A) or TCR cross-linking (Fig. 1B) for the indicated times. Activation of Rap1 was rapid and transient upon chemokine-receptor or TCR stimulation, as seen in mRFP only expressing cells (Fig. 1 A and B). CXCL12-induced Rap1 activation was greatly reduced in cells that overexpressed RhoH (Fig. 1A). In stark contrast, overexpression of RhoH resulted in enhanced and prolonged activation of Rap1 upon TCR stimulation (Fig. 1B). Transfection efficiency was verified by quantitation of tranfection frequency and mRFP Western blots (Fig. 1A and Fig. S2) and overexpression of RhoH–mRFP did not alter the surface expression of CXCR4 or TCR (Fig. S3). Furthermore, RhoH−/− T cells revealed significantly greater constitutively active Rap1 than WT cells (Fig. 1C). Previously RhoH−/− hematopoeitic stem cells were also reported to have more active Rac1–GTP present in basal state (15). Unlike in WT cells, TCR stimulation of RhoH−/− T cells failed to increase, but slightly reduced Rap1 activation (Fig. 1C). However, incubation of RhoH−/− T cells with CCL21 significantly enhanced Rap1 activation compared to TCR cross-linking (Fig. 1C). The altered pattern of Rap1 activation in the absence or overexpression of RhoH may not be explained by changes in protein expression of C3G or CalDAG–GEFI (Fig. S4). Our data suggest another small GTPase effected by changes in RhoH expression levels.
Fig. 1.

RhoH negatively regulates chemokine-mediated Rap1 activation and positively regulates TCR-mediated Rap1 activation. (A–C) Jurkat cells were transiently transfected with mRFP or RhoH–mRFP, then at various time after stimulation with CXCL12 or CD3 cross-linking, cell lysates were assessed for active Rap1, total Rap-1, and b-actin. Representative blots of Rap1 activation by CXCL12 (A) or CD3 cross-linking (B) are shown and bar graphs of each pair of blots consist of compiled densitometry analysis (Right). (C) Freshly isolated CD4+ T cells from WT and RhoH−/− mice were unstimulated (NS) or stimulated with CD3 cross-linking or CCL21 and Rap1–GTP assessed. Data are means ± SEM, n = 3, and statistically significant, *P < 0.05 vs. WT NS, statistically significant, #P < 0.03 vs. RhoH−/− CD3.
Opposing Roles for RhoH in Chemokine-Receptor and TCR-Induced LFA-1 Activation.
RhoH expression suppresses chemokine-mediated Rap1 activation, whereas it enhances Rap1 activation induced by TCR ligation, suggesting that RhoH overexpression may play a disparate role in LFA-1 adhesion induced by these signals. To assess the LFA-1 adhesiveness of RhoH-overexpressing cells stimulated under each condition, we generated stable Jurkat cell lines expressing control (mRFP) or RhoH (RhoH–mRFP) vector (Fig. 2A). T-cell binding on ICAM-1–coated plates revealed that overexpression of RhoH reduced the basal adhesion of cells greater than fourfold as seen by a decrease in frequency of cells bound to ICAM-1 in the absence of stimulation (Fig. 2 B and C), confirming previous findings (8). Consistent with the results of the Rap1 assay, RhoH-overexpressing cells stimulated with CXCL12 failed to increase cell binding on ICAM-1, whereas TCR cross-linking successfully increased the number of cells bound to ICAM-1 (Fig. 2 B and C). Although the magnitude of cells adhering to ICAM-1 was similar between control and RhoH-overexpressing cells in the presence of TCR cross-linking, the increase in adhesion in the presence of stimulation was four- to fivefold greater in RhoH-overexpressing cells (Fig. 2C). These results were further corroborated using sorted transiently transfected primary human T cells expressing control or RhoH–mRFP (Fig. 2 D–F). The effects of differential expression of RhoH on chemokine signals in T cells were not related to different onset of activation as seen in Fig. 2D. Finally, to assess whether this effect is due to altered Rap1 activation, RhoH–mRFP+ T cells were cotransfected with constitutively active Rap1. Chemokine-induced T-cell adhesion on ICAM-1 substrate was recovered by the expression of constitutively active Rap1 (Fig. 2G), suggesting that the effect of RhoH on T-cell adhesion is a result of impaired Rap1 activation.
Fig. 2.

RhoH negatively regulates chemokine-mediated LFA-1 adhesion and positively regulates TCR-mediated LFA-1 adhesion. (A–C) Stable Jurkat cell lines expressing pcDNA, mRFP, or RhoH–mRFP were generated and verified by anti-mRFP immunoblot. Cells were stimulated with CXCL12 (B) or OKT3 and secondary antibody (C) to induce TCR cross-linking, then assessed in an ICAM-1 adhesion assay. (D–G) Primary human lymphocytes transiently transfected with GFP or RhoH–mRFP were sorted. ICAM-1 adhesion of CXCL12 stimulation (D) or TCR stimulation (E) over time, CXCL12 titration (F) or ICAM-1 adhesion of CXCL12 stimulation of control, RhoH overexpressing, or double positive RhoH–mRFP and Rap1V12–GFP (G). Data are means ± SEM. For B, n = 4 and P < 0.02 and C–G n = 3 independent donors.
Reduced Chemokine-Mediated Migration and Sustained T:APC Conjugation of RhoH-Overexpressing T Cells.
Diminished Rap1 activation and LFA-1 adhesiveness in the presence of RhoH overexpression would impact chemokine-induced migration. Cell migration plots are shown for CXCL12-induced migration of GFP or RhoH–mRFP expressing T cells (Fig. 3A and Movie S1). Quantitative analysis of the velocities of cell migration revealed that overexpression of RhoH reduces the speed of migration in response to chemokine stimulation (Fig. 3B). RhoH-overexpressing cells displayed a polarized morphology with relatively short tails compared with control cells (Fig. S5). Recent evidence suggests that activated Rap1 recruits the Par polarity complex to regulate actin remodeling required for T-cell polarization (16). Therefore, it is possible that the establishment of polarity is inhibited or enhanced depending upon the levels of Rap1 activation and hence RhoH expression.
Fig. 3.
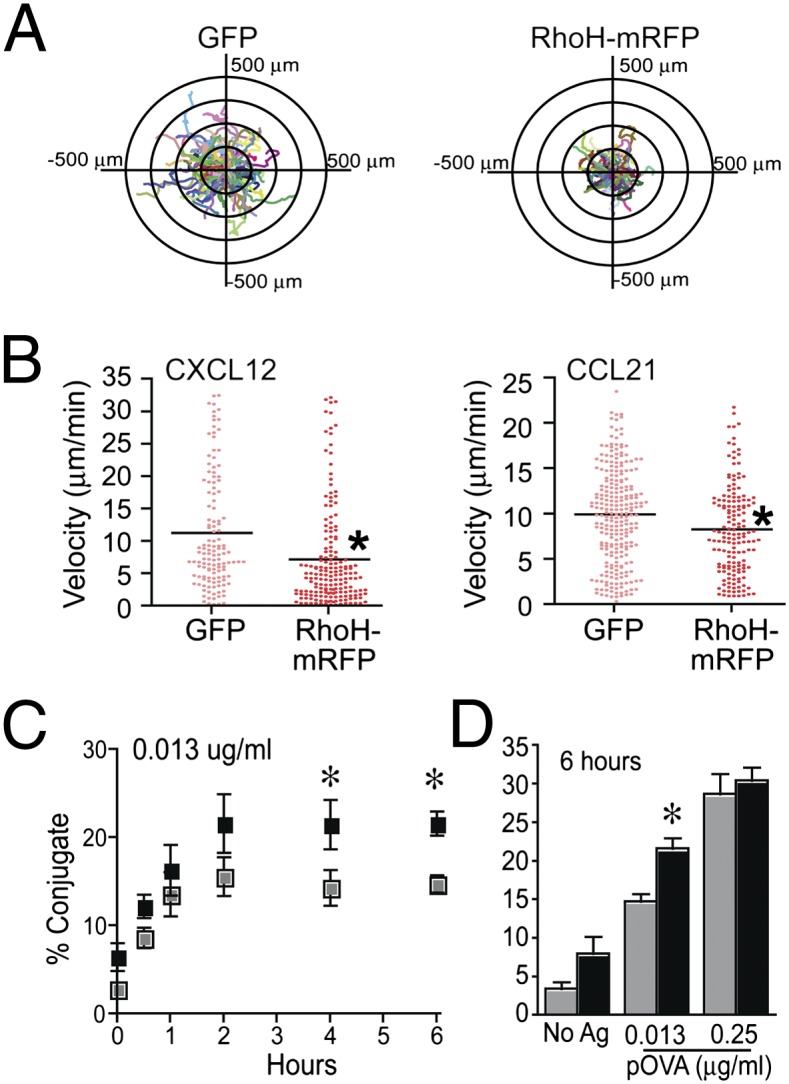
RhoH negatively regulates chemokine-induced T-cell migration and positively regulates sustained T:APC conjugates. (A) Human T-cell blasts were transiently transfected with control vector (GFP) or RhoH–mRFP vector. Cell migration on CXCL12/ICAM-1 was analyzed; corresponding cell tracking plots are shown (A, Movie S1). Images include fluorescent signal to indicate transfected cells overlaid with migration tracks as indicated by colored lines. (B) Velocity (millimeters per minute) of cell migration induced by CXCL12 or CCL21 on ICAM-1 was analyzed. Data are combined from three independent donors and are statistically significant P < 0.001 by Mann–Whitney test. (C and D) DO11.10 TCR transgenic T-cell blasts were transiently transfected with control vector or RhoH–mRFP and assessed in a flow cytometry-based conjugate assay. (C) Representative plot of conjugate formation over time at one antigen dose. (D) Conjugates at 6 h with NoAg or two doses of Ag. Data are means ± SEM, n = 3 and statistically significant P < 0.05.
RhoH overexpression enhanced Rap1 activation and LFA-1 adhesiveness in the presence of TCR signals (Figs. 1B and 2 C and E), suggesting that RhoH-overexpressing T cells may produce more stable T:APC conjugates. A cell:cell conjugation assay revealed that RhoH overexpression leads to an increased frequency of conjugates over time (Fig. 3C). This effect is Ag dose sensitive (Fig. 3 C and D). Furthermore, RhoH overexpression resulted in enhanced dilution of PKH compared to control T cells (Fig. S6). The result suggests that RhoH-overexpressing T cells form stable T-cell adhesion to ICAM-1 in the presence of TCR cross-linking at the earlier time points (Fig. 2E) and maintain prolonged conjugate formation over time through active LFA-1 (Fig. 3C), enabling enhanced signal integration for T-cell activation.
Distinct RhoH Distribution Patterns During Chemokine-Mediated T-Cell Migration vs. T:APC Conjugate Formation.
RhoH lacks intrinsic GTPase activity and has specific amino acid substitutions that lead to RhoH remaining GTP bound, thus the activity of RhoH is not regulated by the typical GTP/GDP exchange cycles (14). Rapid alteration in RhoH localization may serve as an efficient mechanism to enable RhoH to act in opposing ways to signals rapidly being sensed by a T cell. A Rap1–Raichu fluorescence resonance energy transfer (FRET) sensor revealed that rapid Rap1 activation was selectively localized at the leading edge of migrating T cells on CXCL12/ICAM-1–coated substrate (Fig. 4A and Movie S2) and minimal FRET signal is detected in cells on ICAM-1–only coated substrate (Fig. S7 and Movie S3). Due to limitations in the FRET system, we were able to image the global distribution pattern of Rap1 activation but not to the fine subcellular patterning of membrane proximity. In contrast, the RhoH–mRFP signal was excluded from the leading edge of the cell where active LFA-1 was detected, with the majority of RhoH–mRFP localized to the uropod (Fig. 4 B and C and Movie S4). Upon TCR activation, LFA-1, Rap1, and its interacting partners are recruited to the IS (7) (Fig. 4F). To assess RhoH localization during IS formation, conjugates were formed with transiently transfected Jurkat cells. Unlike in migrating T cells, RhoH–mRFP was localized toward the T:APC interface where LFA-1 activation was also detected (9) (Fig. 4 D–F).
Fig. 4.

Rap1 and RhoH are segregated during chemokine-induced migration and localized toward the IS upon TCR activation. (A) Human T-cell blasts were transiently transfected with Raichu–Rap1 FRET sensor, then allowed to migrate on ICAM-1/CXCL12-coated coverslips. CFP and YFP are shown in gray and intensity is pseudocolored below. FRET efficiency is shown in rainbow colors, highest (red) to lowest (blue). (Right) Dynamic Rap1 FRET at select time points; white arrows indicate leading edge of cell (A, Movie S2). (Lower Left) Kymograph of the area selected by the white rectangle over time. (B) Localization of RhoH–mRFP in transiently transfected human T-cell blast migrating in response to CXCL12 is shown every minute. The distribution of fluorescence intensity is shown in a pseudocolor scale, from low (black) to high (red). (Lower panels, Movie S4). (C) Representative AL-57 (activation-dependent LFA-1 mAb) staining (green) of RhoH–mRFP (red) transfected cells. (D and E) Cell conjugates between transiently transfected Jurkat cells and SEE-loaded Raji cells were tracked by contacts between APC and mRFP+ cells and then the localization of mRFP assessed, four representative images each. Localization of mRFP was quantified by the frequency of conjugates displaying a diffuse localization pattern of mRFP versus mRFP recruited toward the APC (E). (F) Total LFA-1 (TS2/4, red) versus active LFA-1 (AL57, green) in a representative T:APC conjugate reveals that active LFA-1 is also found at the T:APC interface.
Enhanced Migration of RhoH-Deficient T Cells.
To assess whether there is a corresponding increase in migration in the absence of RhoH, in vitro migration assays on ICAM-1– and CCL21-coated surface were performed with sorted naïve T cells from WT or RhoH−/− cells (Fig. 5 A and B and Fig. S8). A significant increase in migration velocity and meandering index was detected among RhoH−/− cells (Fig. 5 A and B and Movies S5 and S6). The changes in migration do not track with altered levels of chemokine receptors or integrins (Fig. S8). Thus, the absence of RhoH leads to faster migration and less wandering of the cells.
Fig. 5.

RhoH-deficient T cells migrate in a more directional manner and have altered homing to lymphoid organs. (A and B) Migration of naïve T cells isolated from wild type (WT) or RhoH-deficient (RhoH−/−) mice were assessed on dishes coated with ICAM-1 and CCL21 (Movies S5 and S6). Shown are velocity and meandering index from one representative experiment of three (A). Cell migration plots of cell tracks overlaid with origins from center are shown (B). (C and D) Adoptive transfer of cells 1 d before treatment with FTY720 (C) or anti-CD62L (D) for 12 or 20 h, respectively. The fold change in the homing index, as determined by [carboxy fluorescein succinimidyl ester (CFSE)+sample/PKH+sample]/(CFSE+input/PKH+input) at 12 h over 0 h. These data are means ± SEM, n = 4 experiments with 13 mice/time point for FTY720 and n = 3 with 12 mice/time point for anti-CD62L, peripheral lymph node (pLN), and mesenteric lymph node (mLN). Statistical significance P < 0.05.
Competitive homing assays were done to assess whether this translates into altered homing in vivo. The ratio of RhoH−/−: WT cells from various organs were assessed 1 d posttransfer (Fig. S9). RhoH−/− cells have altered homing among lymphoid tissues, which may be due to faster transit times such that RhoH−/− cells enter and depart lymph nodes more quickly. To assess the rate at which cells enter a lymph node, we used FTY720 as a blockade for cell egress to detect the accumulation of cells in lymph nodes. The fold change in the homing index was greater in the presence of FTY720 consistent with RhoH−/− T cells entering in larger numbers than WT during this time frame (Fig. 5C). As retention signals by CCR7 versus S1P sensing determine the homeostatic retention time of T cells within the lymph node (17), we also assessed the importance of RhoH expression for egress from lymph nodes. There were no differences in cell egress between WT and RhoH−/− mice (Fig. 5D) at a 20-h time point, although this visualizes the sum of cell departure within the time frame rather than the kinetics by which those cells departed. Overall, these data suggest that RhoH expression effects chemokine responsiveness for entry or movement within lymph node rather than egress.
Altered RhoH Expression During T-Cell Activation.
T-cell motility to distinct locations within the lymph node at various times subsequent to antigen exposure are necessary for effector functions to be performed, including help for CD8 and B-cell responses, as well as for efficient egress. Although maintaining RhoH expression is necessary for stable T:APC conjugation and proximal TCR signaling, at the same time, our data showed that high RhoH expression level inhibits chemokine-induced T-cell migration. Therefore, spatial regulation of RhoH may enable initial T-cell activation, yet RhoH function must then be limited to allow T cells to leave APC and migrate following chemokine signals. To assess the expression levels of RhoH during T-cell activation, the levels of mRNA were measured (14). RhoH mRNA levels were rapidly reduced following TCR triggering and before the first cell division (Fig. 6 A and C), whereas IL-2, an indicator of activation, was up-regulated (Fig. 6 B and D). In addition, there was a further significant reduction in RhoH mRNA as cells went through later divisions (Fig. 6A). Also, T-cell activation induces degradation of RhoH protein (18). Limited synthesis of more protein alongside degradation of the existing protein would combine to result in an overall reduction in RhoH expression. This may serve as a mechanism to turn off activation and enable cells to sense and respond to the chemokine milieu to adopt a migratory phenotype once more.
Fig. 6.

RhoH expression decreases at the transcriptional level as T-cell activation progresses. CFSE+ T cells were stimulated with plate bound CD3/CD28 and assayed for RhoH (A) and IL-2 (B) transcripts normalized to GAPDH. Unstimulated cells (T0), undivided (0), one to two divisions (1/2), three to four divisions (3/4), and five divisions onward (5+) are shown as a fold change relative to unstimulated. (C and D) OT-II TCR transgenic CD4 T cells were stimulated with OVA323–339 presented by irradiated RhoH−/− splenocytes and RNA was isolated at indicated times. RNA analysis by the comparative CT method was normalized to CD3d, a T-cell–specific gene. Data are means ± SEM, n = 3 and considered statistically significant by Student’s t test, P < 0.05.
Discussion
The crucial role of Rap1 in LFA-1 activation during T-cell migration and priming is well established (2, 5, 19). It was, however, unexpected to find disparate downstream effects of RhoH overexpression on chemokine and TCR-mediated Rap1 activation. In this study, we have demonstrated that RhoH impacts the outcome of signals received by a T cell as a negative regulator of chemokine-mediated migration while promoting TCR-signal–induced conjugate formation with APCs. Our data establish a previously unappreciated role for RhoH in the activation of Rap1 and provide insight into the dynamic regulation of LFA-1 adhesion in the stop and go movement of lymphocytes. Thus, expression of RhoH may serve as a key functional determinant that dictates T-cell migration, yet may maintain a naïve T cell in a state whereby it may rapidly respond upon cognate antigen recognition.
It was reported that the transcription level of RhoH, given the lack of typical GTP exchange mechanisms (9, 14), controls regulation of its cellular function. Although maintenance of RhoH expression in naïve T cells is essential for proximal TCR signaling (12), down-regulation of RhoH mRNA is shown in Jurkat cells treated with phorbol myristate acetate (PMA) and Th1 cells stimulated through the TCR (14). Consistent with the study, our data show rapid down-regulation of RhoH mRNA upon antigen stimulation of naïve T cells and further reduction as cells proceed through division. This may provide a mechanism for T cells to become more responsive to chemokine-induced migration (by removing an inhibitory signal for chemotaxis) as well as a tuning mechanism to control T-cell activation (by removing a positive signal for conjugate formation).
In the presence of chemokine stimulation, RhoH inhibits LFA-1 adhesion, whereas Rap1 activation enhances LFA-1 affinity. Therefore, recruitment of activated Rap1 (a positive regulator for LFA-1 activation) to the front and redistribution of RhoH (a negative regulator) to the rear of cells may be essential for the spatial regulation of LFA-1 activation and deactivation during T-cell migration. In the presence of TCR stimulation, however, RhoH promotes Rap1 activation, thus RhoH, Rap1, and active LFA-1 are located toward the APC. The combination of reduced RhoH expression and differential localization determine the outcome when T cells receive migratory signals or reencounter activating signals.
The mechanisms by which RhoH differentially regulates Rap1 activation remain unclear. RhoH displays inhibitory effects on many of the pathways induced by other Rho GTPases, including NF-κB, MAP kinase p38 (14), and Zap70 (9), as well as LFA-1–mediated adhesion (8). One way in which RhoH may perform dual functions in T-cell migration and activation is by altering levels of other Rho GTPases (10, 14, 15). A role for RhoH downstream of CXCR4 signaling is identified by a failure to activate PAK1 in response to strong stimulation via CXCR4 in the absence of RhoH (13), further linking RhoH with regulation of the cytoskeleton by Rac GTPases. However, constitutive activation of Rac1 increases proliferation of thymocytes in DN3 and an accelerated transition from DN3 to DN4 (20), whereas RhoH-deficient cells reveal diminished signaling and a block at this stage even in the presence of activated Rac1 (21), suggesting that regulation by RhoH is more complicated than only cytoskeletal effects. Defects in thymic selection, alongside studies of proximal TCR signals implicate RhoH as an important player in conjunction with Zap70 (9, 21). Zap70 is also reported to be involved in outside-in signaling for high-affinity LFA-1–mediated adhesion (22). Zap70 binds to immunoreceptor tyrosine-based activation motifs (ITAMs) of the CD3 chains by its tandem SH2 domains. Interestingly, RhoH contains an ITAM-like motif (9), raising the possibility that, given appropriate circumstances, Zap70 may interact with RhoH. Whether any of these mechanisms are a component of the effects of RhoH on Rap1 activation remain to be determined.
Our data suggest that RhoH expression serves as a sensing mechanism for T cells to stop and go in response to the signals they have received. Intravital imaging studies describe several distinct phases of T-cell motility upon antigen challenge (23). The exact dynamics of these phases may differ depending upon the cell numbers, route of antigen challenge, antigen quantity and quality, and presence of costimulatory signals, but the ability of T cells to switch between periods of minimal motility and highly migratory states is thought essential for full T-cell activation and effector function (24, 25). RhoH expression levels may remain high during early phases after cell entry into a lymph node to enhance TCR-mediated adhesion by LFA-1. The distribution of RhoH–mRFP is altered as a cell switches from migration to a phase of activation. A consequence of T-cell activation is a decrease in the level of RhoH expression of which a sufficient reduction leads to an enhanced ability of the cell to respond to chemokine-driven migratory signals. Thus, activated cells regain a high level of motility, undergo division, perform effector functions in other locations within a lymph node, as well as access sites of egress to depart and perform effector functions at the site of injury (24). Furthermore, the efficiency of down-modulation of RhoH expression could impact T:APC dwell times thus strong signals lead to rapid loss of mRNA, whereas weaker TCR engagements or low antigen dosage may lead to slower kinetics of mRNA reduction. The quality of the TCR interaction would then change the RhoH/Rap1 balance. Rapid regulation of RhoH would enable a cell to counterbalance overstimulation or alter the threshold of activation. An inappropriate T:APC dwell time may lead to death by overactivation, whereas lack of RhoH may result in such a short contact time as to prevent a primary response. A possible drawback in the current study, however, is that the majority of Rap1 assay reported here has been performed either after overexpressing RhoH protein or by using RhoH-deficient T cells. Therefore, we cannot completely exclude the possibility that some of the outcomes of our experiment may have resulted from cellular reactions caused by the nonphysiological protein expression level. Suppression of endogenous RhoH with small interfering RNA (siRNA) would be an excellent alternative approach. Unfortunately, we were unable to find an antibody showing a specific RhoH signal in Western blot. Therefore, without a proper assay to confirm the protein knockdown, siRNA data may not directly support our conclusion because we cannot prove whether the experimental data resulted entirely from the knockdown of RhoH or were due to nonspecific reactions by the siRNA treatment. Another constraint in the RhoH study that prevented us from fully analyzing Rap1 activation in RhoH-deficient T cells is the limited number of naïve T cells that we can isolate from the RhoH−/− mouse. As shown in the current study and by other investigators (9), RhoH regulates key signals in T-cell functions and development. Thus, RhoH−/− mice manifest striking defects at two important T-cell development transitions, DN3 to DN4 and DP to SP (9). Because of these defects, RhoH−/− mice suffer severe T-cell lymphopenia. Due to this phenotype, comprehensive analysis of RhoH-deficient T cells for their Rap1 activation in various conditions has not been possible.
The mechanisms of stop and go signals are alluded to in many studies, including the balance between the strength of TCR and migratory signals received. In vitro some chemokine signals override ongoing TCR signals, whereas for others, the TCR signal is dominant (26). Furthermore, chemokine-receptor internalization and desensitization may allow TCR ligation to occur. The affinity of TCR is 1–100 mM (27), whereas chemokine-receptor affinity, such as CCR7 and CXCR4, is 3–15 nM (28). Some chemokine receptors also serve in a costimulatory capacity (29). Chemokine-induced integrin adhesiveness may destabilize the IS if this signal occurs distal from the site of TCR engagement or it may synergize with TCR to enhance activation if ligated within the IS. Up-regulation of negative regulators of activation such as CTLA4 may also alter this balance. Indeed CTLA4 expression has a negative impact on the TCR stop signal and coligation of CD3 and CTLA4 provides a positive migratory cue in vitro (30). The development of autoimmunity in CTLA4-deficient mice may, in part, be due to the sustained dwell time of T:APC interactions leading to activation by self-antigens. One intracellular mechanism to regulate stop and go signals is the counterplay between PKCθ and Wiskott–Aldrich syndrome protein (WASP) (31). PKCθ causes periodic disruptions of the IS in vitro and PKCθ−/− T cells make more stable contacts with APC in vivo. Cytoskeletal-associated WASP protein is necessary to counteract PKCθ and stabilize the IS (31). The mechanisms of stop and go signaling, including RhoH, will be key determinants in the functional outcome of a CD4 response in the face of antigenic challenge.
Materials and Methods
Primary T cells were isolated from whole blood by the polymorph layering technique and cultured (32). Mouse CD4 T cells were isolated by negative selection, and T-cell blasts from DO11.10 or OT-II were generated (33, 34). Transient transfection was performed per manufacturer protocol (Lonza). Detailed methods may be found in SI Materials and Methods. The Human Research Studies Review Board and University of Rochester Committee on Animal Care approved these studies.
Supplementary Material
Acknowledgments
We thank Rachel Spoonhower, Pranita Sarangi, and Nicole Morin for technical assistance; Dr. Jim Miller for reagents and manuscript comments; Dr. Andrea Sant for the Mel-14 hybridoma; and Nathan Laniewski for cell sorting. This project was supported by National Institutes of Health Grants HL087088 and HL018208 (to M.K.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1114214109/-/DCSupplemental.
References
- 1.Cyster JG. Chemokines, sphingosine-1-phosphate, and cell migration in secondary lymphoid organs. Annu Rev Immunol. 2005;23:127–159. doi: 10.1146/annurev.immunol.23.021704.115628. [DOI] [PubMed] [Google Scholar]
- 2.Hogg N, Laschinger M, Giles K, McDowall A. T-cell integrins: More than just sticking points. J Cell Sci. 2003;116:4695–4705. doi: 10.1242/jcs.00876. [DOI] [PubMed] [Google Scholar]
- 3.Vigl B, et al. Tissue inflammation modulates gene expression of lymphatic endothelial cells and dendritic cell migration in a stimulus-dependent manner. Blood. 2011;118:205–215. doi: 10.1182/blood-2010-12-326447. [DOI] [PubMed] [Google Scholar]
- 4.Bromley SK, et al. The immunological synapse. Annu Rev Immunol. 2001;19:375–396. doi: 10.1146/annurev.immunol.19.1.375. [DOI] [PubMed] [Google Scholar]
- 5.Dustin ML, Bivona TG, Philips MR. Membranes as messengers in T cell adhesion signaling. Nat Immunol. 2004;5:363–372. doi: 10.1038/ni1057. [DOI] [PubMed] [Google Scholar]
- 6.Bos JL, de, Rooij J, Reedquist KA. RAP1 signalling: Adhering to new models. Nat Rev Mol Cell Biol. 2001;2:369–377. doi: 10.1038/35073073. [DOI] [PubMed] [Google Scholar]
- 7.Katagiri K, Maeda A, Shimonaka M, Kinashi T. RAPL, a Rap1-binding molecule that mediates Rap1-induced adhesion through spatial regulation of LFA-1. Nat Immunol. 2003;4:741–748. doi: 10.1038/ni950. [DOI] [PubMed] [Google Scholar]
- 8.Cherry LK, Li X, Schwab P, Lim B, Klickstein LB. RhoH is required to maintain the integrin LFA-1 in a nonadhesive state on lymphocytes. Nat Immunol. 2004;5:961–967. doi: 10.1038/ni1103. [DOI] [PubMed] [Google Scholar]
- 9.Gu Y, et al. RhoH GTPase recruits and activates Zap70 required for T cell receptor signaling and thymocyte development. Nat Immunol. 2006;7:1182–1190. doi: 10.1038/ni1396. [DOI] [PubMed] [Google Scholar]
- 10.Gu Y, Jasti AC, Jansen M, Siefring JE. RhoH, a hematopoietic-specific Rho GTPase, regulates proliferation, survival, migration, and engraftment of hematopoietic progenitor cells. Blood. 2005;105:1467–1475. doi: 10.1182/blood-2004-04-1604. [DOI] [PubMed] [Google Scholar]
- 11.Gu Y, Zheng Y, Williams DA. RhoH GTPase: A key regulator of hematopoietic cell proliferation and apoptosis? Cell Cycle. 2005;4:201–202. doi: 10.4161/cc.4.2.1490. [DOI] [PubMed] [Google Scholar]
- 12.Wang H, Zeng X, Fan Z, Lim B. RhoH modulates pre-TCR and TCR signalling by regulating LCK. Cell Signal. 2011;23:249–258. doi: 10.1016/j.cellsig.2010.09.009. [DOI] [PubMed] [Google Scholar]
- 13.Wang H, Zeng X, Fan Z, Lim B. RhoH plays distinct roles in T-cell migrations induced by different doses of SDF1 alpha. Cell Signal. 2010;22:1022–1032. doi: 10.1016/j.cellsig.2010.02.005. [DOI] [PubMed] [Google Scholar]
- 14.Li X, et al. The hematopoiesis-specific GTP-binding protein RhoH is GTPase deficient and modulates activities of other Rho GTPases by an inhibitory function. Mol Cell Biol. 2002;22:1158–1171. doi: 10.1128/MCB.22.4.1158-1171.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chae HD, Lee KE, Williams DA, Gu Y. Cross-talk between RhoH and Rac1 in regulation of actin cytoskeleton and chemotaxis of hematopoietic progenitor cells. Blood. 2008;111:2597–2605. doi: 10.1182/blood-2007-06-093237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gérard A, Mertens AE, van der Kammen RA, Collard JG. The Par polarity complex regulates Rap1- and chemokine-induced T cell polarization. J Cell Biol. 2007;176:863–875. doi: 10.1083/jcb.200608161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pham TH, Okada T, Matloubian M, Lo CG, Cyster JG. S1P1 receptor signaling overrides retention mediated by G alpha i-coupled receptors to promote T cell egress. Immunity. 2008;28:122–133. doi: 10.1016/j.immuni.2007.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schmidt-Mende J, Geering B, Yousefi S, Simon HU. Lysosomal degradation of RhoH protein upon antigen receptor activation in T but not B cells. Eur J Immunol. 2010;40:525–529. doi: 10.1002/eji.200939556. [DOI] [PubMed] [Google Scholar]
- 19.Kinashi T, Katagiri K. Regulation of immune cell adhesion and migration by regulator of adhesion and cell polarization enriched in lymphoid tissues. Immunology. 2005;116:164–171. doi: 10.1111/j.1365-2567.2005.02214.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gomez M, Tybulewicz V, Cantrell DA. Control of pre-T cell proliferation and differentiation by the GTPase Rac-I. Nat Immunol. 2000;1:348–352. doi: 10.1038/79808. [DOI] [PubMed] [Google Scholar]
- 21.Dorn T, et al. RhoH is important for positive thymocyte selection and T-cell receptor signaling. Blood. 2007;109:2346–2355. doi: 10.1182/blood-2006-04-019034. [DOI] [PubMed] [Google Scholar]
- 22.Evans R, Lellouch AC, Svensson L, McDowall A, Hogg N. The integrin LFA-1 signals through ZAP-70 to regulate expression of high-affinity LFA-1 on T lymphocytes. Blood. 2011;117:3331–3342. doi: 10.1182/blood-2010-06-289140. [DOI] [PubMed] [Google Scholar]
- 23.Bousso P. T-cell activation by dendritic cells in the lymph node: Lessons from the movies. Nat Rev Immunol. 2008;8:675–684. doi: 10.1038/nri2379. [DOI] [PubMed] [Google Scholar]
- 24.Mempel TR, Henrickson SE, Von Andrian UH. T-cell priming by dendritic cells in lymph nodes occurs in three distinct phases. Nature. 2004;427:154–159. doi: 10.1038/nature02238. [DOI] [PubMed] [Google Scholar]
- 25.Bromley SK, Mempel TR, Luster AD. Orchestrating the orchestrators: Chemokines in control of T cell traffic. Nat Immunol. 2008;9:970–980. doi: 10.1038/ni.f.213. [DOI] [PubMed] [Google Scholar]
- 26.Bromley SK, Peterson DA, Gunn MD, Dustin ML. Cutting edge: Hierarchy of chemokine receptor and TCR signals regulating T cell migration and proliferation. J Immunol. 2000;165:15–19. doi: 10.4049/jimmunol.165.1.15. [DOI] [PubMed] [Google Scholar]
- 27.Davis SJ, Davies EA, Tucknott MG, Jones EY, van der Merwe PA. The role of charged residues mediating low affinity protein-protein recognition at the cell surface by CD2. Proc Natl Acad Sci USA. 1998;95:5490–5494. doi: 10.1073/pnas.95.10.5490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ricart BG, John B, Lee D, Hunter CA, Hammer DA. Dendritic cells distinguish individual chemokine signals through CCR7 and CXCR4. J Immunol. 2011;186:53–61. doi: 10.4049/jimmunol.1002358. [DOI] [PubMed] [Google Scholar]
- 29.Molon B, et al. T cell costimulation by chemokine receptors. Nat Immunol. 2005;6:465–471. doi: 10.1038/ni1191. [DOI] [PubMed] [Google Scholar]
- 30.Schneider H, et al. Reversal of the TCR stop signal by CTLA-4. Science. 2006;313:1972–1975. doi: 10.1126/science.1131078. [DOI] [PubMed] [Google Scholar]
- 31.Sims TN, et al. Opposing effects of PKCtheta and WASp on symmetry breaking and relocation of the immunological synapse. Cell. 2007;129:773–785. doi: 10.1016/j.cell.2007.03.037. [DOI] [PubMed] [Google Scholar]
- 32.Morin NA, et al. Nonmuscle myosin heavy chain IIA mediates integrin LFA-1 de-adhesion during T lymphocyte migration. J Exp Med. 2008;205:195–205. doi: 10.1084/jem.20071543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sanchez-Lockhart M, et al. Cutting edge: CD28-mediated transcriptional and posttranscriptional regulation of IL-2 expression are controlled through different signaling pathways. J Immunol. 2004;173:7120–7124. doi: 10.4049/jimmunol.173.12.7120. [DOI] [PubMed] [Google Scholar]
- 34.Zuckerman LA, Pullen L, Miller J. Functional consequences of costimulation by ICAM-1 on IL-2 gene expression and T cell activation. J Immunol. 1998;160:3259–3268. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.